

Abstract
The tracheal system of Drosophila melanogaster is an interconnected network of gas-filled epithelial tubes that develops during embryogenesis and functions as the main gas-exchange organ in the larva. Larval tracheal cells respond to hypoxia by activating a program of branching and growth driven by HIF-1α/sima-dependent expression of the breathless (btl) FGF receptor. By contrast, the ability of the developing embryonic tracheal system to respond to hypoxia and integrate hard-wired branching programs with sima-driven tracheal remodeling is not well understood. Here we show that embryonic tracheal cells utilize the conserved ubiquitin ligase dVHL to control the HIF-1 α/sima hypoxia response pathway, and identify two distinct phases of tracheal development with differing hypoxia sensitivities and outcomes: a relatively hypoxia-resistant ‘early’ phase during which Sima activity conflicts with normal branching and stunts migration, and a relatively hypoxia-sensitive ‘late’ phase during which the tracheal system uses the dVHL/sima/btl pathway to drive increased branching and growth. Mutations in the archipelago (ago) gene, which antagonizes btl transcription, re-sensitize early embryos to hypoxia, indicating that their relative resistance can be reversed by elevating activity of the btl promoter. These findings reveal a second type of tracheal hypoxic response in which Sima activation conflicts with developmental tracheogenesis, and identify the dVHL and ago ubiquitin ligases as key determinants of hypoxia sensitivity in tracheal cells. The identification of an early stage of tracheal development that is vulnerable to hypoxia is an important addition to models of the invertebrate hypoxic response.
Keywords: Drosophila, Trachea, Hypoxia, dVHL, Archipelago, Sima, FGF signaling
Introduction
The development and survival of an organism are dependent on its ability to adapt to changing environmental conditions. Responses to some environmental changes, for example in nutrient availability, temperature, or oxygen concentration, involve alterations in patterns of gene expression that allow the organism to survive periods of environmental stress. In metazoan cells, the cellular response to reduced oxygen is mediated primarily by the HIF (hypoxia inducible factor) family of transcription factors, which are heterodimers composed of α and β subunits belonging to the bHLH Per-ARNT-Sim (bHLH-PAS) protein family (reviewed in Kaelin and Ratcliffe, 2008). The HIF-1 αβ heterodimer is the primary oxygen-responsive HIF in mammalian cells and binds to a specific DNA sequence termed hypoxia response element (HRE) present in the promoters of target genes involved in energy metabolism, angiogenesis, erythropoiesis, and autophagy (Manalo et al., 2005). HIF-1 activity is inhibited under normoxic conditions by two hydroxylase enzymes that use dioxygen as a substrate for catalysis to hydroxylate specific proline or aspartate residues in the HIF-1α subunit (reviewed in Kaelin and Ratcliffe, 2008). These modifications limit HIF-1 activity by either reducing HIF-1α levels or inhibiting its ability to activate HRE-containing target promoters. One of these inhibitory mechanisms involves the 2-oxoglutarate/Fe(II)-dependent HIF-1 prolyl hydroxylase (HPH), which attaches a hydroxyl group onto each of two conserved proline residues in the oxygen-dependent degradation domain (ODD) of mammalian HIF-1α. These modifications create a binding site in the HIF-1α ODD for the Von Hippel-Lindau (VHL) protein, the substrate adaptor component of a ubiquitin ligase that subsequently polyubiquitinates HIF-1α and targets it for degradation by the proteasome (reviewed in Kaelin, 2005). This degradation mechanism operates constitutively in normoxia and is epistatic to otherwise wide spread expression of HIF-1α mRNA. HIF-1α protein is also modified by a second oxygen-dependent hydroxylase termed Factor Inhibiting HIF (FIH) that hydroxylates an asparagine residue in the HIF-1α C-terminal activation domain (reviewed in Kaelin, 2005). This blocks interaction with the CBP/p300 transcriptional co-factor and thus further restricts expression of HIF-1 responsive genes. These parallel O2-dependent hydroxylation mechanisms by HPH and FIH ensure that HIF-1α levels and activity remain low in normoxic conditions. However as oxygen levels become limiting in the cellular environment, rates of hydroxylation decline and HIF-1α is rapidly stabilized in a form that dimerizes with HIF-1β, translocates to the nucleus, and promotes transcription of HRE-containing target genes.
Evidence suggests that invertebrate homologs of HIF-1 are also regulated in response to changes in oxygen availability (reviewed in Gorr et al., 2006). In the fruit fly Drosophila melanogaster, the HPH homolog fatiga (fga) has been shown to genetically antagonize the HIF-1α homolog similar (sima) during development (Centanin et al., 2005). The Drosophila VHL homolog dVHL has also been shown to be capable of binding to human HIF-1α and stimulating its proteasomal turnover in vitro (Aso et al., 2000). In addition, the Drosophila genome encodes a well-characterized HIF-1β homolog tango (tgo) (Sonnenfeld et al., 1997), and two potential FIH homologs (CG13902 and CG10133; Berkeley Drosophila Genome Project) that have yet to be analyzed functionally. Spatiotemporal analysis of sima activation using sima-dependent hypoxia-reporter transgenes has shown that exposure to an acute hypoxic stress induces Sima most strongly in cells of the larval and embryonic tracheal system (Arquier et al., 2006; Lavista-Llanos et al., 2002), while induction of reporter activity in other tissues requires more chronic exposure to low oxygen (Lavista-Llanos et al., 2002). The larval tracheal system is composed of an interconnected network of polarized, epithelial tubes that duct gases through the organism (reviewed in Ghabrial et al., 2003). As the trachea acts as the primary gas-exchange organ in the larva, it is thus a logical site of hypoxia sensitivity. During larval stages, specific cells within the tracheal system called ‘terminal cells’ respond to hypoxia by initiating new branching and growth that results in the extension of fine, unicellular, gas-filled tubes toward hypoxic tissues in a manner somewhat analogous to mammalian angiogenesis (Guillemin et al., 1996; Jarecki et al., 1999). Studies have shown that sima and its upstream antagonist fga function within terminal cells to regulate this process (Centanin et al., 2008). sima is necessary for terminal cell branching in hypoxia and its ectopic activation, by either transgenic overexpression or loss of fga, is sufficient to induce excess branching even in normoxia. These phenotypes have been linked to the ability of sima to promote expression of the breathless (btl) gene (Centanin et al., 2008), which encodes an FGF receptor (Klambt et al., 1992) that is activated by the branchless (bnl) FGF ligand (Sutherland et al., 1996). This receptor/ligand pair is known to act via a downstream MAP-kinase signaling cascade to promote cell motility and tubular morphogenesis in a variety of systems (reviewed in Lubarsky and Krasnow, 2003). Excessive activation of this pathway within tracheal cells by transgenic expression of btl is sufficient to drive excess branching (Lee et al., 1996b). Reciprocally, misexpression of the bnl ligand in certain peripheral tissues is sufficient to attract excess terminal cell branching (Jarecki et al., 1999). Indeed production of secreted factors such as Bnl may be a significant part of the physiologic mechanism by which hypoxic cells attract new tracheal growth. Sima-driven induction of btl in conditions of hypoxia thus allows larval terminal cells to enter what has been termed an ‘active searching’ mode (Centanin et al., 2008) in which they are hyper-sensitized to signals emanating from nearby hypoxic non-tracheal cells.
The role of the btl/bnl pathway in tracheal development is not restricted to hypoxia-induced branching of larval terminal cells. It also plays a critical, earlier role in the initial development of the embryonic tracheal system from the tracheal placodes, groups of post-mitotic ectodermal cells distributed along either side of the embryo that undergo a process of invagination, polarization, directed migration, and fusion to create a network of primary and secondary tracheal branches (reviewed in Ghabrial et al., 2003). btl and bnl are each required for this process via a mechanism in which restricted expression of bnl in cells outside the tracheal placode represents a directional cue for the migration of btl-expressing cells within the placode. Accordingly, btl expression is normally highest in pre-migratory and migratory embryonic fusion cells (Ohshiro and Saigo, 1997). In contrast to the larval hypoxic response, sima does not appear to be required for morphogenesis of the embryonic tracheal system (Ohshiro and Saigo, 1997). Rather, developmentally programmed signals in the embryo dictate a stereotyped pattern of btl and bnl expression that leads to a similarly stereotyped pattern of primary and secondary tracheal branches (Centanin et al., 2008). The btl/bnl pathway thus responds to developmental signals to drive a fixed pattern of branching in the embryo, while in the subsequent larval stage it responds to hypoxia-dependent sima activity to facilitate the homeostatic growth of larval terminal cells and tracheal remodeling.
