

SUMMARY
E. coli RecE protein is part of the classical RecET recombination system that has recently been employed in powerful new methods for genetic engineering. RecE binds to free dsDNA ends and processively digests the 5′-ended strand to form 5′-mononucleotides and a 3′-overhang that is a substrate for single strand annealing promoted by RecT. Here, we report the crystal structure of the C-terminal nuclease domain of RecE at 2.8 Å resolution. RecE forms a toroidal tetramer with a central tapered channel that is wide enough to bind dsDNA at one end, but is partially plugged at the other end by the C-terminal segment of the protein. Four narrow tunnels, one within each subunit of the tetramer, lead from the central channel to the four active sites, which lie about 15 Å from the channel. The structure, combined with mutational studies, suggests a mechanism in which dsDNA enters through the open end of the central channel, the 5′-ended strand passes through a tunnel to access one of the four active sites, and the 3′-ended strand passes through the plugged end of the channel at the back of the tetramer.
INTRODUCTION
Homologous recombination is a fundamental cellular process for the repair of dsDNA breaks and the generation of genetic diversity (Kuzminov, 1999). A central pathway for homologous recombination in all cells involves RecA protein, or Rad51 in eukaryotes, which polymerizes on a region of ssDNA exposed at a site of damage to form a helical nucleoprotein filament that promotes homologous recombination by a strand invasion mechanism (Lusetti and Cox, 2002; Bell, 2005). Phage-derived recombination systems such as RecET, encoded within a cryptic rac prophage found in certain strains of E. coli, and Redαβ of bacteriophage γ, promote homologous recombination via a more simplified pathway known as single strand annealing (Stahl et al., 1997; Kolodner et al., 1994). These recombination systems consist of a 5′-3′ exonuclease, RecE or Redα (λ exonuclease), that resects the DNA end created at the break to form a 3′-overhang, and a second protein, RecT or Redβ (β protein), that loads onto the overhang to promote its annealing with a complementary strand of ssDNA (Kmiec and Holloman, 1981; Muniyappa & Radding, 1986; Hall et al., 1993). The exonuclease and recombinase proteins of each system form a specific protein-protein interaction that may serve to load the recombinase onto the 3′-overhang as it is generated by the exonuclease (Muyrers et al., 2000; Radding et al., 1971). Due to their simplicity, high efficiency, and ability to work at short regions of homology, RecET and Redαβ have been employed in powerful new methods for genetic engineering, termed “recombineering” or “ET cloning” (Poteete, 2001; Muyrers et al., 2001; Copeland et al., 2001). Genes encoding the proteins of this recombination system are found in a wide variety of bacterial and phage genomes, and are currently being exploited for genetic engineering in organisms such as Mycobacterium tuberculosis (van Kessel and Hatfull, 2007). Due to their highly processive natures, the exonuclease enzymes are also being exploited in novel methods for nanopore DNA sequencing (Branton et al., 2008).
RecE and λ exonuclease are Mg2+-dependent enzymes that bind tightly to dsDNA ends and processively digest the 5′-ended strand to form 5′-mononucleotides and a 3′-ended ssDNA tail (Little, 1967; Joseph and Kolodner, 1983b). Hallmark features of these enzymes include their liberation of 5′-mononucleotides and ssDNA as the exclusive products, a total lack of activity on circular DNA substrates, very low activity on ssDNA, and high processivities. The crystal structure of λ exonuclease revealed a toroidal trimer with a central channel that is tapered from about 30 Å on one side to about 15 Å on the other (Kovall and Matthews, 1997). The funnel-shaped channel suggests a mode of action in which the trimer tracks along the duplex with dsDNA entering on one side, and ssDNA exiting on the other. The three active sites of the trimer each bind a Mg2+ and are located within a cleft on each subunit that is exposed to the central channel. The portion of λ exonuclease that forms the active site exhibits a fold dubbed the “restriction endonuclease-like” fold that is also seen in several type II restriction endonucleases, the nuclease domain of the RecB subunit of the RecBCD helicase-nuclease complex, the MutH DNA repair endonuclease, and several other nucleases (Kovall and Matthews, 1998; Singleton et al., 2004; Lee et al., 2005). The proteins within this diverse group of nuclease enzymes likely share a common evolutionary origin and related catalytic mechanisms (Kovall and Matthews, 1999).
RecE is a much larger protein than λ exonuclease (866 vs. 226 amino acids), but limited proteolysis and genetic studies have identified a ~300-residue C-terminal domain of RecE that has all of the activities of the full-length protein (Chang and Julin, 2001; Chu et al., 1989; Muyrers et al, 2000). This domain of RecE has essentially no detectable overall sequence homology with λ exonuclease, but segments of RecE and RecB containing the presumed active site residues can be aligned to one another (Aravind et al., 1999; Chang and Julin, 2001). Phylogenetic analyses indicate that RecB and λ exonuclease belong to distinct families within the endonuclease-like superfamily, and that RecE is more closely related to the RecB family (Aravind et al., 2000). Although RecE and λ exonuclease have essentially the same function, given the extensive differences in their amino acid sequences, there is some question as to how similar their structures will be, particularly at the quaternary level. To gain further insight into the structure and mechanism of this class of processive exonuclease enzymes, we have determined the crystal structure of the C-terminal nuclease domain of RecE. The structure of RecE reveals a toroidal tetramer that forms a central channel of similar size and shape as that seen in the λ exonuclease trimer. Comparison of the structures of RecE and λ exonuclease reveals common structural features that appear to be fundamental to their modes of action.
RESULTS
RecE Forms a Stable Tetramer in Solution
For structural studies of RecE we initially expressed and purified residues 564–866 (RecE564), which form the C-terminal nuclease domain identified by limited proteolysis (Chang and Julin, 2001). The available data indicate that the first ~550 residues of RecE are not structured and are dispensable for activity. Previous biophysical analyses of full-length RecE protein revealed that it forms an oligomer, but the precise number of subunits was not established (Joseph and Kolodner, 1983a). To definitively determine the oligomeric state of RecE564, analytical ultracentrifugation was performed (Figure 1). Sedimentation velocity indicated a single 6.3 S species with an estimated mass of 144 ± 7 kDa, compared to the calculated mass of 138.0 kDa for a tetramer of RecE564. A frictional coefficient of 1.6 indicated a non-spherical shape, consistent with a toroidal structure. To further verify the mass of this species, sedimentation equilibrium was performed at three different rotor speeds and sample concentrations ranging from 1.2 to 12 μM. A single-species fit was sufficient to model the observed data, and resulted in an experimental molecular weight of 137 ± 5 kDa, again consistent with a stable tetramer of RecE564. These results establish that the oligomeric state of the C-terminal nuclease domain of RecE, and by inference of the full-length protein, is a stable tetramer in solution. This is in notable contrast to λ exonuclease, which forms a trimer.
Figure 1. RecE Protein Forms a Stable Tetramer in Solution.
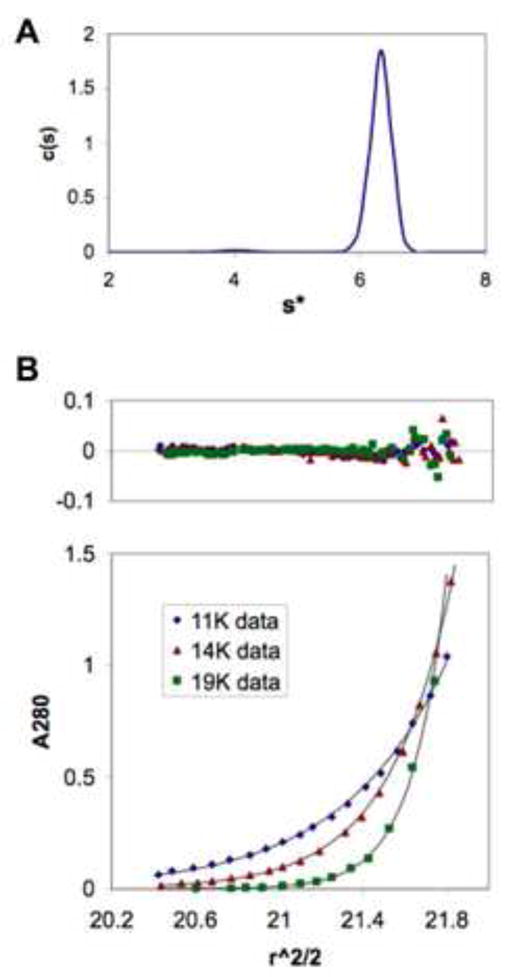
(A) Sedimentation coefficient distribution plot from a sedimentation velocity run of RecE564. A sharp peak corresponding to a single 6.3 S species is observed. The MW of this species estimated from the velocity data is 144 ± 7 kDa, consistent with a tetramer of RecE564 (calculated mass 138.0 kDa).
