

Summary
The occupancy of endothelial protein C receptor (EPCR) by protein C switches the protease activated receptor 1 (PAR-1)-dependent signaling specificity of thrombin from a permeability enhancing to a barrier protective response in vascular endothelial cells. In this study, the modulatory effects of thrombin and thrombin receptor agonist peptides (TRAP) on TNF-α-stimulated HUVECs in the absence and presence of the catalytically inactive protein C-S195A were evaluated by monitoring the expression of cell surface adhesion molecules (VCAM-1, ICAM-1 and E-selectin), adhesion of freshly isolated neutrophils to cytokine-stimulated endothelial cells, regulation of the Rho family of small GTPases and the activation of nuclear factor-κB (NF-κB) pathway. The analysis of results indicate that both thrombin and TRAP initiate proinflammatory responses in endothelial cells, thus neither PAR-1 agonist influenced the proinflammatory effects of TNF-α in the absence of the protein C mutant. Interestingly, however, the occupancy of EPCR by the protein C mutant switched the PAR-1-dependent signaling specificity of thrombin, thus leading to thrombin inhibition of the expression of all three adhesion molecules as well as the binding of neutrophils to TNF-α-activated endothelial cells. Furthermore, similar to activated protein C, both thrombin and TRAP activated Rac1 and inhibited the activation of RhoA and NF-κB pathways in response to TNF-α in cells pretreated with protein C-S195A. Based on these results we conclude that when EPCR is ligated by protein C, the cleavage of PAR-1 by thrombin initiates antiinflammatory responses, thus leading to activation of Rac1 and inhibition of RhoA and NF-κB signaling cascades in vascular endothelial cells.
Keywords: thrombin, EPCR, PAR-1, protein C, cell adhesion, NF-κB, Rho
Introduction
Thrombin is the final protease of the clotting cascade that cleaves fibrinogen to form insoluble fibrin clots to stop the bleeding at the site of a vascular injury (1). In addition to its procoagulant role, thrombin also functions as a potent anticoagulant when it binds to its endothelial cell receptor thrombomodulin to activate protein C to activated protein C (APC) (2). Thrombin is an allosteric enzyme with an elaborate structure that exerts diverse biological effects by interacting with many soluble molecules in circulation (3, 4) as well as with various receptors on the surface of vascular and nonvascular cells (5, 6). Recent results from several laboratories have indicated that the direct cellular effects of thrombin are primarily mediated through its interaction with and subsequent cleavage of a sub-family of G-protein coupled receptors called protease activated receptors (PARs) that are expressed on target cells in various organs (5). So far, four members of the PAR family (PAR-1, PAR-2, PAR-3 and PAR-4) have been cloned and characterized (5). The protease cleavage of PARs on various cell types exposes new N-termini in the exodomain of these receptors that bind intramolecularly to the second membrane-spanning loop of the receptors, thereby activating them and initiating intracellular signaling events under different pathophysiological conditions (5). Since human umbilical vein endothelial cells (HUVECs) express all four PARs (7–9), these cells have been extensively used in in vitro model systems to understand the PAR-cleavage dependent intracellular signaling mechanisms of thrombin (also other proteases in the circulation) and to identify the down-stream signaling molecules that may be modulated by the activation of each receptor type (5, 9, 10). The consensus that has emerged, based on numerous studies using HUVECs and other cell types, is that the cleavage of PAR-1 by thrombin initiates potent inflammatory responses in endothelial cells (7, 11–13), including the up-regulation of cell surface adhesion molecules (14), induction of hyperpermeability (15) and the activation of both RhoA GTPase and nuclear factor κB (NF-κB) pathways (12, 16). Interestingly, using the same cellular model systems, it has been demonstrated that APC inhibits the thrombin-mediated up-regulation of these inflammatory pathways by cleavage of the same receptor (PAR-1) in stimulated endothelial cells (12, 13, 16, 17). The PAR-1-dependent protective cellular effects of APC, which are dependent on the interaction of the protease with endothelial protein C receptor (EPCR), are believed to contribute to the beneficial therapeutic effect of APC in reducing the rate of mortality in the severely septic patients (17, 18). The mechanism of the opposing PAR-1 cleavage-dependent cellular signaling responses that are evoked by both thrombin and APC is not understood. Noting that thrombin can cleave PAR-1 with 3–4 orders of magnitude higher efficiency than APC to elicit proinflammatory responses in endothelial cells (5, 19), the hypothesis that APC can exert an antiinflammatory response through the cleavage of the same receptor has stimulated intense debate within the scientific community as to whether or not the protective effect of APC can be mediated through the cleavage of PAR-1 (20, 21).