Under normal circumstances, developing Drosophila tissues do not begin to experience hypoxia until the first larval stage, when organismal growth and movement begin to consume more oxygen than can be provided by passive diffusion alone (Manning and Krasnow, 1993). As a consequence, the first hypoxic challenge normally occurs after the btl/bnl-dependent elaboration of the primary and secondary embryonic branches is complete. Thus, the ability of the larval tracheal system to drive new branching and remodeling via sima and btl represents the response of a developed ‘mature’ tracheal system to reduced oxygen availability. By contrast the effect of hypoxia on embryonic tracheal development, which requires tight spatiotemporal control of Btl signaling to pattern the tracheal network, is not as well understood. Given that the trachea does not function as a gas-exchange organ until after fluid is cleared from the tubes at embryonic stage 17 (Tsarouhas et al., 2007), it may be that the transcriptional response of embryonic tracheal cells to hypoxia (Lavista-Llanos et al., 2002) leads to mainly metabolic changes rather than to a btl-driven program of tubulogenesis and remodeling. However, if the embryonic tracheal system does utilize the sima pathway to induce hypoxia-dependent changes in btl gene transcription, then hypoxic exposure of embryos might be predicted to produce a situation of competing developmental and homeostatic inputs that converge on the btl/bnl pathway. The ability of tracheal cells to integrate such signals may then determine whether or not the embryonic tracheal system is able to adapt to oxygen stress, or whether embryonic tracheal development represents a sensitive period during which the organism's ability to respond to changes in oxygen levels is inherently limited by a pre-programmed pattern of developmental gene expression.
Here we show that the embryonic tracheal system utilizes the dVHL/sima pathway to respond to hypoxia, but that the type and severity of resulting phenotypes depend on the developmental stage of exposure. Hypoxic challenge while embryonic tracheal cells are responding to developmentally programmed btl/bnl migration signals disrupts tracheal development and results in fragmented and unfused tracheal metameres. In contrast, hypoxic challenge at a somewhat later embryonic stage after fusion is complete results in overgrowth of the primary tracheal branches and the production of extra secondary branches. Interestingly, we find that the threshold of hypoxia required to induce tracheal phenotypes in the early embryo is higher than that required to induce excess branching phenotypes in later embryonic stages, indicating that tracheal patterning events in the embryo are relatively resistant to hypoxia. Genetic analysis indicates that both types of hypoxic tracheal phenotypes — stunting and overgrowth — require sima and can be phenocopied in normoxia by reducing expression of the HIF-1α ubiquitin ligase gene dVHL specifically within tracheal cells. Moreover, we find that reduced dVHL expression in the larval trachea leads to excess terminal cell branching in a manner quite similar to that observed in fga mutants. Molecular and genetic data indicate that excess btl transcription is a major cause of hypoxia-induced tracheal phenotypes. Consistent with this, mutations in the archipelago (ago) gene, which antagonizes btl transcription in tracheal fusion cells (Mortimer and Moberg, 2007), synergize strongly with dVHL inactivation to disrupt tracheal migration and branching. Interestingly, ago mutations also lower the threshold of hypoxia required to elicit tracheal phenotypes in the ‘early’ embryo, suggesting that the relative activity of the btl promoter can affect hypoxic sensitivity. These findings show that the dVHL/sima pathway plays an important role in tracheal development, and identify two distinct phases of embryonic development that show different phenotypic outcomes of activating this pathway: an early phase during which sima activity conflicts with developmental control of tracheal branching and migration, and a later phase during which the tracheal system uses the dVHL/sima/btl pathway to adapt to hypoxia by increasing its future capacity to deliver oxygen to target tissues.
Materials and methods
Stocks, genetics and statistics
The dVHL open reading frame was cloned as a PCR product into the EcoRI site of the pSymp vector (Giordano et al., 2002) and used to generate UAS-dVHLi stocks (D. Rennie, Massachusetts General Hospital Transgenic Drosophila Core). Other strains used in this study were: ago1, ago3 (Moberg et al., 2001), UAS-agoΔF (Mortimer and Moberg, 2007), btl-Gal4 (Shiga et al., 1996), UAS-sima (Lavista-Llanos et al., 2002), UAS-btl:GFP (Dammai et al., 2003), UAS-Adf1RNAi (Vienna Drosophila RNAi Center), UAS-pigeonRNAI (NIG-Fly, National Institute of Genetics, Japan), bnlP1, btldev1, btlEY01638, trh10512, actin-Gal4, UAS-Ras85D.V12, UAS-pnt.P1, w1118, sima07607, Df(3R)BSC502, esg-lacZ, en-Gal4, GMR-Gal4 and Exel6060 (all from the Bloomington Drosophila Stock Center). Embryos were genotyped using the TM6B,P{iab-2(1.7) lacZ}6B,Tb1, TM6B,P{w+mC=35UZ}DB1,Tb1, SM6b,P{eve-lacZ8.0}SB1 and CyO,P{ry+t7.2=lArB} A208.1M2 ‘blue’ balancers, and the CyO,P {ActGFP}JMR1 balancer (all from the Bloomington Drosophila Stock Center). Hypoxia treatments were performed using 0.5% O2: 99.5% N2 gas in a hypoxia chamber (Billups-Rothenberg). O2 concentration within the chamber was monitored by an electronic O2 sensor (RKI Instruments, Inc., Union City, CA; see Supplementary data). Statistical comparisons were performed using Student’s t-test.
Embryo immunohistochemistry and antibodies
Embryos were staged and fixed as described previously (Mortimer and Moberg, 2007) and incubated with the following primary antibodies: mouse anti-Tango (1:2, Developmental Hybridoma Studies Bank; DSHB), mouse mAb2A12 (1:5, DSHB), rat anti-Trh (1:200, ref), rabbit anti-β-Gal (1:250, Cappel) and rabbit anti-GFP (1:400, Molecular Probes). Secondary antibodies conjugated to HRP, AP, Cy3 and FITC were used as recommended (Jackson ImmunoResearch). Stage 15–16 embryos were analyzed, except where indicated. Embryos from w1118 and UAS-dVHLi strains crossed to actin-Gal4/CyO,P{ActGFP}JMR1 were collected at indicated stages and sorted by the absence of GFP. Whole embryo extracts were prepared in sample buffer and resolved on 12% SDS-PAGE prior to Western blotting with anti-dVHL (Arquier et al., 2006) and anti-β-tubulin (1:2000, Santa Cruz Biotechnology).
RNA analysis
RNA in situ hybridization was performed as described (Merabet et al., 2005). DIG labeled riboprobes were synthesized from PCR fragments of btl or bnl cDNA and visualized with anti-DIG-AP (1:2000, Roche) or anti-DIG-Biotin (1:500, Sigma) followed by TSA-Biotin amplification (PerkinElmer) and incubation with SA-FITC. For dVHL expression analysis, total RNA was isolated from staged embryos and reverse-transcribed using random hexamer primers (Invitrogen) with Superscript II Reverse Transcriptase (Invitrogen). dVHL and β-tubulin cDNAs were then amplified with gene-specific primers.
Results
Stage-specific effects of hypoxia on embryonic tracheogenesis
To determine how the embryonic tracheal system responds to hypoxia, wild type embryos were placed in a reduced oxygen environment according to the scheme depicted in Fig. 1A. Two different hypoxic treatments were used: 0.5% O2 for 5 h, or 1% O2 for 4 h. In one set of embryos (denoted ‘early’), hypoxic treatment was initiated at stage 11 (6–8 h after egg laying; AEL) when dorsal trunk (DT) branches are actively migrating, and in the other (denoted ‘late’) it was initiated at stage 15 (13–15 h AEL) when DT fusion is complete. Following these treatments, embryos were returned to normoxia (21% O2) and allowed to develop to embryonic stage 16, at which time tracheal architecture was visualized with the mAb2A12 tracheal lumen antibody (Figs. 1B-F). As has been described elsewhere (DiGregorio et al., 2001; Douglas et al., 2001), embryonic development was arrested by the stronger 0.5% O2 treatment but resumed upon re-exposure to normoxia. The weaker hypoxic treatment only led to a slight delay in embryonic development (data not shown). With very high penetrance (Fig. 1G, Table 1 and Supplemental Table V), ‘early’ exposure to 0.5% O2 severely stunted DT branch formation and fusion such that adjacent metameres appear as unconnected lumenal fragments (Fig. 1C). Structures that form after DT fusion, for example the lateral trunk (LT), were less severely affected. At an organismal level, ‘early’ hypoxia also resulted in complete lethality prior to hatching (data not shown). In contrast to the stunting effect of ‘early’ hypoxia on tracheal growth, ‘late’ exposure to 0.5% O2 induced a convoluted tube overgrowth phenotype throughout the tracheal system (Fig. 1D) at high penetrance (Fig. 1G, Table 1 and Supplemental Table V). A similar hypoxia-induced tube overgrowth phenotype has been reported previously (Arquier et al., 2006). The major primary and secondary branches of these embryos are highly convoluted and sinuous, and show visceral branch (VB) and dorsal branch (DB) duplications (Figs. 1E and F). Unlike the ‘early’ 0.5% O2 treatment, these ‘late’ embryos showed no significant reduction in organismal viability (data not shown). Interestingly, when these experiments were repeated under the weaker hypoxic condition of 1% O2, the overall penetrance of tracheal phenotypes in ‘early’ embryos dropped considerably (from 97% [n=27] to 12% [n=42]; see Table 1) while the penetrance of tracheal overgrowth in ‘late’ embryos remained quite high (Fig. 1G and Table 1). However, comparing DT length in ‘late’ embryos exposed to 1% or 0.5% O2 (Fig. 1H) reveals a progressive increase in tube length relative to normoxic controls (21 ± 1.1% longer in 1% O2 [n=4] and 46±3.1% longer in 0.5% O2 [n=4]). Thus stronger hypoxic challenges produce a progressively stronger tracheal growth phenotype in the ‘late’ embryo. Overall, these patterns of tracheal sensitivity to ‘early’ and ‘late’ hypoxia suggest that hypoxic activation does not always lead to tracheal overgrowth, but can in fact also stunt tracheal branching during a specific window of ‘early’ embryonic development. However, the ‘early’ embryonic tracheal system is relatively resistant to these effects, while the ‘late’ embryonic tracheal system is sensitized to graded activation of the hypoxic response pathway.