(B) Equilibrium sedimentation curves observed for RecE564 at three different rotor speeds. The upper panel shows the residuals of a fit of the data to a single species of mass 137 ± 5 kDa, again consistent with a tetramer.
Crystallization and Structure Determination
We were able to crystallize RecE564, but the crystals diffracted x-rays to a maximum resolution of only 3.5 Å at the synchrotron, with severe anisotropy. In genetic studies, shorter C-terminal fragments of RecE, beginning as far into the amino acid sequence as residue 606, exhibit full activity in recombination assays in vivo (Muyrers et al., 2000; Chu et al., 1989). Therefore, we expressed and purified residues 606–866 of RecE (RecE606), and obtained crystals of the protein that diffract to 2.8 Å resolution at the synchrotron. During the course of this work, we discovered that the construct we used to express RecE606 contained a P658L mutation relative to the sequence of recE from E. coli K12. Fortuitously, this mutation improves the diffraction of the crystals by about 1 Å resolution relative to crystals of RecE606 without the mutation. As measured using a fluorescence-based exonuclease assay, the P658L mutation does not affect the enzymatic activity of either RecE564 or RecE606 (Figure 2). However, the shorter RecE606 fragment, with or without the P658L mutation, exhibits significantly lower exonuclease activity than RecE564 in this assay, as has been reported previously using a gel-based assay (Muyrers et al., 2000). Importantly, despite its lower exonuclease activity in vitro, RecE606 actually exhibits 4-fold increased activity in recombination assays in vivo, as compared to longer fragments of the protein (Muyrers et al., 2000).
Figure 2. Exonuclease Activities of RecE Proteins.
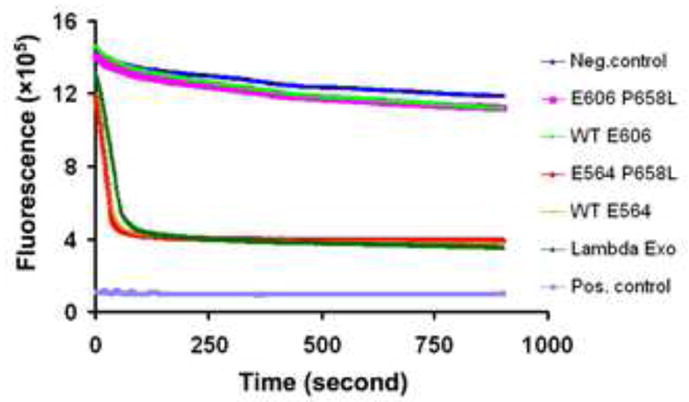
The plot shows the decrease in fluorescence of PicoGreen due to the digestion of a linear 2.7 KB dsDNA fragment upon addition of exonuclease enzyme. The different curves are for reactions with different exonuclease enzymes, as indicated at right. The upper blue curve is the negative control, measured from a reaction with storage buffer substituted for exonuclease enzyme. The positive control (lowest curve) shows the fluorescence of the heat-denatured 2.7 KB dsDNA fragment at ½ concentration to mimic the low signal of the ssDNA product. Notice that the exonuclease activity of RecE564 is similar to that of λ exonuclease, and much higher than that of RecE606. The P658L mutation, which dramatically improves the crystals of the RecE606 protein, has essentially no effect on the exonuclease activity of either RecE564 or RecE606.
Figure 4. Close-up Stereo View Comparing the Active Sites of RecE and RecB.
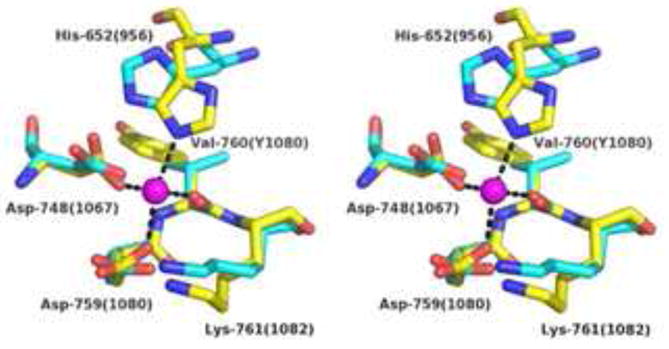
The structures of the RecB C-terminal domain (PDB code 1W36; Singleton et al., 2004) and RecE were aligned by least squares superposition (RMSD = 1.6 Å for 82 pairs of Cα atoms). The Ca2+ ion from the structure of RecB is shown as a magenta sphere. Residues of RecB that coordinate the Ca2+ are shown in stick form with carbon yellow, nitrogen blue, and oxygen red. Dashed lines indicate direct bonds to the Ca2+ ion. Notice that the coordination geometry is octahedral, with two of the six positions on the Ca2+ not occupied by protein atoms. Residues of RecE that superimpose on the ligand residues of RecB are shown in stick form with carbon in cyan. Labels indicate residues of RecE, with the corresponding residue numbers of RecB in parenthesis. Notice that the positions of the ligand residues of RecE are consistent with the octahedral geometry observed for RecB if slight reorientations of the side chains are allowed, especially for His-652.
Crystals of RecE606 belong to the tetragonal space group P4212, with cell dimensions of a = b = 123 Å, c = 64 Å, one monomer per asymmetric unit, and a solvent content of 72%. The crystal structure was determined by single wavelength anomalous diffraction (SAD) from crystals of the selenomethionine protein (Figure S1). The final structure includes residues 612–664 and 699–864 of RecE and was refined at 2.8 Å resolution to crystallographic R- and free R-factors of 28.9% and 31.1%, respectively (Table S1).
Structure of the RecE Monomer
RecE folds into a structure with a central five-stranded mixed β-sheet surrounded by nine α-helices and a smaller three-stranded anti-parallel β-sheet (Figure 3). As expected, the C-terminal portion of RecE, residues 725–845, exhibits a fold that is found in λ exonuclease, the RecB nuclease domain, and several other nuclease enzymes (Kovall and Matthews, 1998; Singleton et al., 2004). This common core fold includes all eight β-strands and helices C, G, and H of RecE (Figure S2). In addition, helices A, B, and I of RecE are also present in λ exonuclease, and helix E of RecE is also present in RecB. Although the structural alignment between RecE and λ exonuclease includes two more α helices than that between RecE and RecB, RecE aligns more closely to RecB. For the core fold that forms the active site, the rmsd between RecE and λ exonuclease is 2.7 Å for 98 pairs of Cα atoms, while the rmsd between RecE and RecB is 1.6 Å for 82 pairs of Cα atoms. Interestingly, RecE and λ exonuclease both have an extended loop that crosses over the active site to create a hole in the structure of about 5–10 Å in diameter. This loop connects helices B and C in RecE and is in a structurally equivalent position in λ exonuclease. Based on the structure of the RecE tetramer described below, it appears that the 5′-ended strand of the DNA substrate is likely to be threaded through this hole (from the back of Figure 3A) as it enters the active site.
Figure 3. Structure of the RecE Monomer.
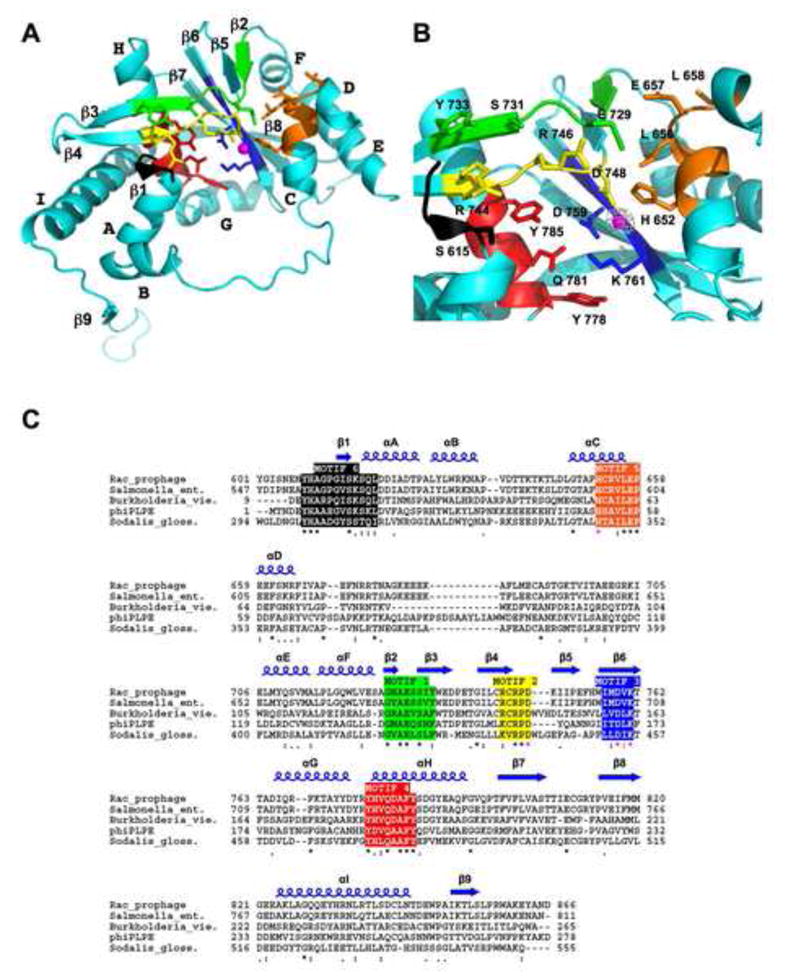
(A) Ribbon diagram of the RecE monomer with the six conserved sequence motifs highlighted in different colors as defined in panel C. The position of the active site Mg2+ is indicated by a magenta sphere. Side chains of highly conserved active site residues are shown in stick form. The orientation of the monomer is approximately the same as for the cyan subunit of the tetramer in Figure 5a.