In a recent study, we demonstrated that both PAR-1 and EPCR are associated with caveolin-1 within lipid rafts of endothelial cells (22) and that the occupancy of EPCR by either APC or a sub-physiological concentration of the zymogen protein C leads to dissociation of EPCR from caveolin-1 that culminates in the recruitment of PAR-1 to a protective signaling pathway independent of the protease that cleaves the receptor (8). Specifically, we discovered that the occupancy of EPCR changes the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in stimulated cultured endothelial cells (8). In this study, we monitored the PAR-1 cleavage-dependent regulation of down-stream inflammatory molecules by both APC and thrombin in response to TNF-α in endothelial cells treated with the catalytically inactive S195A mutant of protein C. The results indicate that when EPCR is occupied by its ligand, the cleavage of PAR-1 by either thrombin or APC inhibits all in vitro indices of inflammatory responses in TNF-α-stimulated endothelial cells, thus both proteases inhibit the expression of cell surface adhesion molecules, prevent the binding of freshly isolated neutrophils to activated endothelial cells, and inhibit the activations of both RhoA and NF-κB pathways mediated by the inflammatory cytokine. Based on these results, we conclude that the cleavage of PAR-1 by thrombin in intact endothelium expressing EPCR would initiate potent protective intracellular responses in the presence of physiological concentrations of the zymogen protein C.
Materials and methods
Expression and purification of wild-type protein C and the Ser-195 to Ala substitution mutant of protein C (PC-S195A) have been described (8). The antibody blocking the activation of PAR-1 (H-111) was purchased from Santa Cruz Biologics (Santa Cruz, CA). The function-blocking anti-EPCR antibody (clone RCR-252) was purchased from Cell Sciences (Canton, MA). Both antibodies were used at 25 µg/mL. Tumor necrosis factor-α (TNF-α) was purchased from R&D System (Minneapolis, MN). The thrombin receptor agonist peptides SFLLRN and TFLLRN were purchased from Bachem Bioscience (Torrance, CA). The anti-NF-κB p65 antibody detecting the endogenous levels of total NF-κB p65 and the anti-phospho-NF-κB p65 antibody detecting the activated Ser-536 phosphorylated form of NF-κB p65 were purchased from Cell Signaling Tech. (Danvers, MA). The kits for monitoring the activation of RhoA and Rac1 GTPases were obtained from Cytoskeleton (Denver, CO).
Adhesion assay and analysis of expression of cell adhesion molecules
Freshly isolated neutrophil adherence to thrombin or TNF-α-stimulated immortalized human umbilical vein endothelial (EA.hy926) cells (provided by Dr. C. Edgell from University of North Carolina at Chapel Hill, NC) in the absence and presence of PC-S195A (80 nM) was evaluated by the fluorescent labeling of neutrophils as described (23, 24). The expression of vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1) and E-selectin on endothelial cells was determined by a whole-cell ELISA as described previously (23). In all experiments described below cells were first treated with PC-S195A followed by stimulation with the proinflammatory molecules. In the presence of function-blocking antibodies, cells were first incubated with the appropriate antibody (25 µg/mL) for 30 min, followed by incubation with PC-S195A for 30 min and then stimulation with proinflammatory molecules.
Permeability assay
EA.hy926 cell permeability in response to PAR-1 agonist peptides SFLLRN and TFLLRN (0–500 µM) in the absence and presence PC-S195A (80 nM) was quantitated by the spectrophotometric measurement of the flux of Evans blue-bound albumin across functional EA.hy926 cell monolayers using a modified 2-compartment chamber model as described (8). Prior to stimulation with the agonist peptides, cells were first treated with PC-S195A for 30 min.
Measuring RhoA and Rac1 activation
EA.hy926 cells were grown to confluence in 6-well culture dishes and starved overnight in the serum free medium. Determination of RhoA and Rac1 activations (RhoA-GTP or Rac1-GTP) was monitored using commercially available kits. Briefly, conditioned cells (see figure legends) were lysed using a cell lysis buffer provided by the manufacturer. After determining the protein concentration by a Bio-Rad assay, 50 µg of lysates were saved for western quantitation of the total RhoA. The remaining sample was incubated with 20 µg of the GST fusion protein RBD (rhotekin Rho-binding domain), which is bound to the colored glutathione-sepharose beads, at 4 °C with rotation for 1 h. The RBD protein motif binds specifically to the active GTP-bound form of RhoA. Beads were washed, resuspended in loading buffer and proteins were separated on 12% SDS-PAGE followed by transferring to a PVDF membrane and western blotting using an anti-RhoA monoclonal antibody as described by the manufacturer. The same procedures were employed to monitor Rac1 activation except that appropriate cellular lysates were incubated with GST-PAK PBD (Rac1 effector protein, p21 activated kinase 1) beads that bind specifically to the active GTP-bound form of Rac1. The total and activated Rac1 were detected by western blotting using an anti-Rac1 monoclonal antibody provided in the kit as described (25).
Measuring NF-κB activation
EA.hy926 cells were grown to confluence in 6-well culture dishes and subjected to starvation overnight in the serum free medium. The NF-κB pathway activation in the conditioned cell lysates (treated with different PAR-1 agonists and activated by TNF-α as described under figure legends) was monitored by western blotting employing two antibodies that are specific for either NF-κB p65 (as an index of total cellular NF-κB p65) or its phosphorylated form (as an index of the NF-κB pathway activation).