Fig. 1.

Tracheal morphogenesis defects following exposure to hypoxia. (A) Schematic representation of severity and timing of hypoxic challenges, relative to migration of dorsal trunk (DT) fusion cells (in red). (B—F) Lateral images of w1118 embryos stained with the tracheal lumen marker mAb2A12. Unless otherwise indicated, embryos are shown anterior left, and dorsal up. (B) Tracheal architecture of a normoxic embryo. The DT, dorsal branches (DBs), lateral trunk (LT) and visceral branches (VBs) are indicated. (C—F) Hypoxia treated embryos showing characteristic phenotypes following exposure to 0.5% O2. (C) Early hypoxic exposure leads to defects in tracheal morphogenesis. (D) Sinuous overgrowth seen following late hypoxic exposure. (E, F) Late hypoxic exposure also causes duplications of the (E) VB and (F) DB within given segment. ‘Extra’ branches are indicated. (G) Quantification of penetrance of tracheal defects in the indicated hypoxic conditions and genotypes (*p<0.001 relative to w1118 embryos in 0.5% O2 [blue bars]). (H) Quantification of DT length in the indicated hypoxic conditions relative to normoxic control, showing a graded hypoxic response (*p<0.005; error bars are ±standard error of the mean [SEM]).
Table 1.
| Genotype | Total penetrance (%)a | Migration defects (%)b | n |
|---|---|---|---|
| Hypoxia-induced embryonic tracheal phenotypes | |||
| Normoxia | |||
| wt | 9 | 3 | 61 |
| 0.5% O2 | |||
| wt, stage 11 | 99 | 86 | 45 |
| sima07607, stage 11 | 16* | 8* | 39 |
| wt, stage 15 | 97 | 0 | 27 |
| sima07607, stage 15 | 19* | 0 | 26 |
| 1% O2 | |||
| wt, stage 11 | 12 | 5 | 42 |
| ago3/+, stage 11 | 41** | 9 | 58 |
| wt, stage 15 | 74 | 0 | 34 |
| ago3/+, stage 15 | 71 | 0 | 35 |
| Tracheal RNAi phenotypes | |||
| btl-Gal4:UAS-Adf1i | 15 | 10 | 59 |
| btl-Gal4:UAS-pigi | 16 | 8 | 25 |
| btl-Gal4:UAS-dVHLi4B2 | 60*** | 16 | 32 |
| btl-Gal4:UAS-dVHLi11A2 | 50*** | 18 | 34 |
| btl-Gal4:UAS-dVHLi34B3 | 68*** | 32*** | 48 |
| btl-Gal4:UAS-dVHLi4B6 | 54*** | 33*** | 33 |
| btl-Gal4:UAS-dVHLi31A2 | 61*** | 38*** | 40 |
| actin-Gal4:UAS-dVHLi34B3 | 48*** | 24*** | 29 |
Refers to tube overgrowth, missing or duplicated secondary branches, excess terminal cell branching, and migration defects (if evident).
Refers to interruptions of the DT or LT, or failure of DB midline fusion.
p<0.01 relative to wt 0.5% O2 values.
p<0.01 relative to wt 1% values.
p<0.01 relative to btl-Gal4:UAS-Adf1i.
To test whether sima is responsible for both ‘early’ and ‘late’ hypoxic tracheal phenotypes, embryos homozygous for the sima07607 loss-of-function allele (Centanin et al., 2005) were exposed to the ‘strong’ 0.5% O2 hypoxic challenge at ‘early’ and ‘late’ time points. In both cases lack of wild type sima strongly suppressed the penetrance of the hypoxia-induced tracheal phenotypes (Fig. 1G and Table 1), indicating that activation of the sima hypoxia response pathway in early stage embryos blocks tracheal cell migration and fusion, while in later stage embryos it promotes tracheal overgrowth and excess secondary branching.
dVHL is required to suppress the tracheal hypoxic response
By analogy to mammalian HIF-1α, dVHL-dependent degradation is predicted to be one of the major mechanisms that restricts sima activity in normoxia. Indeed analysis of fga mutants suggests that preventing dVHL from acting on Sima protein specifically within larval tracheal cells is a required element of the organismal response to hypoxia (Centanin et al., 2008). To explore its possible role in regulating the embryonic hypoxic response, dVHL expression was assayed during embryogenesis by reverse transcriptase-PCR (RT-PCR, Fig. 2A). dVHL mRNA is detected throughout embryonic development, beginning prior to the onset of tracheal development (stages 1–8, Fig. 2A lane 1) and continuing throughout early (stages 9–11), mid (stages 12–14) and late (stages 15–17) tracheogenesis (Fig. 2A lanes 2–4, respectively), with an apparent peak of expression during mid tracheogenesis. This is consistent with a role for dVHL in regulating sima, which is ubiquitously expressed during embryogenesis (Nambu et al., 1996). Because of a lack of available genomic alleles of dVHL, we consequently used a transgenic RNA interference (RNAi) approach to test the role of dVHL in tracheal morphogenesis. A dVHL RNAi transgene was constructed by inserting a DNA fragment corresponding to the single dVHL exon into the pSymp vector (Giordano et al., 2002). Flanking UAS sites allow for Gal4-driven production of a dVHL dsRNA that is processed by the Drosha/Dicer pathway into small interfering RNAs (Kim et al., 2006). Multiple UAS-dVHLi lines were generated and tested for knockdown efficiency by Western blot of dVHL protein in animals expressing the ubiquitous ‘driver’ actin-Gal4 (Fig. 2B). A range of knockdown efficiencies was observed, ranging from strong (line i31A2) to mild reduction (lines i11A2 and i34B3) of dVHL protein levels (Fig. 2B).
Fig. 2.

Tracheal-specific knockdown of dVHL leads to defects in embryonic tracheal morphogenesis. (A) RT-PCR analysis of dVHL (top) and β-tubulin (bottom) expression during embryonic stages 1–8 (lane 1), 9–11 (lane 2), 12–14 (lane 3) and 15–17 (lane 4). (B) Western blot analysis of dVHL (top panel) levels in whole embryo extracts from stage 13–16 control embryos (wt) and embryos expressing the indicated combination of actin-Gal4 and UAS-dVHLi lines. α-β-Tubulin is used a loading control (bottom panel). (C—G) Lateral and (H) dorsal images of embryos stained with the tracheal lumen marker mAb2A12. (C) Normal tracheal architecture in a btl-Gal4-UAS-Adf1 control RNAi embryo (D—F) Embryos of the indicated genotypes showing the range of tracheal defects seen following btl-Gal4:UAS-dVHLi knockdown. (G,H) btl-Gal4 driven dVHL knockdown also causes duplications of (G) VBs and (H) DBs as indicated. (I, J) Lateral images of (I) w1118 and (I) btl-Gal4:UAS-dVHLi31A2 embryos stained with α-Tgo to mark tracheal cells. (J) Magnified view of a dVHLi embryonic trachea showing DT interruption, and missing (asterisk) or duplicated (arrows) DBs.