(B) Close up view of the active site of RecE. The grey cage of electron density shows a 5σ difference Fourier map (FMn2+ - Fnat) calculated at 3.7 Å from data collected on a crystal soaked with 25 mM MnCl2.
(C) Alignment of five different RecE sequences showing six conserved sequence motifs, which are numbered based on a scheme used previously for the RecB family (Aravind et al., 2000). The top line labeled “Rac_prophage” shows the sequence of E. coli RecE protein. The multiple sequence alignment was performed with the ClustalW algorithm. Invariant (*), highly conserved (:), and conserved (.) residues are indicated by the symbols below the sequence. Residues that coordinate the Mg2+ are indicated with these symbols in magenta. Secondary structures of RecE are indicated above the sequence.
A 34-residue segment of RecE located between helices D and E, residues 665–698, is not well resolved in the electron density and is not included in the final refined structure. However, sparse electron density shows that it extends about 25 Å into the solvent region to contact the next layer of molecules in the crystal (Figure S1). These residues form an insertion in RecE that is not present in RecB or λ exonuclease (Figure S3). In the RecE tetramer, this extended segment is in position to interact with the “downstream” portion of the incoming DNA substrate (Figure 6E). The P658L mutation, which dramatically improves the crystal diffraction but does not affect enzymatic activity, is located at the tight turn between helices C and D, just preceding the 34-residue extended segment (Figure 3a). The very C-terminal portion of RecE, residues 848–864, forms an extended segment that is also not present in RecB or λ exonuclease. In the tetramer, the C-terminal segment packs against the adjacent subunit to form an integral part of the subunit interface (Figure 5a).
Figure 6. View of the Central Channel and Active Site Tunnels in the RecE Tetramer.
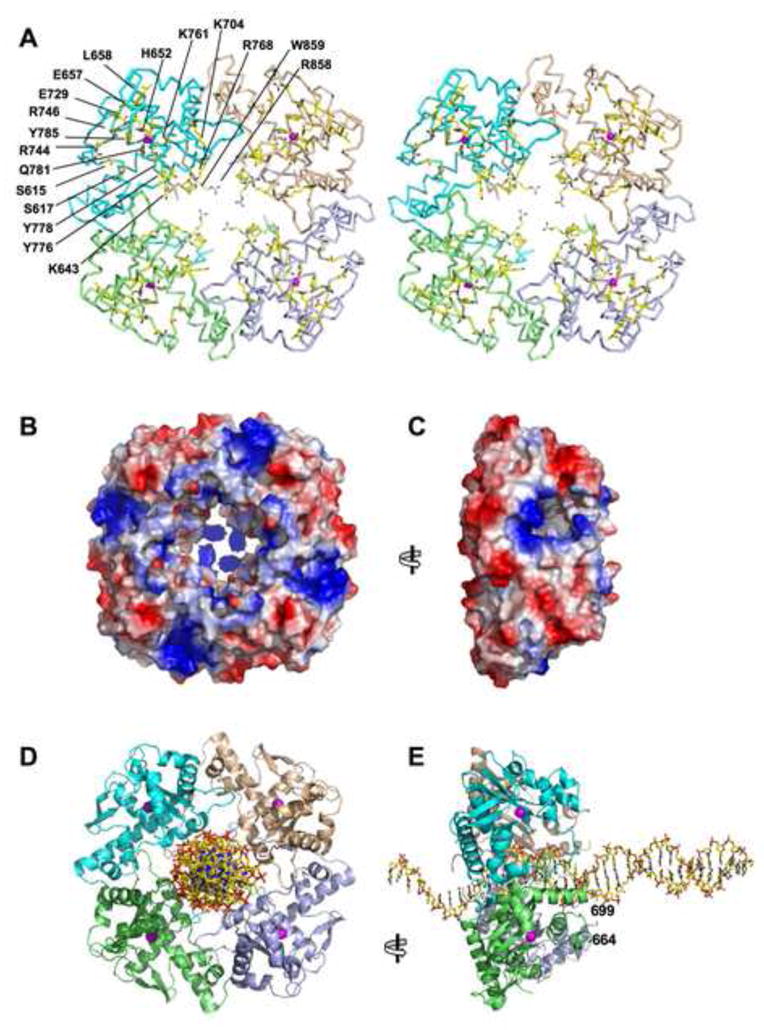
(A) Stereo Cα trace of the RecE tetramer with individual subunits colored as in Figure 5. Side chains lining the central channel and the active site tunnels are highlighted in stick form with carbon yellow, oxygen red, and nitrogen blue. The rear of the central channel is plugged by the C-terminal segment of each subunit, which includes Arg-858 and Trp-859. Side chains of Lys-704 and Lys-643 are not included in the final refined model, but are shown here to indicate their positions on the backbone.
(B) Surface view of the RecE tetramer colored according to electrostatic potential, with blue strongly positive and red strongly negative based on a charge-smoothed potential calculated in PYMOL (Delano Scientific LLC). Notice the weakly positive electrostatic potential of the central channel, except at the back where Arg-858 gives it a strong positive potential. Side chains of residues 637–646, 700–701, and 704 are not included in the final model, but are included here to indicate their presence on the surface.
(C) View from the left of panel B, looking into the active site portal of the upper, front subunit. Notice the strong positive electrostatic potential around the rim of the active site portal.
(D) View of the RecE tetramer with B form DNA modeled manually into the central channel.
(E) Side view of the model for the RecE-DNA complex, with dsDNA entering the central channel at right, and ssDNA exiting the channel at left. Residues flanking the disordered loop of the green subunit are indicated by the numbers. Notice that this loop and the equivalent loops of the other three subunits are in position to interact with the downstream portion of the incoming DNA substrate.
Figure 5. Structure of the RecE Tetramer and Comparison to the λ Exonuclease Trimer.
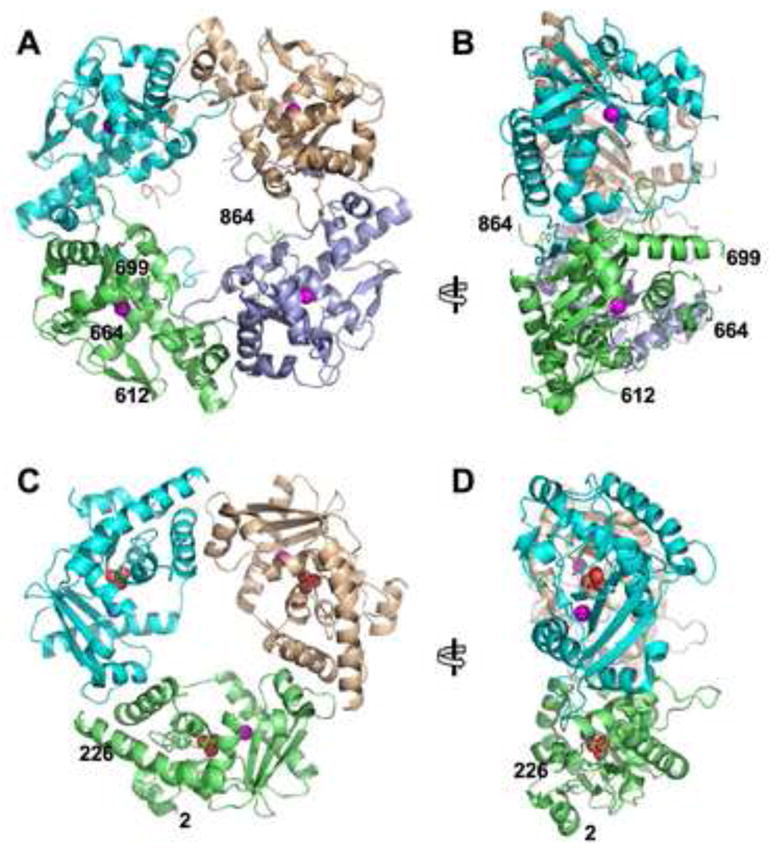
(A) View of the RecE tetramer looking down into the central channel. The active site Mg2+ of each subunit is indicated by a magenta sphere. The N- and C-terminal residues of the model (612, 864) and the residues that flank the disordered loop between helices D and E (664, 699) are indicated on the green subunit.