Statistical Analysis
Results are expressed as mean ±SEM, and t-Tests (paired or independent) were used to assess data. Differences were considered statistically significant at p values of <0.05. Statistics were performed using the software package SPSS version 14.0 (SPSS, Chicago, IL). All experiments were repeated at least three times. All experiments were reproduced at least 2–3 times.
Results
Thrombin Inhibits TNF-α-mediated Leukocyte Adhesion to Endothelial Cells if EPCR is Occupied by Protein C
It is known that APC suppresses the TNF-α- or thrombin-mediated interaction of neutrophils with endothelial cells by inhibiting the expression of cell surface adhesion molecules VCAM-1, ICAM-1 and E-selectin (14,23). Consistent with previous results, we discovered that both thrombin (20 nM) and TNF-α (10 ng/mL) up-regulated the expression of all three cell surface adhesion molecules in EA.hy926 cells by a concentration and time dependent manner and APC exerted a potent inhibitory effect that reached its maximum level following 4 h of APC treatment (data not presented). We investigated the signaling effect of thrombin on the expression of the adhesion molecules as well as on the interaction of neutrophils with the TNF-α-stimulated endothelial cells under conditions in which EPCR was occupied by PC-S195A. Interestingly, the thrombin-mediated expression of all three adhesion molecules was inhibited if EA.hy926 cells were incubated with PC-S195A prior to their activation by thrombin (Fig. 1A). Thus, the expression levels of all three adhesion molecules in the thrombin-activated endothelial cells in the presence of PC-S195A was decreased to the control level [unstimulated PC-S195A treated cells] (Fig. 1A). The thrombin + PC-S195A mediated down-regulation of cell adhesion molecules required the activation of PAR-1 by thrombin since a function-blocking anti-PAR-1 antibody abrogated the effect of thrombin (Fig. 1A). Consistent with an antiinflammatory role for PAR-1 activation by thrombin in the presence of PC-S195A, the PAR-1 agonist peptides also potently inhibited the proinflammatory effect of TNF-α in endothelial cells pretreated with the protein C mutant zymogen (Fig. 1B, shown for TFLLRN only). Further studies were initiated to determine whether the inhibition of the expression of these adhesion molecules by thrombin correlates with the inhibition of binding of neutrophils to TNF-α-stimulated EA.hy926 cells. The results presented in Fig. 1C demonstrate that, similar to APC, thrombin potently blocks the adhesion of neutrophils to TNF-α-stimulated endothelial cells by EPCR and PAR-1-dependent pathways if the cell monolayers were treated with PC-S195A prior to incubation with thrombin. These results clearly suggest that the cleavage of PAR-1 by thrombin elicits antiinflammatory responses in endothelial cells when EPCR is occupied by protein C. Essentially identical results were obtained if primary HUVECs (data not presented) or primary human pulmonary artery endothelial cells were used in these experiments (26). Thus, the occupancy of EPCR changes the PAR-1-dependent signaling specificity of thrombin in endothelial cells of both arterial and venular beds (26).
Figure 1. Analysis of inhibition of thrombin or TNF-α-induced adhesion molecules in EA.hy926 cells treated with PC-S195A.

(A) The expression of adhesion molecules VCAM-1 (white bars), ICAM-1 (grey bars) and E-selectin (black bars) in confluent EA.hy926 cells was analyzed by ELISA after treating monolayers with either APC (20 nM) or thrombin (20 nM) in the absence and presence of PC-S195A (80 nM) and a function-blocking anti-PAR-1 antibody. Cells were first treated with PC-S195A for 30 min and then treated with APC or thrombin for 4 h. In the presence of PAR-1 function-blocking antibody, cells were first treated with the antibody for 30 min, followed by PC-S195A for 30 min and finally incubation with thrombin for 4 h. The data in the first column represents cells treated with thrombin alone. The data in the last column PC-S195A (control) represents cells treated with PC-S195A alone. (B) The same as (A) except that the TNF-α-mediated (10 ng/mL for 4 h) expression of adhesion molecules was analyzed before or after treating the monolayers with PC-S195A (80 nM) followed by TFLLRN (100 µM for 4 h). The data in the last column (PC-S195A) suggests that the mutant zymogen by itself does not exhibit a protective activity in response to TNF-α. (C) TNF-α-mediated (10 ng/mL for 4 h) adhesion of freshly isolated neutrophils to endothelial cells was analyzed after sequential treatment of monolayers with either APC (20 nM for 4 hr) or thrombin (20 nM for 4 h) ± PC-S195A (80 nM) in the absence and presence of function-blocking antibodies to either PAR-1 or EPCR as described in panel (A).