To determine what role dVHL plays in embryonic tracheal development, UAS-dVHLi transgenes were expressed in embryos using either the ubiquitous actin-Gal4 driver or the tracheal cell-XSspecific btl-Gal4 driver (Shiga et al., 1996). Because each had a similar effect on tracheal patterning (Table 1), btl-Gal4 was used for subsequent experiments. All dVHLi lines had effects on tracheal development that were not observed in control embryos expressing RNAi against the neuronal genes Adf1 or pigeon (Fig. 2C and Table 1) or in embryos expressing dVHLi in non-tracheal tissue with the en-Gal4 driver (data not shown). Expression of the strongest dVHLi line, i31A2, produced migration defects in a significant percentage of embryos (Fig. 2D) that resemble the effect of ‘early’ hypoxic challenge on DT migration, but are more severe in that they also include defective DB/VB migration and interruptions of the LT. Examination of the fusion cell marker esg-lacZ in this background reveals that individual LacZ-positive fusion cells are detectable in tracheal segments that display migration defects (Supplemental Fig. S4), indicating that loss of dVHL does not prevent cells from adopting the fusion cell fate. However, because the altered morphology of these btl-Gal4;dVHLi31A2 makes it difficult to determine absolute DT fusion cell numbers, we cannot be sure that fusion cells are specified normally in all segments. By contrast to i31A2, expression of the more mild dVHLi knockdown lines leads to an intermediate phenotype characterized by fewer migration and fusion defects per embryo, particularly in the DT and LT (Fig. 2E), and sinuous overgrowth of the primary and secondary branches that resembles the ‘late’ response to hypoxia (Fig. 2F). dVHL knockdown also produces overgrown and intertwined secondary branches (Figs. 2G, H) and secondary branch duplication (Fig. 2J) similar to that observed in wild type embryos exposed to ‘late’ hypoxia (see Supplemental Table V for a summary of embryonic phenotypes). In all combinations of btl-Gal4 driver and UAS-dVHLi transgene, a majority of animals develop through larval stages and die as pupae (Fig. 4B and Supplemental Table IV), indicating that persistent knockdown of dVHL in tracheal cells eventually leads to organismal death. This pupal lethality is specific to tracheal expression and is not seen in pupae expressing dVHLi with other tissue specific Gal4 drivers (e.g. en-Gal4 or GMR-Gal4; KHM and NTM unpublished). During the 3rd instar, these larvae show increases in thick terminal branches (TTBs) (Figs. 3A, C) that can be phenocopied by exposing larvae to 1% O2 [Fig. 3B; TTBs quantified in Supplemental Table III as in (Centanin et al., 2008)]. They also show duplication of larval tertiary branches (see LG branches in Fig. 3E), and failure of lateral trunk fusion associated with cells terminating in multiple, fine extensions (Fig. 3F). dVHL is thus required within tracheal cells to pattern embryonic and larval tracheal development, and loss of the gene in these cells is sufficient to mimic the systemic effect of hypoxia on the embryonic and larval tracheal systems.
Fig. 4.
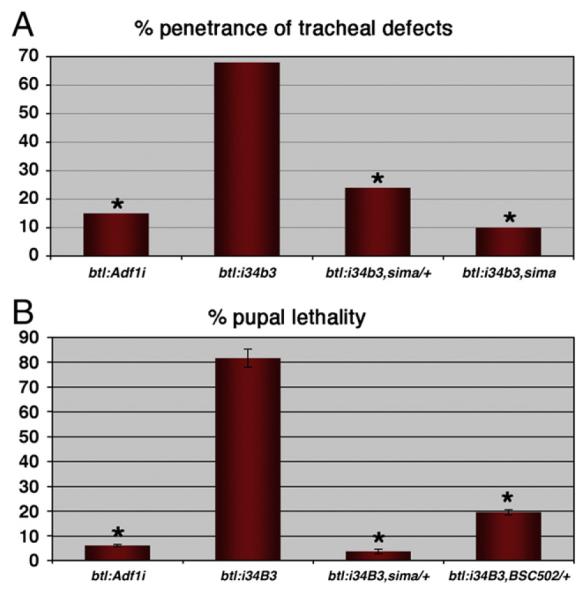
dVHL knockdown phenotypes genetically require sima. (A) Penetrance of tracheal defects in embryos with btl-Gal4 driven expression of UAS-Adf1i or the UAS-dVHLi34B3 transgene. dVHLi phenotypes are suppressed by sima alleles in a dosedependent manner (*p<0.001 relative to btl-Gal4:UAS-dVHLi34B3). (B) Frequency of pupal lethality in the indicated genotypes. btl-Gal4:UAS-dVHLi34B3 lethality is dominantly suppressed by sima or Df(3R)BSC502 (*p<0.005 relative to btl-Gal4:UAS-dVHLi34B3; error bars are ±SEM).
Fig. 3.

Tracheal cell-specific dVHL knockdown causes defects in terminal cell branching and larval tracheal morphology. (A—C) Bright-field dorsal images of third instar larvae showing ramified branches of a dorsal branch terminal cell from tracheal segment 3 (Tr3). Thick terminal branch (TTB) number is increased by (B) exposure to 1% O2 or (C) btl-Gal4 driven expression of UAS-dVHLi31A2. (D—F) Lateral images of third instar larvae. (D) Image of a single segment from a normoxic larva. The ganglionic branch (GB), transverse connective (TC), lateral trunk (LT), and LT terminal cells LF, LG and LH are indicated. btl-Gal4:UAS-dVHLi31A2 expression leads to duplications of lateral trunk terminal cells. (E) Two LG branches are indicated. (F) btl-Gal4 driven dVHLi31A2 knockdown also causes defects in migration/fusion of secondary branches. The LT branches from adjacent placodes should fuse at the point indicated by the asterisk, but instead fail to fuse leading to the ramification of multiple fine tracheal branches.
dVHL genetically antagonizes sima in the embryonic trachea
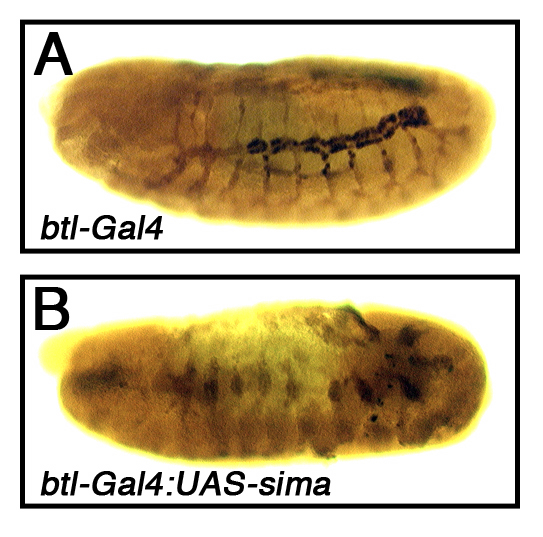
Stronger dVHL knockdown correlates with more disruptive effects on primary and secondary branch migration and fusion in a manner similar to ‘early’ hypoxia, while less efficient knockdown produces a higher penetrance of tracheal overgrowth and excess branching in a manner similar to ‘late’ hypoxia. Given the graded tracheal phenotypes produced by 0.5% and 1% O2 exposure, this range of dVHLi phenotypes seems very likely to reflect variable efficiency of dVHL knockdown. These data thus suggest that chronic activation of sima in the embryonic tracheal placodes impairs subsequent tracheal migration and fusion events, while more mild activation of sima leads to excess tracheal cell branching and growth later in embryonic development. Analysis of tracheal architecture in btl-Gal4,UAS-sima embryos confirms that overexpression of exogenous sima is sufficient block placode branching and migration in the embryo (Supplemental Fig. 1). To test whether endogenous sima is in fact required to promote both types of dVHLi tracheal phenotypes, the btl-Gal4 driver was used to drive the dVHLi lines in the tracheal systems of sima07607/+ or sima07607/sima07607 embryos (Fig. 4), which can survive up to and through the pupal phase (Centanin et al., 2005 and our observations). Initial analysis on was performed on multiple dVHLi lines, all of which gave a similar result (Fig. 4A and data not shown). As expected based on the evolutionarily conserved relationship between VHL and HIF-1α, removing one copy of sima significantly reduced the penetrance of dVHLi34B3 tracheal phenotypes, from 68% (n=48) to 24% (n=53) (Fig. 4A and Supplemental Table I). Moreover, removing the remaining wild type copy of sima further suppressed the frequency of dVHLi34B3 tracheal phenotypes to background levels equivalent to that seen in control btl-Gal4,UAS-Adf1i RNAi embryos, indicating that sima is absolutely required for dVHLi tracheal phenotypes. Reducing sima gene dosage by half is also sufficient to completely suppress the pupal lethality of the btl-Gal4,UAS-dVHLi34B3 genotype back to adult viability (Fig. 4B and Supplemental Table IV). Heterozygosity for a genomic deletion removing the sima locus (Df(3R)BSC502) is also sufficient to suppress dVHLi34B3 pupal lethality (Fig. 4B and Supplemental Table IV) suggesting the ability of the sima07607 allele to suppress dVHLi phenotypes is due to loss of sima rather than some cryptic mutation in the background. These strong genetic interactions between dVHL and sima validate the specificity of the UAS-dVHLi system as a technique to antagonize dVHL activity in vivo, and indicate that sima is a major effector of the morphological changes that occur in the embryonic tracheal system in response to either reduced oxygen availability or loss of dVHL.