(B) Side view of the RecE tetramer, from the left of panel A. The open end of the central channel, to which dsDNA would bind, faces to the right.
(C) View of the λ exonuclease trimer looking down into the central channel (PDB code 1AVQ; Kovall ad Matthews, 1997). The position of the active site Mg2+ is indicated by a magenta sphere, and the phosphate bound near the active site is shown with oxygen atoms as red spheres.
(D) Side view of λ exonuclease, from the left of panel C. As in panel B, the open end of the central channel to which dsDNA would bind faces to the right.
Structure of the RecE Active Site
As expected based on sequence comparisons and mutational studies (Chang & Julin, 2001), Asp-748, Asp-759, and Lys-761 of RecE come together to form the presumed active site on the enzyme (Figure 3b). These residues are located on the loop preceding β-strand 5 and on β-strand 6. Although Mg2+ is present at 2 mM in the crystals, electron density at its expected location is not observed, possibly due to the presence of 100 mM malate, which was required for crystallization. To confirm the position of the active site Mg2+, a 3.7 Å data set was collected on a crystal soaked with 25 mM MnCl2. The resulting difference Fourier map (FMn2+ - Fnat) showed an 8 σ peak, clearly the highest peak in the map, at the expected position for the Mn2+ (Figure 3b). Similar to λ exonuclease, the Mn2+ is coordinated by the carboxylate groups of Asp-748, Asp-759 and the backbone carbonyl oxygen of Val-760. In RecE, the Mn2+ is also coordinated by a fourth ligand, His-652, which is located on α-helix C. An equivalent residue (His-956) is found in RecB, where it coordinates the Ca2+ ion bound in the active site (Singleton et al., 2004). In λ exonuclease, a highly conserved glutamate (Glu-85), which could potentially be a fourth ligand, is found at the equivalent position (Kovall & Matthews, 1997; Aravind et al., 2000). Thus, the active site of RecE, in addition to its fold, more closely resembles RecB than λ exonuclease, despite its closer functional similarity to the latter. Although the lower resolution of the Mn2+-soaked RecE structure does not permit a detailed analysis of the coordination geometry, structural alignment with RecB suggests that RecE uses the same octahedral coordination geometry observed for RecB, with four sites on the metal occupied by protein ligands and two sites available for potential interactions with the DNA substrate and/or a hydrolytic water molecule (Figure 4).
In an alignment of five different RecE sequences sharing about 30 % sequence identity with one another (Figure 3c), six highly conserved sequence motifs emerge. As seen in Figure 3 where the motifs are color coded on the structure of RecE and in the sequence alignment, all six of the conserved motifs cluster around the active site. Three of these motifs (Motifs II, III, and IV) are also conserved in RecB and λ exonuclease family members, as has been noted (Aravind et al., 2000). The residues that coordinate the Mg2+ in RecE come from Motif II (Asp-748), Motif III (Asp-759 and Val-760) and Motif V (His-652). Several other residues of these motifs, though not directly coordinated to the Mg2+, are also highly conserved. Lys-761 of Motif III, which is essentially invariant among the entire superfamily of nuclease enzymes, is within 3 Å of the Mg2+, and has been suggested to stabilize the negatively charged pentacoordinate phosphate intermediate and/or activate the hydrolytic water molecule (Kovall and Matthews, 1999). Also invariant among RecB and λ exonuclease family members is Gln-781 of Motif IV, which is hydrogen bonded to the catalytic Lys-761 residue and thus may play a role in precisely orienting it or in modulating its reactivity. Glu-729 of Motif I is essentially invariant in RecE and RecB family members, but does not appear to be present in λ exonuclease. In the RecE structure Glu-729 is more distant from the Mg2+ (8.8 Å), but is within 4 Å of the active site histidine residue (His-652).
Other residues of RecE that are highly conserved include Ser-731 and Tyr-733 of Motif I, Arg-744 and Arg-746 of Motif II, Tyr-778 and Tyr-785 of Motif IV, and Leu-656, Glu-657, and Pro-658 of Motif V (Figure 3). These residues are all within about 15 Å of the Mg2+, and are likely to play key roles in substrate binding and/or catalysis. Finally, a sixth motif that is highly conserved within the RecE family is located at the very N-terminal end of the crystallized RecE606 construct, but is not resolved in the electron density. Motif VI includes a YHA sequence, residues 608–610, that is invariant among RecE sequences. Based on the structure, the YHA sequence would be positioned within or preceding β-strand 1 (colored black in Figure 3), about 20 Å from the active site. Although the YHA sequence appears to be unique to RecE family members, Ser-615 and Ser-617 of Motif VI, which are located at the N-terminal end of helix A, are also conserved in RecB and λ exonuclease family members and are located at similar positions in their structures.
In the structure of λ exonuclease, a phosphate ion acquired from the buffers used in protein purification is bound to a pocket near the active site, where it is coordinated by Arg-28, Ser-35, Ser-117, and Gln-157 (residue numbering of λ exonuclease). λ exonuclease has much higher activity on DNA substrates that contain a 5′-phosphate group (Little, 1967; Mitsis & Kwagh, 1999), and the phosphate observed in the crystal structure has been proposed to mark the site on the protein for binding to the terminal 5′-phosphate of the dsDNA substrate (Subramanian et al., 2003). Electron density is not observed for such a phosphate in the RecE structure, even though phosphate was present during protein purification. Although Gln-157 and Ser-35 of λ exonuclease are conserved in RecE (as Gln-781 and Ser-617, respectively), Arg-28 and Ser-117 are not. Consistent with these structural observations, the activity of RecE is not sensitive to the presence of a 5′-phosphate group on the DNA substrate (Joseph & Kolodner, 1983b).
Structure of the RecE Tetramer
In the crystal, RecE forms a C4-symmetric tetramer with overall dimensions of 95 × 95 × 45 Å (Figure 5). Since the molecular 4-fold axis of the tetramer is coincident with the crystallographic 4-fold, the tetramer has perfect 4-fold symmetry. As viewed perpendicular to the 4-fold axis (Figure 5b), the tetramer has a remarkably flat surface on one side (the left side), and a much more featured surface on the other side. The 34-residue segment between helices D and E that is not included in the model would project out about 25 Å to the right of the tetramer to give it an even more featured appearance (Figure S1). A total of 2,760 Å2 of solvent-accessible surface area is buried at each subunit interface, which is formed by a roughly equal mixture of hydrophobic, ion pair, and hydrogen bonding interactions. A prominent feature of the interface is the extended C-terminal segment, residues 848–864, which packs against the neighboring subunit in the tetramer, in part through parallel β-sheet hydrogen bonding with β-strand 8. Other regions of RecE that are located at the subunit interface include helix B and the extended segment that follows it, which pack against helix E and the β7-β8 hairpin of the neighboring subunit. In general, the residues buried at the subunit interface are not highly conserved among RecE sequences, making it difficult to predict if all RecE proteins will form a similar tetramer. However, the RecE sequences are highly divergent, such that essentially all of the highly conserved residues are near the active site. All of the RecE proteins end at about the same amino acid position (±4 residues), suggesting that the extended C-terminal segment that helps to form the tetramer in E. coli RecE may be a common feature among them.