The barrier disruptive and protective effects of thrombin are mediated through modulation of the Rho family of GTPases
Both PAR-1 agonist peptides SFLLRN and TFLLRN elicited a barrier disruptive response in EA.hy926 cells that could be effectively reversed to a potent protective response if the endothelial cells were treated with PC-S195A prior to their stimulation by the PAR-1 agonist peptides (Figs. 2A and B). It has been previously demonstrated that the barrier disruptive effect of thrombin and the opposite barrier protective effect of APC are mediated through these proteases differentially modulating the cytoskeletal proteins actin and myosin via the activation of the Rho family of small GTPases (12, 27). Thus, thrombin and other proinflammatory molecules increase the levels of the phosphorylated myosin light chain through the activation of RhoA, thereby inducing the formation of actin stress fibers that can lead to cellular contraction and disruption of the endothelial barrier function (12, 27). On the other hand, the barrier protective effect of APC is mediated through the APC activation of another member of the Rho family of GTPases called Rac1 whose activity is associated with cytoskeletal remodeling that counteracts the barrier disruptive effects of the proinflammatory molecules (Fig. 3A, lane 10) (12,27). To determine whether the ligation of EPCR by protein C alters the down-stream signaling specificity of thrombin, the ability of both thrombin and APC to modulate the two proteins of the Rho family of GTPases was analyzed in the TNF-α-stimulated endothelial cells. Consistent with the hypothesis that protein C occupancy of EPCR switches the signaling specificity of thrombin from a proinflammatory to an antiinflammatory response, thrombin in the presence of PC-S195A, but not in its absence, activated Rac1 and inhibited the activation of RhoA in TNF-α-stimulated endothelial cells (Fig. 3A, lanes 4 and 5). The switch in the signaling specificity of thrombin was both PAR-1 and EPCR dependent since the function-blocking antibodies against either receptor eliminated the protective effect of thrombin (Fig. 3A, lanes 8 and 9). Further studies with both PAR-1 agonist peptides SFLLRN and TFLLRN confirmed these results (Fig. 3B), clearly demonstrating that the ligation of EPCR with the protein C mutant reverts the RhoA-dependent proinflammatory activity of thrombin to a Rac1-dependent antiinflammatory response. The rationale for using both agonist peptides was based on the observation that, in addition to the activation of PAR-1, SFLLRN can also activate PAR-2 (28). On the other hand, TFLLRN is known to be specific for PAR-1 activation with no effect on PAR-2 (28). As expected, the anti-PAR-1 antibody did not influence the results with either SFLLRN or TFLLRN as both agonist peptides initiated protective responses in both the absence and presence of the antibody when EPCR was occupied by the PC-S195A mutant zymogen (Fig. 3B, lanes 5 and 8). It is worth noting that PC-S195A did not change the PAR-1-dependent signaling specificity of thrombin with higher concentrations of thrombin (>50 nM), possibly suggesting that thrombin cleaves other cell surface receptors under these conditions.
Figure 2. The barrier permeability effect of the PAR-1 agonist peptides in EA.hy926 cells in the absence and presence of PC-S195A.

(A) The effect of increasing concentrations of SFLLRN (3 h incubation) on the permeability of EA.hy926 cells was monitored before (○) or after pretreatment of endothelial cells for 30 min with PC-S195A (●) (80 nM). (B) The same as (A) except that TFLLRN was used as the PAR-1 agonist peptide. Note that these cells were not treated with TNF-α, but the assay is measuring the cell permeability mediated by the thrombin receptor agonist peptides alone.
Figure 3. Regulation of activation of Rho GTPases and NF-κB by thrombin and PAR-1 agonist peptides.