dVHL suppresses btl expression in the embryo
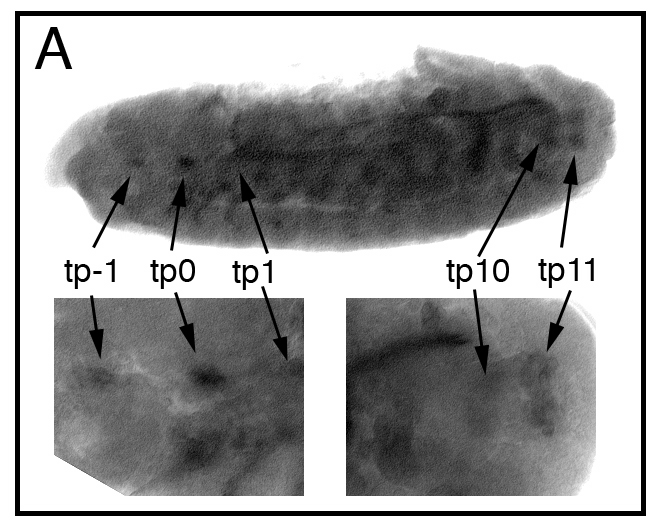
By analogy to the larval tracheal system in which sima-driven expression of the btl FGF receptor induces excess growth and branching of terminal cells (Centanin et al., 2008), we next sought to determine whether both aspects of dVHLi embryonic tracheal phenotype — defective migration and sinuous overgrowth and branching — were also dependent on Btl/FGF signaling. Recessive lethal alleles of the btl receptor (btldev1) (Kennison and Tamkun, 1988), the bnl ligand (bnlP1) (Sutherland et al., 1996), and the trh bHLH-PAS transcription factor responsible for induction of btl expression in the tracheal placode (trh10512) (Wilk et al., 1996), were placed in the background of btl-Gal4,UAS-dVHLi34B3. Embryonic tracheal architecture was analyzed with the mAb2A12 antibody and the fraction of embryos showing either migration defects or excess growth/branching defects was calculated as a percentage of the total (Fig. 5A and Supplemental Table I). Loss of one copy of either btl or bnl led to a significant reduction in the penetrance of dVHLi34B3 tracheal phenotypes from 68% (n=48) to 26% (n=54) and 25% (n=55) respectively, comparable to heterozygosity for the sima07607 allele. Tracheal phenotypes that result from reduced dVHL activity thus display equal sensitivity to reductions in Btl signal strength or to reduced activity of the btl transcriptional activator Sima. This sensitivity might reflect a role for dVHL as an antagonist of a pathway that operates in parallel to Btl or a role for dVHL as a direct antagonist of Btl (Hsu et al., 2006), although dVHL inactivation had little effect on the levels or trafficking of a Btl:GFP fusion protein (Supplemental Fig. 2). Precedent with the larval function of the HPH fga suggests that dVHL may act through Sima to control btl transcription in the embryo more directly (Centanin et al., 2008). Consistent with this hypothesis, expression of a wild type btl transgene specifically in tracheal cells produces tracheal overgrowth (Fig. 5B), DT breaks (Mortimer and Moberg, 2007), and excess larval TTBs (Supplemental Table III) that resemble dVHLi tracheal-specific knockdown phenotypes. A constitutively active btl allele (btlλ) has previously been shown to block early migration events in the embryonic tracheal system and also lead to secondary branch duplication (Lee et al., 1996b). Moreover, expression of either of two downstream effectors of Btl — Ras85D (Lee et al., 1996a) or the pnt transcription factor (Klaes et al., 1994) — disrupts tracheal branching in the embryo (Figs. 5C, D). To test whether dVHL restricts btl transcription in vivo, we first made use of the prior observation that ubiquitous overexpression of sima in the embryo is sufficient to induce specific groups of non-tracheal cells to form ectopic btl-positive placodes (Centanin et al., 2008). sima can induce two ectopic placodes (tp1 and tp0) anterior to the first tracheal placode (tp1), and occasionally a third (tp11) distal to the last placode (tp10). These cryptic placodes also appear in response to trh (Wilk et al., 1996) and appear to be primed to adopt a tracheal fate by spatially restricted signals like vvl (Boube et al., 2000; Zelzer and Shilo, 2000a) that sensitize the btl promoter to other trans-activators. Significantly, ubiquitous knockdown of dVHL leads to the appearance of an ectopic btl-positive placode at the tp0 position (Figs. 5F, G). At lower frequency btl-expressing cells also appear at the tp1 and tp11 positions as well (Supplemental Fig. 3). Loss of dVHL is thus capable of mimicking the effect of trh or sima overexpression on patterns of btl transcription. Expression of Trh protein in this genetic background remains restricted to placodes 1–10 (Fig. 5P), again supporting the notion that expression of dVHLi leads specifically to sima-mediated phenotypes. To test whether reduced dVHL activity also results in higher levels of btl transcription in differentiated tracheal cells, UAS-dVHLi/+ and UAS-dVHLi/btlGal4,UAS-GFP embryos at either stage 11 (pre-migratory) or stage 15 (post-migratory) were hybridized with a btl RNA probe and developed using a tyramide-amplification protocol (Merabet et al., 2005). Samples from each stage were processed together and genotyped afterward by GFP expression. At stage 11, levels of btl transcript are elevated in tracheal placodes expressing the dVHLi knockdown transgene compared to control embryos (Figs. 5I vs. 5M). Similarly stage 15 embryos carrying the dVHLi knockdown transgene show increased levels of btl transcript in cells of the DT, the DBs, and the transverse connectives (Figs. 5K vs. 5O). dVHL thus restricts btl transcription in tracheal cells at multiple stages of embryonic development. In view of the genetic requirement for btl in dVHLi tracheal phenotypes, these data indicate that Sima-driven elevation in btl expression and activity is a major cause of both the ‘early’ and ‘late’ tracheal responses to hypoxia.
Fig. 5.

dVHLi expression leads to ectopic btl transcription. (A) Penetrance of tracheal defects in embryos with btl-Gal4 driven expression of UAS-Adf1i control RNAi or the UAS-dVHLi34B3 transgene in tracheal cells. btl-Gal4:UAS-dVHLi34B3 phenotypes are dominantly suppressed by alleles of the FGF pathway components btl or bnl, but not by alleles of the transcription factor trh (*p<0.001 relative to btl-Gal4:UAS-dVHLi34B3). (B—D) Lateral images of embryos stained with the tracheal lumen marker mAb2A12. btl-Gal4:UAS-dVHLi34B3 phenotypes are phenocopied by btl-Gal4 driven overexpression of (B) btl, or FGF downstream pathway components (C) Ras85D.V12 (a constitutively active form of Ras85D), or (D) the MAPK transcriptional effector pnt.P1. (E—G) Lateral images of embryos hybridized with a btl anti-sense probe. (E) Control actin-Gal4 embryos display the normal complement of ten btl-positive tracheal placodes (tp1 through tp10). (F, G) actin-Gal4 driven expression of dVHLi31A2 causes ectopic transcription of btl in cryptic tracheal placodes (indicated as tp0). Control UAS-dVHLi31A2 stage 11 (H, I) and stage 15 (J, K) embryos hybridized with a btl anti-sense probe and stained with α-GFP. btl is expressed in all cells of stage 11 tracheal placodes, but only in migrating tracheal cells of stage 15 embryos. UAS-dVHLi31A2/btl-Gal4,UAS-GFP stage 11 (L, M) and stage 15 (N, O) embryos hybridized with a btl anti-sense probe and stained with α-GFP, showing elevated btl expression. Embryos were genotyped by expression of GFP. Arrowhead indicates DT. (P) Stage 11 actin-Gal4:UAS-dVHLi31A2 embryo stained with α-Trh. Trh staining shows the wild type pattern of ten Trh positive tracheal placodes (tp1 through tp10).