Looking down the 4-fold axis (Figure 5a), the RecE tetramer forms a central channel that is tapered from about 30 Å on one side, the front as viewed in Figure 5a, to about 10 Å on the other side. At the back of the tetramer, the central channel is partially plugged by the C-terminal residues 858–864 of each subunit. In particular, the side chains of Arg-858 and Trp-859 project towards the 4-fold axis of the tetramer to result in a narrowing of the channel to about 10 Å (Figure 6a). The depth of the channel is approximately 40 Å, enough to accommodate one complete turn of B-form DNA (Figure 6e). Based on the tapered dimensions of the central channel and the presence of extended loops on the side at which the central channel is open, it is most reasonable to conclude that the DNA substrate would bind to the open end central channel at the front of the RecE tetramer as viewed in Figure 5a. It is conceivable that if the C-terminus of the protein were to become unraveled, the DNA could enter the channel from the opposite site. However, the C-terminal residues 854–864 appear to be fairly well anchored to the neighboring subunit in the tetramer via a network of hydrophobic, hydrogen bonding, and ion pair interactions. Somewhat surprisingly, the central channel has only a weakly positive electrostatic potential (Figure 6b). Side chains of RecE that line the surface of the channel and are thus in position to interact with the DNA substrate through electrostatic or hydrogen bonding interactions include Glu-765, Gln-767, Arg-768, Thr-771, and Asp-775 of helix G, as well as Arg-858 and Trp-859 of the C-terminal plug (Figure 6a). Though not included in the final refined model, the side chains of Lys-643 (of the loop connecting helices B and C) and Lys-704 (at the beginning of helix E), as well as the 34-residue segment between helices D and E, are near the opening of the channel and are thus in position to interact with the incoming dsDNA substrate.
The four active sites of the tetramer are located about 15 Å from the central channel (28 Å from the 4-fold axis), and about midway (~20 Å) along its depth (Figures 5–6). The active sites are exposed to the central channel by a ~10 Å wide tunnel through each subunit, the roof of which is formed by the loop connecting helices B and C (Figure 6a). The tunnel is wide enough to allow passage of one strand of a DNA substrate bound within the central channel, but not the entire duplex. Thus, the structure suggests a mechanism in which the terminal 3–5 base pairs of the dsDNA substrate are unwound to allow the 5′-ended strand to pass through a tunnel to access one of the four active sites, while the 3′-ended strand passes through the narrow opening at the back of the channel. A number of conserved residues, including Ser-615 and Ser-617 of Motif VI, and Tyr-776, Tyr-778, Gln-781, and Tyr-785 of Motif IV, line the surface of the active site tunnel (Figure 6a), and are thus in position to interact with the 5′-ended strand of the DNA substrate as it passes through the tunnel. Interestingly, the four residues of Motif IV, three of which are tyrosine, form a ladder in which the side chains point out from the same face of helix H and are spaced ~3.8 Å from one another. The conserved tyrosine residues could potentially form stacking interactions with the bases of the 5′-ended strand of the DNA substrate as it moves through the active site. Each active site tunnel forges all the way through its subunit to form a portal at the outer surface of the tetramer (Figure 6c). A number of positively charged residues, including Arg-744 and Arg-746 of Motif II, line the rim of the portal to give it a strongly positive electrostatic potential (Figure 6c). This positively charged region could be a site for binding the terminal 5′-phosphate group that is generated at each cycle of the exonuclease reaction, which would help to position the scissile phosphodiester bond of the DNA substrate correctly within the active site. The portal itself could facilitate release of the 5′-mononucleotides that are liberated from the 5′-ended strand of the DNA substrate as it is processively digested.
Structure-activity analysis
In order to probe the roles of the different regions of RecE in DNA-binding and catalysis, 24 different residues of RecE564 were mutated to alanine, and the exonuclease (kcat) and DNA-binding (Kd) activities of the purified proteins were determined (Figure 7 and Table 1). The regions of the protein that were targeted in these experiments include the conserved active site motifs, the extended loop that is disordered, residues lining the central channel, and the C-terminal plug. Mutation of His-652 of Motif V to alanine completely abolishes activity, consistent with the notion that this residue, like Asp-748 and Asp-759 (Chang & Julin, 2001), is essential for binding and positioning the active site Mg2+. Mutation of several other residues of the conserved active site motifs, including Glu-729 of Motif I, Arg-746 of Motif II, Lys-761 of Motif III, Tyr-778 and Tyr-785 of Motif IV, Glu-657 of Motif V, and Tyr-608 of Motif VI, results in a >20-fold reduction in kcat, indicating that at least one residue from each of the six active site motifs is required for efficient catalysis. From these data it is apparent that a fairly large network of interactions between the DNA substrate and active site residues of RecE is important for precisely orienting the scissile bond of the DNA substrate relative to the Mg2+ center and/or translocation of the enzyme along the 5′-ended strand of the duplex. Surprisingly, mutation of Gln-781 of Motif IV, a residue that is very near the catalytic lysine (Lys-761) and is essentially invariant not only in RecE but also in the RecB and λ exonuclease families, results in only a 3-fold reduction in kcat. Mutations of the conserved residues of the active-site motifs have only a modest effect on DNA-binding, resulting in at most a 2 to 3-fold increase in Kd.
Figure 7. Effects of Mutations on RecE Exonuclease and DNA Binding Activities.

(A) Raw fluorescence time course for exonuclease reactions with WT and three mutant RecE564 proteins. The exonuclease activity is observed in real time as a decrease in the fluorescence of PicoGreen. Reactions were performed as described in Experimental Procedures.
(B) Fluorescence values from each time course in panel A were converted to %DNA digested after applying a correction factor to account for the non-linear dependence of PicoGreen fluorescence on dsDNA concentration and comparison to values from the positive and negative control reactions (Figure 2) at equivalent timepoints. The rate of digestion in nucleotides per second (kcat) is determined from the slope of the linear portion of each reaction and is reported in Table 1.
(C) Fluorescence anisotropy DNA-binding experiment. A 25 bp DNA duplex containing a 5′-fluorescein at one end was incubated with varying concentrations of WT or mutant RecE564 protein and the fluorescence anisotropy was measured as described in Experimental Procedures. Dissociation constants (Kd) reported in Table 1 were determined from a fit of the data in each curve to an equation that assumes a 1:1 binding stoichiometry.
Table 1.
Effects of mutations on RecE exonuclease and DNA-binding activities.
| Protein or mutant of RecE564 | kcat (sec−1) | Kd (nM) |
|---|---|---|
| WT (RecE564) | 18.8 ± 1.9 | 70 ± 2 |
| P658L (RecE606) | 0.42 ± 0.01 | 110 ± 20 |
| λ exonuclease | 9.3 ± 0.4 | 161 ± 2 |
| Motif I | ||
| E729A | 0.03 ± 0.08 | 57 ± 3 |
| Motif II | ||
| R744A | 6.6 ± 0.7 | 95 ± 8 |
| R746A | 0.3 ± 0.1 | 90 ± 10 |
| Motif III | ||
| K761A | 0.16 ± 0.04 | 130 ± 10 |
| Motif IV | ||
| Y778A | 0.2 ± 0.2 | 130 ± 20 |
| Q781A | 6.2 ± 0.4 | 70 ± 20 |
| Y785A | 0.4 ± 0.1 | 98 ± 9 |
| Motif V | ||
| H652A | 0.02 ± 0.04 | 130 ± 10 |
| E657A | 0.8 ± 0.1 | 90 ± 10 |
| P658L | 14.4 ± 0.1 | 80 ± 10 |
| Motif VI | ||
| Y608A | 0.6 ± 0.1 | 160 ± 10 |
| H609A | 2.1 ± 0.1 | 50 ± 3 |
| S615A | 14.6 ± 0.1 | 100 ± 4 |
| S617A | 4.5 ± 0.3 | 150 ± 20 |
| Disordered loop (residues 665–698) | ||
| F661A | 14.7 ± 0.6 | 118 ± 8 |
| F665A | 16.4 ± 0.3 | 200 ± 20 |
| P669A | 23.2 ± 1.3 | 109 ± 8 |
| T675A | 23.6 ± 0.9 | 650 ± 30 |
| Central channel | ||
| K643A | 22.3 ± 0.8 | 210 ± 6 |
| K704A | 24.6 ± 2.2 | 330 ± 50 |
| R768A | 25.5 ± 0.6 | 310 ± 30 |
| C-terminal plug | ||
| R858A | 28.3 ± 1.9 | 77 ± 5 |
| W859A | 27.8 ± 2.0 | 350 ± 20 |
| Δ858–866 | 6.4 ± 0.5 | 660 ± 80 |
Values of kcat in nucleotides per second per DNA end were measured for each purified protein by the fluorescence-based assay as demonstrated in Figure 7a,b and described in Experimental Procedures. Values of Kd (nM) were determined for each protein by fluorescence anisotropy as demonstrated in Figure 7c and described in Experimental Procedures. All standard deviations are based on measurements from at least three independent experiments.