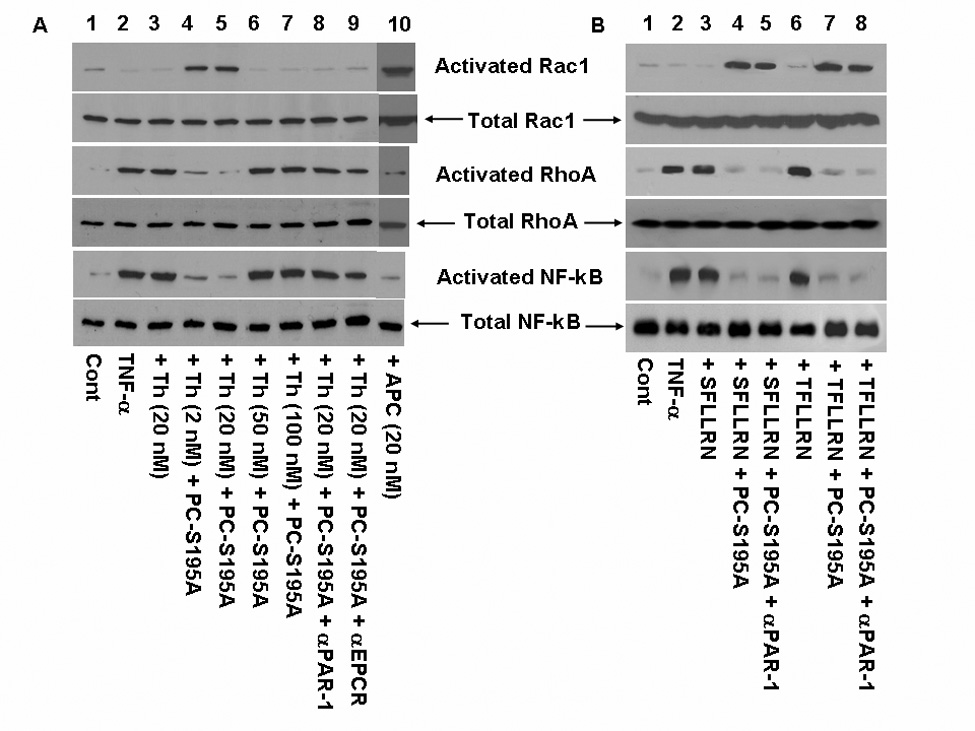
(A) TNF-α-mediated (10 ng/mL for 1 h) activation of Rho GTPases and NF-κB before and after treatment of endothelial cells with either APC (20 nM) or thrombin ± PC-S195A (80 nM) was analyzed by western-blotting using specific antibodies as described under “Materials and methods”. Lane 1, not treated control; lane 2, TNF-α only; lane 3, cells were incubated with 20 nM thrombin for 30 min followed by addition of TNF-α; lane 4, cells were treated with PC-S195A for 30 min followed by incubation with 2 nM thrombin for 30 min and then stimulation with TNF-α; lane 5, the same as lane 4 except that 20 nM thrombin was used; lane 6, the same as lane 4 except that 50 nM thrombin was used; lane 7, the same as lane 4 except that 100 nM thrombin was used; lane 8, the same as lane 5 except that prior to treatment with PC-S195A, the cell monolayer was incubated with anti-PAR-1 antibody (25 µg/mL for 30 min); lane 9, the same as lane 8 except that the cell monolayer was treated with anti-EPCR antibody; and lane 10, the same as lane 3 except that cells were treated with 20 nM APC followed by stimulation with TNF-α. (B) The same as (A) except that thrombin agonist peptides (100 µM) were used as the PAR-1 agonist to monitor the activation of RhoA or Rac1 GTPases in the absence and presence of PC-S195A. Lane 1, not treated control; lane 2, TNF-α only; lane 3, SFLLRN + TNF-α; lane 4, PC-S195A + SFLLRN + TNF-α; lane 5, anti-PAR-1 antibody + PC-S195A + SFLLRN + TNF-α; lane 6, TFLLRN + TNF-α; lane 7, PC-S195A+ TFLLRN + TNF-α; lane 8, anti-PAR-1 antibody + PC-S195A + TFLLRN + TNF-α.
The occupancy of EPCR by protein C inhibits NF-κB activation
The proinflammatory cytokines, LPS and thrombin are known to stimulate endothelial cells by activating the cytosolic NF-κB, thereby leading to its translocation into the nucleus and induction of the target genes by binding to their upstream promoters (14, 16). Thus, the NF-κB pathway activation upregulates the expression of endothelial cell adhesion molecules and APC has been shown to inhibit this proinflammatory pathway (14, 16, 17). To understand the PAR-1-dependent modulatory effect of thrombin on the NF-κB pathway, the endothelial cells were treated with thrombin or thrombin + PC-S195A followed by activation by TNF-α and analysis of cell lysates by western-blotting employing two specific antibodies that detect either the total cellular NF-κB p65 or its Ser-536 phosphorylated form which represents the activated form of the transcription factor that has been translocated into the nucleus. The results presented in Fig. 3A indicated that, TNF-α activates the NF-κB pathway in endothelial cells and that a prior treatment of the cell monolayer with APC potently inhibits this pathway (Fig. 3A, lane 10). While thrombin also activates the NF-κB pathway in endothelial cells, nevertheless, thrombin at two concentrations of 2 and 20 nM exhibited a potent inhibitory effect in response to TNF-α when the cell monolayers were pretreated with PC-S195A prior to activation by the cytokine (Fig. 3A, lanes 4 and 5). This protective effect was abolished at higher concentrations (50–100 nM) of thrombin (Fig. 4A, lanes 6 and 7). The antiinflammatory effect of thrombin + PC-S195A was EPCR and PAR-1-dependent since the function-blocking antibodies to either receptor abrogated the protective effect of thrombin (Fig. 3A, lanes, 8 and 9). The PAR-1 agonist peptides SFLLRN and TFLLRN potently inhibited the activation of NF-κB in the cytokine stimulated endothelial cells in the presence of PC-S195A (Fig. 3B, lanes 4 and 7). In agreement with the anti-PAR-1 antibody inhibiting the cleavage of PAR-1 by thrombin, but not its activation by the agonist peptides, the antiinflammatory effects of the agonist peptides were not blocked by the anti-PAR-1 antibody (Fig. 3B, lanes 5 and 8).
Figure 4.