dVHL and ago synergize to control embryonic tracheogenesis
The sima and trh transcription factors are each capable of interacting with the btl promoter to induce btl expression in cultured cells (Centanin et al., 2008; Ohshiro and Saigo, 1997). Previous work has shown that trh alleles dominantly suppress tracheal migration defects resulting from ectopic expression of btl in DT fusion cells lacking the archipelago (ago) gene, which encodes an F-box protein that binds Trh and recruits it into a SCF ubiquitin ligase complex for polyubiquitination and proteasome-dependent degradation (Moberg et al., 2001; Mortimer and Moberg, 2007). Thus the observations that dVHLi tracheal phenotypes require sima but are only minimally sensitive to trh gene dosage (Fig. 5A and Supplemental Table I) and that Trh expression is not altered by loss of dVHL (Fig. 5P), suggests that the ago and dVHL ubiquitin ligases restrict btl expression by genetically distinct pathways. To test if dVHL and ago alleles might then collaborate to deregulate btl-dependent branching and migration events, the frequency of the two types of dVHLi embryonic tracheal phenotypes — stunted migration or ectopic branching and sinuous overgrowth — were examined in a background heterozygous for the ago3 strong loss-of-function allele (Moberg et al., 2001). Two ‘weaker’ dVHL lines (i11A2 and i4B2) that gave a higher frequency of sinuous overgrowth and a somewhat lower frequency of migration defects were used for this analysis. In both cases, addition of the ago allele shifts the most frequent tracheal phenotype from sinuous overgrowth to stunted branch migration (Figs. 6A, B) and increases the overall fraction of embryos with migration defects to 70–75% (Fig. 6C and Supplemental Table II). Moreover, embryos trans-heterozygous for the ago3 allele and a genomic deficiency that removes the dVHL locus (Exel6060/+;ago3/+) show an elevated frequency of tracheal fusion and migration defects compared to either lesion alone (Figs. 6D, E). The dVHL deficiency also dominantly enhances the number of DT breaks per affected ago1/ago3 embryo from 1.2±0.09 (n=39) to 2.5±0.19 (n=41) (p < 1×10−6) (Figs. 6F, G). Thus, dVHL and ago act synergistically to control migration and fusion events in the developing tracheal system. Interestingly, examination of L3 larvae that co-express dVHLi31A2 and a dominant-negative form of ago (UAS-agoΔF) (Mortimer and Moberg, 2007) in tracheal cells shows no enhancement of TTB branching beyond that observed with dVHL knockdown alone (Supplemental Table III). The synergy between ago and dVHL may thus be specific to the early embryonic tracheal system. To test whether the functional interaction between dVHL and ago is indeed specific to a particular developmental phase, the ‘weaker’ dose of 1% O2 was used as a switch to activate the dVHL/sima transcriptional program at ‘early’ or ‘late’ embryonic time points (according to the scheme depicted in Fig. 1) in either wild type (+/+) embryos or ago3/+ embryos. Tracheal architecture was then analyzed with mAb2A12. As described above (Fig. 1G), ‘early’ exposure to 1% O2 produces tracheal phenotypes at fairly low penetrance (12%). Significantly, reducing the dose of ago leads to a more than 3-fold increase in the penetrance of tracheal phenotypes in response to this 1% O2 treatment (Fig. 6H and Table 1). This includes an approximate doubling of migration defects (from 5% to 9% of embryos), appearance of duplicated secondary branches (from 0% to 5% of embryos), and an increase in sinuous overgrowth (from 2% to 34% of embryos). Notably, this enhancement is specific to the ‘early’ time point: exposure to 1% O2 at the ‘late’ embryonic time point produced the same penetrance of tracheal phenotypes in ago3/+ and control +/+ embryos (Fig. 6H and Table 1). Moreover, ago heterozygosity did not affect the extent of DT growth induced in response to ‘late’ 1% O2 (21±3.1% longer in +/+, n=4, [see yellow bar in Fig. 1H]; 20±2.6% longer in ago3/+, n=4), demonstrating that a phenotype that is a dose-sensitive readout of pathway activity is also unaffected by lowered ago activity. Reducing ago activity thus specifically sensitizes the ‘early’ embryonic tracheal system to architectural changes in response to milder doses of hypoxia. As ago restricts btl transcription in the developing embryonic tracheal system (Mortimer and Moberg, 2007), this stage-specific synergy between ago and hypoxia appears to define a window of development during which activation of the dVHL/sima pathway induces a program of gene expression that conflicts with normal btl/bnl migration cues. Removing a copy of ago is predicted to enhance this conflict by elevating Trh activity and btl transcription.
Fig. 6.

ago interacts with dVHL in tracheal morphogenesis. (A—D) Lateral views of embryos stained with the lumenal marker mAb2A12. The btl-Gal4:UAS-dVHLi11A2 phenotype (A) is enhanced by reducing the genetic dosage of ago (B). (C) Enhancement of the penetrance of migration defects in btl-Gal4:UAS-dVHLi embryos by the ago3 allele (*p<0.05 compared to btl-Gal4:UAS-dVHLi alone). (D—G) Alleles of ago also interact with a deletion (Exel6060) uncovering the dVHL locus. (D) Lateral view of an Exel6060/+;ago3/+ embryo stained with mAb2A12 displaying defects in tracheal morphogenesis. (E) Quantification of the trans-heterozygous interaction between ago and Exel6060 in tracheal formation (*p<0.001 compared to wt). (F) Lateral view of an Exel6060/+;ago1/3 embryo stained with mAb2A12. (G) Frequency of embryos with 1, 2, or 3+ DT breaks shows enhancement of the ago1/3 tracheal phenotype by Exel6060. (H) Penetrance of all classes of tracheal defects (breaks, overgrowth, missing or duplicated branches) in w1118 and ago3/+ embryos following exposure to 1% O2. (*p<0.005 relative to w1118).
Discussion
Hypoxia-induced remodeling of tracheal terminal cells represents the response of a developed larval tracheal system to reduced levels of O2 in the environment. By contrast, the response of the developing embryonic tracheal system to systemic hypoxia has not been as well characterized. In light of the observation that embryonic tracheal cells display hypoxia-induced activation of a Sima-reporter (Lavista-Llanos et al., 2002) and that sima promotes btl expression in larval tracheal cells (Centanin et al., 2008), embryonic exposure to hypoxia may thus produce a situation in which hard-wired btl/bnl patterning signals in the embryo come into conflict with the type of sima/btl-driven plasticity of tracheal cell branching seen in the larva. Here we examine the effect of hypoxia on embryonic tracheal branching and migration. We find that hypoxia has dramatic effects on the patterns of morphogenesis of the primary and secondary tracheal branches. Surprisingly, varying the timing and severity of hypoxic challenge is able to shift the outcome from severely stunted tracheal branching to excess branch number and enhanced branch growth. Genetic and molecular data indicate that both classes of phenotypes, stunting and overgrowth, involve regulation of sima activity and btl transcription by dVHL, and that the effects of hypoxia on tracheal development can be mimicked in normoxia by tracheal-specific knockdown of dVHL. This observation confirms a central role for dVHL in restricting the hypoxic response in vivo, and identifies a role for dVHL as a required inhibitor of sima and btl during normal tracheogenesis.
Since Trh and Sima/HIF-1α share a similar consensus DNA binding site (Jiang and Crews, 2007; Gorr et al., 2004; Crews and Fan, 1999), it is likely that the tracheal phenotypes elicited by either hypoxia or dVHL knockdown are to some degree the product of a combined ‘Trh/Sima-like’ transcriptional activity in tracheal cells. This conclusion is supported both by the general phenotypic similarity (i.e. migration and overgrowth defects) between hypoxia/dVHL knockdown and trh overexpression (Mortimer and Moberg, 2007), by the modest ability of trh alleles to suppress dVHLi phenotypes, and by the previously demonstrated overlap of transcriptional activity between Trh and human HIF-1α (Zelzer et al., 1997). Indeed, Trh is well-established as a required activator of developmental btl expression. However, because the excess Btl activity that occurs in hypoxia or in the absence of dVHL occurs independently of a change in Trh expression (Fig. 5P), it thus appears to be mediated largely by increased sima activity.
Our analysis suggests that there are two distinct developmental ‘windows’ of embryogenesis during which hypoxia has opposite effects on tracheal branching. The first corresponds to a period immediately before and during primary branch migration that is relatively insensitive to hypoxia. Embryos in this stage show a minimal response to 1% O2, but show a nearly complete arrest of migration in 0.5% O2. Interestingly, a prior study found that similarly staged embryos (stage 11) respond to complete anoxia by prolonged developmental arrest, from which they can emerge and resume normal development (Wingrove and O’Farrell, 1999). These somewhat paradoxical results — that acute hypoxia is more detrimental to development than chronic anoxia — might be explained by the observation that chronic exposure to low O2 induces Sima activity throughout the embryo while acute exposure activates Sima only in tracheal cells (Lavista-Llanos et al., 2002). The former scenario may result in coordinated developmental and metabolic arrest throughout the organism, while in the latter scenario developmental patterns of gene expression in non-tracheal cells may proceed such that tracheal cells emerging from an ‘early’ hypoxic response find an embryonic environment in which developmentally hard-wired migratory signals emanating from non-tracheal cells have ceased.