Mutation of several conserved residues within the disordered loop that projects out from the central channel of RecE (residues 665–698) has only a minimal effect on kcat, but mutation of the highly conserved Thr-675 of this loop reduces the affinity of RecE for the dsDNA substrate by almost 10-fold, which is the greatest effect on DNA binding seen for any of the 24 mutations. Thus, while the residues of the extended loop do not appear to be critical for ongoing digestion of the dsDNA substrate once a processive reaction gets started (as measured by the exonuclease assay with saturating amounts of RecE), these data indicate that the extended loop could be important for initial recognition and binding of RecE to dsDNA ends. Similarly, mutation of three positively charged residues that line the central channel, Lys-643, Lys-704, and Arg-768, has a minimal effect on kcat, but weakens the affinity of RecE for dsDNA ends significantly (3 to 5-fold increases in Kd).
Lastly, deletion of the C-terminal residues of RecE (858–866) that plug the central channel at the back of the tetramer results in significant effects on both catalysis (3-fold reduction in kcat) and DNA binding (9-fold increase in Kd). These data suggest that the residues of the C-terminal plug interact with the 3′-ended strand of the DNA-substrate both in the pre-initiation complex and during ongoing catalysis. The data for mutations of Trp-859 and Arg-858 of the C-terminal plug indicate that the side chain of Trp-859 may play a particularly significant role in binding, while that of Arg-858 is less significant. Interestingly, the R858A and W859A single mutations actually result in a significant (~50%) increase in kcat relative to wild type. A similar effect is seen for other residues of RecE within or near the central channel, such as Lys-704, Thr-675, and Arg-768. These data suggest that while mutations that weaken the interactions with the dsDNA substrate may disrupt the initial recognition of dsDNA ends, they may allow the RecE tetramer to translocate faster along the DNA substrate once a reaction gets started.
DISCUSSION
We report the first crystal structure of RecE protein, a highly processive 5′-3′ exonuclease that is part of a two-component recombination system found in several bacterial and phage genomes. We have determined the crystal structure not of full length RecE protein, which is 866 amino acids, but rather of a C-terminal fragment of the protein, residues 606–866. Although the RecE606 fragment has considerably lower exonuclease activity than RecE564, the stable domain of RecE identified by limited proteolysis, it is important to note that RecE606 is fully functional in recombination assays in vivo (Muyrers et al., 2000). We were able to crystallize the longer RecE564 fragment, but the crystals diffracted x-rays weakly and anisotropically. Thus, removal of residues 564–605 was necessary to obtain crystals of sufficient quality to solve the structure at atomic resolution (2.8 Å). In the crystal structure, the N-terminal residues of RecE606 are involved in extensive crystal packing interactions that would clearly be disrupted if the additional residues of RecE564 were present. This provides a possible explanation for the significantly improved diffraction of crystals of RecE606.
What is the reason for the reduced activity of RecE606 as compared to RecE564? Since residues 564–605 are included in the stable fragment of RecE that is resistant to limited proteolysis, they are likely to be a folded part of the structure. However, this region E. coli RecE is not conserved or even present in many other RecE proteins. Based on the position of the most N-terminal residue (Pro-612) in the structure of RecE606, residues 564–605 would be located at the outer surface of the tetramer, near the active site portal. A highly conserved YHA sequence within Motif VI of RecE (residues 608–610) is present at the N-terminal end of the crystallized RecE606 fragment, but is not resolved in the electron density. Mutation of Tyr-608 or His-609 to alanine reduces catalytic activity significantly, suggesting that both of these residues play important roles in catalysis. Conceivably, the absence of residues 564–605 could disrupt the folding of the YHA motif in the RecE606 fragment, thereby explaining its reduced activity.
During the course of this work, we discovered that the constructs we used to express the RecE proteins contained a P658L mutation. Therefore, we expressed and purified RecE606 and RecE564 proteins without this mutation, and showed that the mutation does not affect the exonuclease activities of either protein. Remarkably, crystals of the RecE606 protein without the P658L mutation diffracted x-rays to only ~4.2 Å on our home x-ray source, a full 1 Å lower in resolution than crystals of RecE606 with the mutation. How could the P658L mutation lead to such a significant improvement in crystal diffraction? Pro-658 is a highly conserved residue within Motif V of RecE, which also includes the histidine residue (His-652) that coordinates the Mg2+. We have determined the crystal structure of the RecE606 protein without the P658L mutation to 4.2 Å resolution, and at this resolution the two structures are essentially indistinguishable (data not shown). In the structure of the P658L mutant, Leu-658 is located in the tight turn between helices C and D, which is near the flexible 34-residue segment that forms the only crystal contacts along the c axis of the crystal (Figure S1). It is conceivable that the mutation somehow stabilizes this region of the protein so that better crystal contacts are formed. It is interesting to note that although Pro-658 is highly conserved among RecE sequences, leucine is actually found at this position in RecB and λ exonuclease (Figure S3).
Structural comparison of RecE and λ Exonuclease
Despite RecE having essentially the same function as λ exonuclease, the structure of the RecE monomer more closely resembles that of RecB, which functions as a single subunit in the RecBCD helicase-nuclease complex. This is evident from the significantly lower rmsd of superimposing the core region of RecE to RecB (1.6 Å) than to λ exonuclease (2.7 Å). Moreover, the active site residues of RecE are more conserved in RecB than in λ exonuclease. For example, the histidine residue that coordinates the active site metal in RecE and RecB is replaced by a glutamate in λ exonuclease, and λ exonuclease does not appear to have the glutamate of Motif I that is essentially invariant in RecE and RecB. Although RecE and λ exonuclease both form toroidal oligomers with central channels of similar size and shape, the regions of the two proteins that come into contact at their respective subunit interfaces are different. Moreover, and strikingly, if we assume that the DNA substrate binds to the open end of the central channel in the RecE and λ exonuclease structures, then it is clear that the subunits of RecE and λ exonuclease are packed into their oligomeric rings in essentially opposite orientations relative to the incoming DNA substrate. This can be been by superimposing the monomers of RecE and λ exonuclease and then translating them into their respective oligomeric rings, as shown in Figure 8. The closer similarity of RecE to RecB and the differences in subunit packing in the RecE and λ exonuclease oligomers suggest that RecE and λ exonuclease have evolved separately, possibly from a common ancestor that was monomeric, to form oligomeric rings for processing dsDNA breaks.
Figure 8. Monomers of RecE and λ exonuclease Pack into their Toroidal Oligomers in Opposite Orientations.
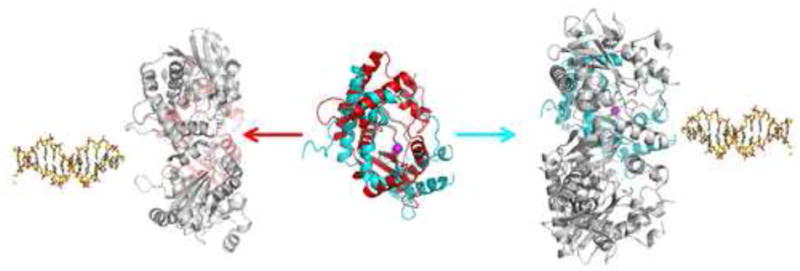
In the center of the figure, the RecE (cyan) and λ exonuclease (red) monomers are superimposed and viewed looking into the active site, which is indicated by the magenta sphere for the Mg2+. The RecE and λ exonuclease oligomers are shown at right and left, respectively, with the colored subunit in the same orientation as in the superposition in the middle. This places the colored subunit at the back of the oligomer in each case, with its active site facing the central channel. The DNA structures indicate the open end of the central channel on each structure, to which the dsDNA substrate would bind. Notice that open end of the central channel of the RecE tetramer faces to the right, while that of the λ exonuclease trimer faces to the left.
Our conclusion that the subunits of RecE and λ exonuclease are packed into their oligomeric rings in opposite orientations relative to the incoming DNA substrate implies the 5′-ended strands of the DNA substrate would approach the active sites of RecE and λ exonuclease from roughly opposite directions. This would seem to require that the active sites of RecE and λ exonuclease, which are evolutionarily related and contain several conserved residues, do the same chemistry on different orientations of the DNA substrate relative to their Mg2+ centers. However, since it is the terminal nucleotide that is cleaved, there may be sufficient conformational flexibility of the 5′-ended strand within the active site, such that the scissile bond of the DNA substrate could actually sit down in similar orientations relative to the Mg2+ center in the two structures. It is also important to note that the RecB C-terminal domain is able to cleave both the 5′- and 3′-ended strands of the DNA substrate, suggesting an inherent adaptability of this fold to accommodate different types or orientations of DNA substrates.