The model of PAR-1 activation by APC and thrombin in the absence and presence of EPCR occupancy in endothelial cells. When EPCR is not occupied, both PAR-1 and EPCR are associated with caveolin-1 in lipid-rafts of endothelial cells (8). In this case, the thrombin activation of PAR-1 signals via Gq and/or G12/13, thereby enhancing the cellular permeability through the activation of RhoA and eliciting proinflammatory responses through activation of NF-κB. When EPCR is occupied by protein C, the receptor dissociates from caveolin-1, thereby coupling PAR-1 to the Gi subfamily of G-proteins (8). In this case, the activation of PAR-1 by thrombin increases the barrier integrity by activating Rac1 GTPase and elicits an antiinflammatory response by inhibiting NF-κB. The EPCR- and PAR-1-dependent protective response of thrombin via Gi-protein may be a direct effect or mediated through the transactivation of S1P1 as has been described for APC (12, 15). See the text for more details. PC, protein C; Cav-1, caveolin-1; Th, thrombin; S1P, sphingosine 1-phosphate; S1P1, sphingosine 1-phosphate receptor.
Discussion
In this study, we demonstrated that thrombin elicits a potent antiinflammatory response in endothelial cells by cleaving PAR-1 when EPCR is ligated by protein C. This is derived from the observation that thrombin effectively inhibited the expression of the adhesion molecules and thus prevented the interaction of neutrophils with TNF-α-stimulated endothelial cells in the presence of PC-S195A by PAR-1 and EPCR dependent mechanisms. Thrombin is known to activate the RhoA member of the small GTPases, thereby causing cytoskeletal rearrangements, actin stress fiber formation and enhanced barrier permeability in endothelial cells by a PAR-1-dependent mechanism (12, 26). The RhoA-dependent barrier disruptive effect of thrombin is counteracted by the protective effect of another GTPase, Rac1, which is known to be induced by APC (12). Rac1 can oppose the effects of RhoA in the cytoskeletal remodeling, thereby protecting endothelial cells from the edemagenic effects of thrombin and proinflammatory cytokines (12). The results of this study clearly show that the occupancy of EPCR by protein C changes the PAR-1-dependent signaling specificity of thrombin, thus the protease potently inhibits the activation of RhoA and enhances the activation of Rac1 in the TNF-α-stimulated endothelial cells. Thrombin and proinflammatory cytokines are known to mediate their direct cellular effect through the activation of NF-κB (14,16). The observation that thrombin in the presence of PC-S195A potently inhibited the activation of NF-κB in the TNF-α-stimulated endothelial cells by a PAR-1-dependent mechanism further confirmed our hypothesis that when EPCR is occupied by its natural ligand, the cleavage of PAR-1 by thrombin initiates antiinflammatory responses in endothelial cells. Based on these results we propose a model for the PAR-1-dependent signaling effect of thrombin in endothelial cells that is presented in Fig. 4. Thus, in the absence of protein C, EPCR is associated with caveolin-1 and PAR-1 is coupled to Gq and/or G12/13 members of the G-protein subfamily (5, 8). On the other hand, when EPCR is bound by protein C (or APC), EPCR dissociates from caveolin-1, thereby coupling PAR-1 to Gi-protein, and thus the cleavage of PAR-1 by thrombin under these conditions elicits antiinflammatory responses in the cytokine-stimulated endothelial cells (8). Further support for this model was provided by the observation that both PAR-1 agonist peptides SFLLRN and TFLLRN reversed the proinflammatory effects of TNF-α in endothelial cells in the presence of PC-S195A, clearly suggesting that PAR-1 activation elicits a protective response if EPCR is bound by its ligand on endothelial cells. Thus the PAR-1-dependent proinflammatory properties that have been attributed to thrombin in in vitro model systems may not be physiologically relevant in intact endothelium expressing EPCR. However, this scenario may be reversed in the injured and endothelial cell-denuded vasculature where subendothelial cells lacking EPCR, but expressing PAR-1 could come in contact with thrombin.