The second type of tracheal response occurs during a later ‘window’ of embryogenesis after btl/bnl-driven primary and secondary branch migration and fusion are largely complete. It involves sinuous overgrowth of the primary and secondary branches (this study and Arquier et al., 2006), and duplication of secondary branches. As in the ‘early’ response, ‘late’ hypoxic phenotypes are controlled by the dVHL/sima pathway, yet unlike the ‘early’ response, these phenotypes occur at high penetrance even at 1% O2. Thus the ‘late’ embryonic tracheal system is relatively sensitized to hypoxia and responds with increased branching in a manner similar to larval terminal cells. Indeed, much as larval branching increases with decreasing O2 levels (Jarecki et al., 1999), we observe that dorsal trunk growth in the late embryo is graded to the degree of hypoxia. The mechanism underlying the differential sensitivity of the ‘early’ and ‘late’ tracheal system may be quite complex. However, we find that tracheogenesis can be sensitized to hypoxia by reducing activity of ago, a ubiquitin ligase component that restricts btl transcription in tracheal cells via its role in degrading the Trh transcription factor (Mortimer and Moberg, 2007). Increasing transcriptional input on the btl promoter thus appears to sensitize ‘early’ tracheal cells to hypoxia. As Sima also controls btl transcription, one explanation of the difference in sensitivity between different embryonic stages may thus lie in differences in the activation state of the btl promoter. If so then the activity of the endogenous btl regulatory network (Boube et al., 2000; Kuhnlein and Schuh, 1996; Llimargas and Casanova, 1997; Mortimer and Moberg, 2007; Ohshiro et al., 2002; Ohshiro and Saigo, 1997; Samakovlis et al., 1996; Zelzer and Shilo, 2000b) may be an important determinant of the threshold of hypoxia required to elicit changes in tracheal architecture.
An organism can have its hypoxic response triggered in two ways, either by systemic exposure of the whole organism to a reduced O2 environment or by localized hypoxia produced by increased O2 consumption in metabolically active tissues. Data from this study and others (Centanin et al., 2008; Jarecki et al., 1999) suggests there may be distinctions between these two triggers. Exposing larvae or embryos to a systemic pulse of hypoxia results in a ‘btl-centric’ response specifically in tracheal cells. Outside of an ‘early’ vulnerable period which corresponds to embryonic branch migration and fusion, elevated Btl activity in embryonic tracheal cells promotes branch duplications and overgrowth similar to that seen in larvae (Centanin et al., 2008). By contrast, tracheal growth induced by localized hypoxia in the larva has been suggested to involve a ‘bnl-centric’ model in which the hypoxic tissue secretes Bnl and recruits new tracheal branching (Jarecki et al., 1999). Whether this type of mechanism operates in embryos, or whether embryos ever experience localized hypoxia in non-tracheal cells, has not been established.
Our data indicate that dVHL is a central player in the hypoxic response pathway in embryonic and larval tracheal cells. A prior study found that injection of dVHL dsRNA into syncytial embryos disrupted normal tracheogenesis (Adryan et al., 2000), but was technically limited in its ability to conduct a detailed analysis of dVHL function in development and homeostasis. We find that dVHL knockdown specifically in tracheal cells mimics the effect of systemic hypoxia on embryonic tracheal architecture and larval terminal cell branching. dVHL knockdown thus phenocopies loss of the HPH gene fga, which normally functions to target Sima to the dVHL ubiquitin ligase in normoxia (Centanin et al., 2005). Moreover, all phenotypes that result from reduced dVHL expression can be rescued by reducing sima activity, suggesting that Sima is the major target of dVHL in the tracheal system. These data support a model in which dVHL, fga, and sima function as part of a conserved VHL/HPH/HIF-1α pathway to control tracheal morphogenesis in embryos and larvae. The btl receptor appears to be an important target of this pathway in embryonic (this study) and larval (Centanin et al., 2008) tracheal cells. Knockdown of dVHL elevates btl transcription in embryonic placodes and tracheal branches, and removal of a copy of the gene effectively suppresses dVHL tracheal phenotypes. Reciprocally, overexpression of wild type btl in embryonic tracheal cells can produce migration defects and sinuous overgrowth (this study and Mortimer and Moberg, 2007), while expression of a constitutively active btl chimera (btlλ) also leads to primary branch stunting and duplication of secondary branches (Lee et al., 1996b). Interestingly, pupal lethality associated with tracheal-specific knockdown of dVHL is not sensitive to the dose of btl, but is dependent on sima. Thus the dVHL/sima pathway may have btl independent effects on tracheal cells in later stages of development.
In addition to sima and Btl/FGF pathway mutants, dVHL also shows very strong genetic interactions with alleles of the ago ubiquitin ligase subunit. The interactions are consistent with the ability of ago to modulate hypoxia sensitivity in the embryo, and suggest a speculative model in which each ligase acts through its own target — Sima or Trh — to regulate btl transcription in tracheal cells. Given that the human orthologs of dVHL and ago are significant tumor suppressor genes, it is intriguing to consider whether their ability to co-regulate tubular morphogenesis in the Drosophila embryo is conserved in mammalian development and disease.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We apologize to those whose work we did not cite due to space constraints. We wish to thank P. Wappner, T. Hsu, S. Sanyal, D. Andrew, M.Csete, and S. Merabet for gifts of reagents and protocols, and S. Burdick for technical assistance. We thank the Developmental Studies Hybridoma Bank, the Bloomington Drosophila Stock Center, and the Drosophila Genomics Resource Center for fly stocks, antibodies and cDNAs. This work was funded by the American Heart Association (Predoctoral Fellowship 0715483B to NTM) and the National Institute of Health (GM079242 to KHM).
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.ydbio.2009.03.001.
References
- Adryan B, Decker HJ, Papas TS, Hsu T. Tracheal development and the von Hippel-Lindau tumor suppressor homolog in Drosophila. Oncogene. 2000;19:2803–2811. doi: 10.1038/sj.onc.1203611. [DOI] [PubMed] [Google Scholar]
- Arquier N, Vigne P, Duplan E, Hsu T, Therond PP, Frelin C, D’Angelo G. Analysis of the hypoxia-sensing pathway in Drosophila melanogaster. Biochem. J. 2006;393:471–480. doi: 10.1042/BJ20050675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso T, Yamazaki K, Aigaki T, Kitajima S. Drosophila von Hippel-Lindau tumor suppressor complex possesses E3 ubiquitin ligase activity. Biochem. Biophys. Res. Commun. 2000;276:355–361. doi: 10.1006/bbrc.2000.3451. [DOI] [PubMed] [Google Scholar]
- Boube M, Llimargas M, Casanova J. Cross-regulatory interactions among tracheal genes support a co-operative model for the induction of tracheal fates in the Drosophila embryo. Mech. Dev. 2000;91:271–278. doi: 10.1016/s0925-4773(99)00315-9. [DOI] [PubMed] [Google Scholar]
- Centanin L, Ratcliffe PJ, Wappner P. Reversion of lethality and growth defects in Fatiga oxygen-sensor mutant flies by loss of Hypoxia-Inducible Factor-alpha/Sima. EMBO Rep. 2005;6:1070–1075. doi: 10.1038/sj.embor.7400528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centanin L, Dekanty A, Romero N, Irisarri M, Gorr TA, Wappner P. Cell autonomy of HIF effects in Drosophila: tracheal cells sense hypoxia and induce terminal branch sprouting. Dev. Cell. 2008;14:547–558. doi: 10.1016/j.devcel.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Crews ST, Fan CM. Remembrance of things PAS: regulation of development by bHLH-PAS proteins. Curr. Opin. Genet. Dev. 1999;9:580–587. doi: 10.1016/s0959-437x(99)00003-9. [DOI] [PubMed] [Google Scholar]
- Dammai V, Adryan B, Lavenburg KR, Hsu T. Drosophila awd, the homolog of human nm23, regulates FGF receptor levels and functions synergistically with shi/dynamin during tracheal development. Genes Dev. 2003;17:2812–2824. doi: 10.1101/gad.1096903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio PJ, Ubersax JA, O’Farrell PH. Hypoxia and nitric oxide induce a rapid, reversible cell cycle arrest of the Drosophila syncytial divisions. J. Biol. Chem. 2001;276:1930–1937. doi: 10.1074/jbc.M003911200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RM, Xu T, Haddad GG. Cell cycle progression and cell division are sensitive to hypoxia in Drosophilamelanogaster embryos. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001;280:R1555–R1563. doi: 10.1152/ajpregu.2001.280.5.R1555. [DOI] [PubMed] [Google Scholar]