If we assume that RecE and λ exonuclease have evolved by separate paths to form oligomeric rings that perform the same function, it is interesting to note several common features of their overall structures that are likely to be fundamental to their modes of action. First, the central channels formed by RecE and λ exonuclease are remarkably similar in size and shape, and are of appropriate dimensions to bind to B-form dsDNA. Both central channels are completely open at one end and partially plugged at the other. In λ exonuclease, the channel is plugged by the N-terminal end of an α-helix (corresponding to helix C in RecE), whereas in RecE, the channel is plugged by the very C-terminal segment of the protein. Thus, different regions of RecE and λ exonuclease have apparently evolved to perform the same functional role. Interestingly, in both structures, the side chain of a positively charged residue, Arg-858 in RecE, and Lys-76 in λ exonuclease, projects toward the central axis of the oligomer at the narrowest part of the channel. Conceivably, this positively charged residue, as well as a nearby tryptophan residue that is present in both structures (Trp-859 in RecE and Trp-80 in λ exonuclease), could be important for binding the 3′-ended strand of the DNA substrate as it passes through the narrow end of the channel. Although single mutations of either residue in RecE have minimal effect on catalysis, removal of both residues by deletion of the C-terminal segment significantly disrupts both catalysis and DNA-binding (Table 1).
Second, RecE and λ exonuclease both have extended segments that project out from the rim of the central channel, in a position to interact with the “downstream” portion of the incoming dsDNA substrate. In λ exonuclease this extended segment, residues 43–50, is formed by a loop between two α helices that correspond to helices A and B of RecE. This loop includes three positively charged residues, Arg-45, Lys-48, and Lys-49, which could form favorable electrostatic interactions with the sugar-phosphate backbone of the incoming DNA substrate. In RecE, the 34-residue segment between helices D and E is located in a very similar position in the tetramer. Although this segment is not well resolved in the crystal structure, sparse electron density shows that it extends out about 25 Å from the rim of the central channel, in a position to interact with the incoming DNA substrate. Consistent with a possible role in DNA-binding, this segment of RecE contains five positively charged residues, a conserved RFIVAP motif (residues 664–669), and an invariant threonine residue (Thr-775). Although mutations of these conserved residues do not disrupt catalysis as measured in our assay with saturating amounts of enzyme, some of the mutations have a significant effect on DNA-binding, indicating that the extended loop could be important for recognition and binding of RecE to dsDNA ends.
Third, even though the subunits of RecE and λ exonuclease appear to be packed into their respective oligomers in opposite orientations relative to the incoming DNA substrate, the active sites of the two proteins are located in very similar positions within the oligomers. In both structures, the active sites are located about 10–15 Å from the rim of the central channel, and are exposed to the channel via a narrow tunnel that forges through each subunit. In both cases, the tunnel that leads from the central channel to the active site is wide enough to allow the passage of ssDNA, but not dsDNA, suggesting that the DNA substrate is unwound by at least 2–3 base pairs prior to nucleolytic digestion. Moreover, in both structures, the tunnel forges all the way through each subunit of the oligomer to form a portal that could facilitate release of the 5′-mononucleotide that is liberated at each cycle of the reaction. In RecE, the portal is located at the outer edge of the side of the tetramer at which the dsDNA is presumed to enter, while in λ exonuclease the portal is located on the other side, the side at which the 3′-ended strand is presumed to exit.
Mechanism For Processive Digestion of dsDNA
Based on the common structural features of RecE and λ exonuclease, which are likely to be fundamental to their modes of action, we propose a general mechanism for how this class of enzymes catalyzes processive digestion of dsDNA. The proposed mechanism is similar to and builds on one proposed previously for λ exonuclease (Kovall and Matthews, 1997). In this mechanism, the dsDNA substrate enters the toroidal oligomer through the open end of the central channel, which is of the appropriate dimensions to accommodate one complete turn of B-form DNA. Upon binding, the strands at the end of the DNA substrate are unwound, with the 5′-ended strand passing through a narrow tunnel to engage with one of the active sites on the oligomer, and the 3′-ended strand passing through the narrow opening at the back of the toroidal oligomer, possibly interacting with Arg-858 and Trp-859 of RecE, or Lys-76 and Trp-80 of λ exonuclease. Upon hydrolysis of the terminal nucleotide on the 5′-ended strand, the released 5′-mononucleotide diffuses out through the active site portal, and the DNA substrate translocates through the channel to position the next nucleotide on the 5′-ended strand within the active site.
Observations from single molecule studies of λ exonuclease support the notion that the terminal base pairs of the dsDNA substrate are unwound prior to cleavage of the 5′-ended strand. In one study, λ exonuclease was observed to digest the substrate at a fairly constant rate of 12 nucleotides per second, except at distinct sites at which the enzyme paused (Perkins et al., 2003). The pause sites exhibited a directional dependence and occurred at a particular GGCGA sequence, which was proposed to interact as ssDNA with residues lining the inner surface of the channel, including Trp-24. In a separate study, the rate of translocation of λ exonuclease along a DNA substrate was shown to depend on the sequence of the DNA, with slower rates correlating with GC-rich sequences (van Oijen et al., 2003). It was concluded that melting of the terminal nucleotide (or nucleotides) was necessary for catalysis and was the rate-limiting step at each cycle of the reaction. These types of experiments have not yet been performed on RecE protein. It would be interesting to see if RecE exhibits a similar behavior.
The structures of RecE and λ exonuclease and the resulting mechanism that is proposed nicely account for the hallmark features of the nuclease activities of these enzymes (Little, 1967; Joseph and Kolodner, 1983b). For example, the total lack of activity on circular DNA substrates is due to the location of the active site within a tunnel on the enzyme. Since in RecE the 5′-ended strand of the DNA substrate must be threaded through the tunnel formed by the loop between helices B and C to access the active site, it is physically not possible for double stranded DNA to gain access to the active site, even if RecE were a monomer. As has been pointed out previously (Breyer and Matthews, 2001), the highly processive nature of these enzymes can be accounted for by the fact that the toroidal oligomer and the DNA substrate are topologically linked by the threading of the 3′-ended strand of the DNA through the central channel of the oligomer, like a bead on a string. The low activity on single-stranded DNA substrates can also be explained. The 5′-end of ssDNA could conceivably diffuse through the central channel to access the active site, consistent with the small amount of activity that is observed. However, cleavage of ssDNA would not be processive, and would therefore be much slower, since it is the other strand of the DNA substrate, the 3′-ended strand, that is responsible for topological linkage to the enzyme.
Based on this model for the action of RecE and λ exonuclease, an interesting question emerges. Do the RecE and λ exonuclease oligomers use all of their active sites during processive digestion of a dsDNA substrate, such as in a sequential type of mechanism? Or does the 5′-ended strand engage with one of the active sites on the oligomer for multiple rounds of nucleolytic hydrolysis, such that in principle only one active site per oliogmer would be sufficient for processive nuclease activity? The structures of RecE and λ exonuclease offer some insight into this question. In the RecE tetramer, the active sites on adjacent monomers are located ~40 Å from one another, and are accessed from the central channel through relatively narrow tunnels on each subunit. In the λ exonuclease trimer, the three active sites are arranged in a similar fashion. Thus, movement of the terminal nucleotide of the 5′-ended strand of the DNA substrate from one active site to the next on the toroidal oligomer would require substantial conformational rearrangements, as well as breaking of what are likely to be extensive interactions between the protein and DNA substrate. Thus, the structures appear to support a mechanism in which the dsDNA substrate is engaged with one of the active sites on the oligomer for multiple rounds of processive dsDNA digestion.
Interaction with the Single Strand Annealing Protein
RecE and λ exonuclease each form a specific protein-protein interaction with their respective single strand annealing protein, RecT or β protein. The functional interaction with RecT is maintained in RecE606 (Muyrers et al., 2000), indicating that the site on RecE for interaction with RecT resides within the crystallized fragment. Although the role of the protein-protein interaction is not firmly established, it could be for loading of the single strand annealing protein directly onto the 3′-ended strand of the DNA substrate as it is generated by the exonuclease. Such a mechanism would be precisely analogous to the loading of RecA onto the 3′-ended strand of a DNA substrate during processing of a dsDNA break by the RecBCD helicase/nuclease complex (Anderson and Kowalczykowski, 1997). If this is indeed the role of the RecE-RecT interaction, one might expect the site on RecE for binding to RecT to be located on the side of the tetramer at which the 3′-ended strand of the DNA substrate is extruded, as is the case for RecBCD (Singleton et al., 2004). Interestingly however, this face of the RecE tetramer is remarkably flat, with no obvious loop or groove to facilitate a stable protein-protein interaction. The same is true for the λ exonuclease trimer. The structure of RecE does reveal an exposed hydrophobic patch on this side of the tetramer, formed by the side chains of Phe-754, Trp-756, Trp-720, Leu-717, Ile-851, and Met-819. Perhaps this hydrophobic patch could be a site for interaction with RecT.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Detailed methods for protein expression and purification are given in the supplementary information. Briefly, genes encoding residues 564–866 or 606–866 of RecE protein and 1-226 of λ exonuclease were cloned into pET-14b and expressed in E. coli BL21(AI) cells. Mutants of RecE were constructed using the Quickchange procedure (Stratagene). RecE606 protein was purified by Ni2+-affinity chromatography, thrombin proteolysis to remove the N-terminal 6xHis tag, and anion exchange chromatography on Hi-Trap QHP (GE Healthcare). Purified RecE606 protein was dialyzed into 20 mM Tris (pH 7.5), 150 mM NaCl, 10% glycerol, and 1mM dithiothreitol (DTT). RecE564 protein and mutant versions of RecE564 were purified by Ni2+ affinity chromatography and gel filtration on Superdex 200 (GE Healthcare), and dialyzed into 20 mM Tris (pH 7.5), 1 mM DTT. All proteins were concentrated to 5–50 mg/ml and stored at −80°C in small aliquots. Protein concentrations were determined by O.D. at 280 nm using extinction coefficients calculated from their amino acid sequences.