APC exerts its protective effect by the PAR-1-dependent activation of Rac1 and inhibition of the expression of NF-κB in the cytokine stimulated cultured endothelial cells (12, 17). The PAR-1-dependent protective effect of APC has been hypothesized to contribute, at least partially, to the beneficial therapeutic effect of APC in treating severely septic patients (17, 18). Noting that the efficiency of APC toward the cleavage of PAR-1 is 3–4-orders of magnitude lower than that of thrombin (19), the hypothesis that APC can elicit a protective response through the cleavage of PAR-1 is controversial (20, 21). The finding of this study that the cleavage of PAR-1 on the EPCR-bound endothelial cells can elicit the same protective antiinflammatory responses that are attributed to APC adds further complexity to the nature of this question. Nevertheless, there is strong evidence for APC possessing antiinflammatory properties that are not shared by thrombin since the inhibition of protein C activation by a specific monoclonal antibody that blocks protein C activation by thrombin leads to 100% lethality in the LD50 models of animal sepsis (29). Since both thrombin and APC exert their PAR-1-dependent protective effects by an EPCR-dependent mechanism, it is likely that the sepsis-related dysfunctionality of the endothelium with respect to EPCR expression would similarly affect the PAR-1-dependent protective effects of both proteases. And since thrombin is the only known protease responsible for the activation of protein C it follows therefore that wherever APC exists thrombin would also be present, and given its very high catalytic efficiency, thrombin is expected to prevail over APC in the PAR-1 cleavage reaction. How does then APC exert its protective effect in severe sepsis? Several possibilities can be discussed. First, it is possible that both the anticoagulant and direct cellular effects of APC are equally important for the beneficial effect of APC in severe sepsis. Second, it is also possible that PAR-1-dependent endothelial cell signaling does not contribute significantly to the antiinflammatory role of APC, but rather that leukocytes are the major target for the protective effect of APC in severe sepsis. Finally, another possibility that cannot be ruled out is that thrombin partitioned to the vasculature may not be capable of activating PAR-1 on microvascular endothelial cells under normal and/or pathological conditions. In this venue, we recently demonstrated that the interaction of exosite-1 of thrombin with the acidic hirudin-like sequence present on the exodomain of PAR-1 is required for recognition and cleavage of the receptor by thrombin on the surface of endothelial cells (30). Thus, the TM fragment containing epidermal growth factor-like domains 4–6 potently inhibited the activation of PAR-1 by thrombin on HUVECs (30). In this previous study, we discovered that the thrombin-TM4-6 complex is a poorer activator of PAR-1 than APC in endothelial cells (30). More importantly, we noted that the interaction of thrombin with the hirudin-like sequence of the lipid raft-localized PAR-1 was essential for the exposure and recognition of the scissile bond of the receptor (30). Noting that the affinity of thrombin for endothelial cell TM (KD < 1 nM) (31) is much higher than that of PAR-1 (in µM range for the exodomain of the receptor) (32–34) and the effective concentration of TM in the microcirculation can be as high as 500 nM (35, 36), thus it is possible that thrombin partitioned to the microvascular endothelial cell would all bind TM, with the complex being capable of activating only the EPCR-bound protein C but not PAR-1. Thus, in microcirculation, the activation of the endothelial cell PAR-1 may proceed via the APC pathway and noting the membrane lipid raft localization of all three receptors (22), this pathway would be sufficiently efficient since the activation of protein C by the thrombin-TM complex would be mechanistically linked to the activation of PAR-1 by APC, as has been previously proposed (10). If thrombin can elicit EPCR and PAR-1-dependent antiinflammatory responses in microcirculation, then the administration of protein C zymogen to severely septic patients may potentially have the same beneficial effects of APC without increasing the risk of bleeding. In light of these interesting results, further in vivo studies are warranted to determine the extent to which the EPCR-dependent cleavage of PAR-1 by either thrombin or APC contributes to the protective effects of these proteases in the inflammatory disorders.
Acknowledgements
We would like to thank Audrey Rezaie for proofreading of the manuscript.
The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institute of Health HL 68571 and HL 62565 to ARR.
References
- 1.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Ann Rev Biochem. 1988;57:915–956. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 2.Esmon CT. Molecular events that control the protein C anticoagulant pathway. Thromb Haemost. 1993;70:1–5. [PubMed] [Google Scholar]