- Ghabrial A, Luschnig S, Metzstein MM, Krasnow MA. Branching morphogenesis of the Drosophila tracheal system. Annu. Rev. Cell Dev. Biol. 2003;19:623–647. doi: 10.1146/annurev.cellbio.19.031403.160043. [DOI] [PubMed] [Google Scholar]
- Giordano E, Rendina R, Peluso I, Furia M. RNAi triggered by symmetrically transcribed transgenes in Drosophila melanogaster. Genetics. 2002;160:637–648. doi: 10.1093/genetics/160.2.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorr TA, Cahn JD, Yamagata H, Bunn HF. Hypoxia-induced synthesis of hemoglobin in the crustacean Daphnia magna is hypoxia-inducible factor-dependent. J. Biol. Chem. 2004;279:36038–36047. doi: 10.1074/jbc.M403981200. [DOI] [PubMed] [Google Scholar]
- Gorr TA, Gassmann M, Wappner P. Sensing and responding to hypoxia via HIF in model invertebrates. J. Insect Physiol. 2006;52:349–364. doi: 10.1016/j.jinsphys.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Groppe J, Ducker K, Treisman R, Hafen E, Affolter M, Krasnow MA. The pruned gene encodes the Drosophila serum response factor and regulates cytoplasmic outgrowth during terminal branching of the tracheal system. Development. 1996;122:1353–1362. doi: 10.1242/dev.122.5.1353. [DOI] [PubMed] [Google Scholar]
- Hsu T, Adereth Y, Kose N, Dammai V. Endocytic function of von Hippel-Lindau tumor suppressor protein regulates surface localization of fibroblast growth factor receptor 1 and cell motility. J. Biol. Chem. 2006;281:12069–12080. doi: 10.1074/jbc.M511621200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki J, Johnson E, Krasnow MA. Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell. 1999;99:211–220. doi: 10.1016/s0092-8674(00)81652-9. [DOI] [PubMed] [Google Scholar]
- Jiang L, Crews ST. Transcriptional specificity of Drosophila dysfusion and the control of tracheal fusion cell gene expression. J. Biol. Chem. 2007;282:28659–28668. doi: 10.1074/jbc.M703803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr. The von Hippel-Lindau protein, HIF hydroxylation, and oxygen sensing. Biochem. Biophys. Res. Commun. 2005;338:627–638. doi: 10.1016/j.bbrc.2005.08.165. [DOI] [PubMed] [Google Scholar]
- Kaelin WG, Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kennison JA, Tamkun JW. Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc. Natl. Acad. Sci. U. S. A. 1988;85:8136–8140. doi: 10.1073/pnas.85.21.8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Lee YS, Harris D, Nakahara K, Carthew RW. The RNAi pathway initiated by Dicer-2 in Drosophila. Cold Spring Harb. Symp. Quant. Biol. 2006;71:39–44. doi: 10.1101/sqb.2006.71.008. [DOI] [PubMed] [Google Scholar]
- Klaes A, Menne T, Stollewerk A, Scholz H, Klambt C. The Ets transcription factors encoded by the Drosophila gene pointed direct glial cell differentiation in the embryonic CNS. Cell. 1994;78:149–160. doi: 10.1016/0092-8674(94)90581-9. [DOI] [PubMed] [Google Scholar]
- Klambt C, Glazer L, Shilo BZ. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev. 1992;6:1668–1678. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- Kuhnlein RP, Schuh R. Dual function of the region-specific homeotic gene spalt during Drosophila tracheal system development. Development. 1996;122:2215–2223. doi: 10.1242/dev.122.7.2215. [DOI] [PubMed] [Google Scholar]
- Lavista-Llanos S, Centanin L, Irisarri M, Russo DM, Gleadle JM, Bocca SN, Muzzopappa M, Ratcliffe PJ, Wappner P. Control of the hypoxic response in Drosophila melanogaster by the basic helix—loop—helix PAS protein similar. Mol. Cell Biol. 2002;22:6842–6853. doi: 10.1128/MCB.22.19.6842-6853.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Feig L, Montell DJ. Two distinct roles for Ras in a developmentally regulated cell migration. Development. 1996a;122:409–418. doi: 10.1242/dev.122.2.409. [DOI] [PubMed] [Google Scholar]
- Lee T, Hacohen N, Krasnow M, Montell DJ. Regulated Breathless receptor tyrosine kinase activity required to pattern cell migration and branching in the Drosophila tracheal system. Genes Dev. 1996b;10:2912–2921. doi: 10.1101/gad.10.22.2912. [DOI] [PubMed] [Google Scholar]
- Llimargas M, Casanova J. ventral veinless, a POU domain transcription factor, regulates different transduction pathways required for tracheal branching in Drosophila. Development. 1997;124:3273–3281. doi: 10.1242/dev.124.17.3273. [DOI] [PubMed] [Google Scholar]
- Lubarsky B, Krasnow MA. Tube morphogenesis: making and shaping biological tubes. Cell. 2003;112:19–28. doi: 10.1016/s0092-8674(02)01283-7. [DOI] [PubMed] [Google Scholar]
- Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- Manning G, Krasnow MA. Development of the Drosophila tracheal system. In: Bate M, Arias A. Martinez, editors. The Development of Drosophila melanogaster. Cold Spring Harbor Press; Cold Spring Harbor, New York: 1993. pp. 609–685. [Google Scholar]
- Merabet S, Ebner A, Affolter M. The Drosophila Extradenticle and Homothorax selector proteins control branchless/FGF expression in mesodermal bridge-cells. EMBO Rep. 2005;6:762–768. doi: 10.1038/sj.embor.7400462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001;413:311–316. doi: 10.1038/35095068. [DOI] [PubMed] [Google Scholar]
- Mortimer NT, Moberg KH. The Drosophila F-box protein Archipelago controls levels of the Trachealess transcription factor in the embryonic tracheal system. Dev. Biol. 2007;312:560–571. doi: 10.1016/j.ydbio.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu JR, Chen W, Hu S, Crews ST. The Drosophila melanogaster similar bHLH-PAS gene encodes a protein related to human hypoxia-inducible factor 1 alpha and Drosophila single-minded. Gene. 1996;172:249–254. doi: 10.1016/0378-1119(96)00060-1. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Saigo K. Transcriptional regulation of breathless FGF receptor gene by binding of TRACHEALESS/dARNT heterodimers to three central midline elements in Drosophila developing trachea. Development. 1997;124:3975–3986. doi: 10.1242/dev.124.20.3975. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Emori Y, Saigo K. Ligand-dependent activation of breathless FGF receptor gene in Drosophila developing trachea. Mech. Dev. 2002;114:3–11. doi: 10.1016/s0925-4773(02)00042-4. [DOI] [PubMed] [Google Scholar]
- Samakovlis C, Hacohen N, Manning G, Sutherland DC, Guillemin K, Krasnow MA. Development of the Drosophila tracheal system occurs by a series of morphologically distinct but genetically coupled branching events. Development. 1996;122:1395–1407. doi: 10.1242/dev.122.5.1395. [DOI] [PubMed] [Google Scholar]
- Shiga Y, Tanaka-Matakatsu M, Hayashi S. A nuclear GFP/beta-galactosidase fusion protein as a marker for morphogenesis in living Drosophila. Dev. Growth Differ. 1996;38:99–106. [Google Scholar]
- Sonnenfeld M, Ward M, Nystrom G, Mosher J, Stahl S, Crews S. The Drosophila tango gene encodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development. 1997;124:4571–4582. doi: 10.1242/dev.124.22.4571. [DOI] [PubMed] [Google Scholar]
- Sutherland D, Samakovlis C, Krasnow MA. branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87:1091–1101. doi: 10.1016/s0092-8674(00)81803-6. [DOI] [PubMed] [Google Scholar]
- Tsarouhas V, Senti KA, Jayaram SA, Tiklova K, Hemphala J, Adler J, Samakovlis C. Sequential pulses of apical epithelial secretion and endocytosis drive airway maturation in Drosophila. Dev. Cell. 2007;13:214–225. doi: 10.1016/j.devcel.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Wilk R, Weizman I, Shilo BZ. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 1996;10:93–102. doi: 10.1101/gad.10.1.93. [DOI] [PubMed] [Google Scholar]
- Wingrove JA, O’Farrell PH. Nitric oxide contributes to behavioral, cellular, and developmental responses to low oxygen in Drosophila. Cell. 1999;98:105–114. doi: 10.1016/S0092-8674(00)80610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer E, Shilo BZ. Cell fate choices in Drosophila tracheal morphogenesis. Bioessays. 2000a;22:219–226. doi: 10.1002/(SICI)1521-1878(200003)22:3<219::AID-BIES3>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Shilo BZ. Interaction between the bHLH-PAS protein Trachealess and the POU-domain protein Drifter, specifies tracheal cell fates. Mech. Dev. 2000b;91:163–173. doi: 10.1016/s0925-4773(99)00295-6. [DOI] [PubMed] [Google Scholar]
- Zelzer E, Wappner P, Shilo BZ. The PAS domain confers target gene specificity of Drosophila bHLH/PAS proteins. Genes Dev. 1997;11:2079–2089. doi: 10.1101/gad.11.16.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.