- 3.Bode W, Mayr I, Baumann U, et al. The refined 1.9 Å crystal structure of human α-thrombin: interaction with D-Phe-Pro-Arg chlorometheylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dang QD, Vindigni A, Di Cera E. An allosteric switch controls the procoagulant and anticoagulant activities of thrombin. Proc Natl Acad Sci (USA) 1995;92:5977–5981. doi: 10.1073/pnas.92.13.5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 6.Lane DA, Philippou H, Huntington JA. Directing thrombin. Blood. 2005;106:2605–2612. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie E, Saka M, MacKenzie C, et al. Cytokine upregulation of proteinase-activated receptors 2 and 4 expression mediated by p38 MAP kinase and inhibitory kappa B kinase beta in human endothelial cells. Br J Pharmacol. 2007;150:1044–1054. doi: 10.1038/sj.bjp.0707150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae J-S, Yang L, Manithody C, et al. The ligand occupancy of endothelial protein C receptor switches the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110:3909–3916. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci (USA) 2007;104:5662–5667. doi: 10.1073/pnas.0700763104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feistritzer C, Schuepbach RA, Mosnier LO, et al. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- 11.Garcia JGN, Pavalko FM, Patterson CE. Vascular endothelial cell activation and permeability responses to thrombin. Blood Coag Fibrinol. 1995;6:609–626. doi: 10.1097/00001721-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Finigan JH, Dudek SM, Singleton PA, et al. Activated protein C mediates novel lung endothelial barrier enhancement. J Biol Chem. 2005;280:17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 13.Ruf W, Dorfleutner A, Riewald M. Specificity of coagulation factor signaling. J Thromb Haemost. 2003;1:1495–1503. doi: 10.1046/j.1538-7836.2003.00300.x. [DOI] [PubMed] [Google Scholar]
- 14.Joyce DE, Grinnell BW. Recombinant human activated protein C attenuates the inflammatory response in endothelium and monocytes by modulating nuclear factor-kB. Crit Care Med. 2002;30:S288–S293. doi: 10.1097/00003246-200205001-00019. [DOI] [PubMed] [Google Scholar]
- 15.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 16.Joyce DE, Gelbert L, Ciaccia A, et al. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 17.Mosnier LO, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 18.Bernard GR, Vincent JL, Laterre PF, et al. Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Eng J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 19.Ludeman MJ, Kataoka H, Srinivasan Y, et al. PAR1 cleavage and signaling in response to activated protein C and thrombin. J Biol Chem. 2005;280:13122–13128. doi: 10.1074/jbc.M410381200. [DOI] [PubMed] [Google Scholar]
- 20.Esmon CT. Is APC activation of endothelial cell PAR1 important in severe sepsis?: No. J Thromb Haemost. 2005;3:1910–1911. doi: 10.1111/j.1538-7836.2005.01573.x. [DOI] [PubMed] [Google Scholar]
- 21.Ruf W. Is APC activation of endothelial cell PAR1 important in severe sepsis?: Yes. J Thromb Haemost. 2005;3:1912–1914. doi: 10.1111/j.1538-7836.2005.01576.x. [DOI] [PubMed] [Google Scholar]
- 22.Bae J-S, Yang L, Rezaie AR. Receptors of the protein C activation and activated protein C signaling pathways are colocalized in lipid rafts of endothelial cells. Proc Natl Acad Sci (USA) 2007;104:2867–2872. doi: 10.1073/pnas.0611493104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bae J-S, Yang L, Manithody C, et al. Engineering a disulfide bond to stabilize the calcium binding loop of activated protein C eliminates its anticoagulant but not protective signaling properties. J Biol Chem. 2007;282:9251–9259. doi: 10.1074/jbc.M610547200. [DOI] [PubMed] [Google Scholar]
- 24.Akeson AL, Woods CW. A fluorometric assay for the quantitation of cell adherence to endothelial cells. J Immunol Methods. 1993;163:181–185. doi: 10.1016/0022-1759(93)90121-m. [DOI] [PubMed] [Google Scholar]
- 25.Reid T, Bathoorn A, Ahmadian MR, et al. Identification and characterization of hPEM-2, a guanine nucleotide exchange factor specific for Cdc42. J Biol Chem. 1999;274:33587–33593. doi: 10.1074/jbc.274.47.33587. [DOI] [PubMed] [Google Scholar]
- 26.Bae J-S, Rezaie AR. Protease activated receptor 1 (PAR-1) activation by thrombin is protective in human pulmonary artery endothelial cells if endothelial protein C receptor is occupied by its natural ligand. Thromb Haemost. 2008;100:101–109. doi: 10.1160/TH08-02-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaughlin JN, Shen L, Holinstat M, et al. Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J Biol Chem. 2005;280:25048–25059. doi: 10.1074/jbc.M414090200. [DOI] [PubMed] [Google Scholar]
- 28.Blackhart BD, Emilsson K, Nguyen D, et al. Ligand cross-reactivity within the protease-activated receptor family. J Biol Chem. 1996;271:16466–16471. doi: 10.1074/jbc.271.28.16466. [DOI] [PubMed] [Google Scholar]
- 29.Taylor FB, Jr, Chang A, Esmon CT, et al. Protein C prevents the coagulopathic and lethal effects of E coli infusion in the baboon. J Clin Invest. 1987;79:918–925. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bae J-S, Yang L, Rezaie AR. lipid raft localization regulates the cleavage specificity of protease activated receptor 1 in endothelial cells. J Thromb Haemost. 2008;6:954–961. doi: 10.1111/j.1538-7836.2008.02924.x. [DOI] [PubMed] [Google Scholar]
- 31.Ye J, Rezaie AR, Esmon CT. Glycosaminoglycan contributions to both protein C activation and thrombin inhibition involve a common arginine-rich site in thrombin that includes residues arginine 93, 97, and 101. J Biol Chem. 1994;269:17965–17970. [PubMed] [Google Scholar]
- 32.Ishii K, Gerszten R, Zheng YW, et al. Determination of thrombin receptor cleavage. J Biol Chem. 1995;270:16435–16440. doi: 10.1074/jbc.270.27.16435. [DOI] [PubMed] [Google Scholar]
- 33.Jacques SL, LeMasurier M, Sheridan PJ, et al. Substrate-assisted catalysis of the PAR1 thrombin receptor. J Biol Chem. 2000;275:40671–40678. doi: 10.1074/jbc.M004544200. [DOI] [PubMed] [Google Scholar]
- 34.Nieman MT, Schmaier AH. Interaction of thrombin with PAR1 and PAR4 at the thrombin cleavage site. Biochemistry. 2007;46:8603–8610. doi: 10.1021/bi700597p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busch C, Cancilla P, DeBault L, et al. Use of endothelium cultured on microcarriers as a model for the microcirculation. Lab Invest. 1982;47:498–504. [PubMed] [Google Scholar]
- 36.Esmon CT. Cell mediated events that control blood coagulation and vascular injury. Ann Rev Cell Biol. 1993;9:1–26. doi: 10.1146/annurev.cb.09.110193.000245. [DOI] [PubMed] [Google Scholar]