Analytical Ultracentrifugation
Samples of RecE564 used in sedimentation experiments were purified on Superdex 200 (GE Healthcare) with 150 mM NaCl, 20 mM Tris (pH 8.0) as the running buffer. Sedimentation experiments were performed at 20 °C in a Beckman XL-I analytical ultracentrifuge using absorbance optics. 400 μl samples of RecE564 in the buffer from gel filtration were spun at 40,000 rpm at 20 °C in double-sector charcoal-filled epon centerpieces. Data were analyzed using the c(s) and c(M) models in the program Sedfit (Schuck, 2000) to determine differential sedimentation coefficient or apparent mass distributions, respectively. In sedimentation equilibrium experiments, 100 μl samples at concentrations ranging from 1.2 to 12 μM were spun at 11,000, 14,000, and 19,000 rpm until equilibrium was reached. Data were truncated using WinReedit and globally fit using WinNonlin (http://www.rasmb.bbri.org/) as described previously (Herr et al., 1997; Herr et al., 2003).
Exonuclease Assay
Exonuclease activities of purified proteins were measured in real time by monitoring the decrease in fluorescence upon addition of excess enzyme to a pre-equilibrated mixture of PicoGreen and dsDNA (Tolun and Myers, 2003). Reactions (200 μl) were performed at 37 °C in 20 mM Tris (pH 7.5), 10 mM MgCl2, 1 mM DTT, and PicoGreen (Invitrogen) diluted 1/20,000. Reactions were initiated by addition of a saturating amount of RecE or λ exonuclease (40–100 nM oligomer) to 0.5 nM BamHI linearized pUC19 DNA (1 nM ends). Time-based fluorescence values were measured using a FluoroMax-3 spectrofluorometer (Horiba Jobin Yvon) at 484 nm excitation, 522 nm emission, 4 nm slit width, and 1 s−1 data sampling frequency. A reaction mixture prepared without addition of enzyme served as the negative control to account for a slight enzyme-independent decrease in signal, possibly due to photobleaching. A positive control was performed with 0.25 nM heat denatured pUC19 DNA (0.5 nM total ssDNA) to mimic completion of the reaction to 5′-mononucleotides and one strand of ssDNA. Raw fluorescence values were converted to %DNA digested using a standard curve to correct for the non-linear dependence of PicoGreen fluorescence on dsDNA concentration and by comparison to the values from the positive and negative control reactions at equivalent time points. The rate of digestion in nucleotides per second (kcat) was determined from the slope of the linear (initial) portion of each reaction divided by two to account for the fact that at saturation both ends of the DNA duplex are digested simultaneously. Standard deviations are based on values from three independent experiments.
Fluorescence Anisotropy DNA Binding Assay
An oligonucleotide with a 5′-fluorescein, 5′-[FluorT]AGAGCTTAATTGCTGAATCTGGTG-3′ (HPLC purified from Integrated DNA Technologies), was annealed with its complement to form a 25 bp duplex (F25). 10 nM F25 was incubated with varying concentrations (10 – 1000 nM) of purified RecE564 protein (or mutant as indicated) for 20 minutes at 25 °C in buffer containing 20 mM Tris pH 7.5, 10 mM CaCl2, and 1 mM DTT. The fluorescence anisotropy (490 nm excitation, 515 nm emission) of each equilibrated sample (20 μl) was measured at 25 °C using a Spectra Max M5 Microplate Reader (Molecular Devices). Dissociation constants (Kd) were determined from a fit of the data in each curve to the following equation, which assumes a 1:1 binding stoichiometry:
where A is the measured anisotropy, Amin is the minimum anisotropy, Amax is the maximum anisotropy, S is the concentration of un-labeled protein and Y is the concentration of labeled DNA. Standard deviations are based on values from three independent experiments.
Crystallization and X-ray Structure Determination
Crystals of RecE606 (P658L) were grown by the hanging-drop vapor diffusion method at 22°C. The reservoir solution consisted of 30–42% glycerol, 100 mM DL-malic acid (pH 7.0). The hanging drop was prepared by mixing 2 μl of 10 mg/ml RecE606 in storage buffer plus 2 mM MgCl2, and 2 μl of reservoir solution. Crystals were flash frozen in liquid nitrogen and x-ray diffraction data were collected at −180°C at beamline 31-ID of the Advanced Photon Source. Diffraction data for both native and SeMet crystals were collected at the Se absorption peak, λ = 0.97929 Å. Crystals belong to space group P4212 with cell dimensions a = b = 123.2 Å, c = 67.3 Å, 1 molecule per asymmetric unit, and a solvent content of 72%. Data were integrated and scaled with MOSFLM and SCALA of the CCP4 suite (CCP4, 1994). Crystals diffracted x-rays anisotropically to about 2.7 Å along the a and b directions and 3.2 Å along the c direction, based on analysis of the data according to the falloff procedure using plots of mean[F/sdF] vs. resolution (CCP4, 1994). The structure was determined by the single wavelength anomalous diffraction method and solvent flattening using the AutoSol feature of the PHENIX suite (Adams et al., 2002). The resulting electron density map allowed tracing of residues 612–634 and 704–861 of RecE606 using the program COOT (Emsley and Cowtan, 2004). The structure was refined using the simulated annealing, minimization, and individual temperature factor refinement protocols of CNS (Brünger et al., 1998) with a maximum likelihood target, and bulk solvent and anisotropic temperature factor correction options. Alternating rounds of model building and refinement yielded the final model, which consists of residues 612–664 and 699–864 of RecE. Side chains of residues 637–646, 700–701, and 704 of RecE were not resolved in the electron density and were truncated to alanine. Data collection and refinement statistics are shown in Table S1. Structural figures were prepared using PYMOL (Delano Scientific, LLC). Solvent accessible surface area calculations were performed using the AREAIMOL feature of CCP4 with a probe radius of 1.4 Å (CCP4, 1994).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant GM067947 to C.E.B., a predoctoral fellowship from the American Heart Association to J.Z., and funds from the State of Ohio Eminent Scholar Program to A.B.H. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the SGX Collaborative Access Team (SGX-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by SGX Pharmaceuticals, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini NK, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Anderson DG, Kowalczykowski SC. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell. 1997;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Makarova KS, Koonin EV. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CE. MicroReview: “Structure and mechanism of Escherichia coli RecA ATPase” Mol. Microbiol. 2005;58:358–366. doi: 10.1111/j.1365-2958.2005.04876.x. [DOI] [PubMed] [Google Scholar]
- Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, et al. The potential and challenges of nanopore sequencing. Nature Biotech. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breyer WA, Matthews BW. A structural basis for processivity. Protein Sci. 10:1699–1711. doi: 10.1110/ps.10301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chang HW, Julin DA. Structure and function of the Escherichia coli RecE protein, a member of the RecB nuclease domain family. J Biol Chem. 2001;276:46004–46010. doi: 10.1074/jbc.M108627200. [DOI] [PubMed] [Google Scholar]
- Chu CC, Templin A, Clark AJ. Suppression of a frameshift mutation in the recE gene of Escherichia coli K-12 occurs by gene fusion. J Bacteriol. 1989;171:2101–2109. doi: 10.1128/jb.171.4.2101-2109.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLLABORATIVE COMPUTATIONAL PROJECT. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. NUMBER 4. [DOI] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Hall SD, Kane MF, Kolodner RD. Identification and characterization of the Escherichia coli RecT protein, a protein encoded by the recE region that promotes renaturation of homologous single-stranded DNA. J Bacteriol. 1993;175:277–287. doi: 10.1128/jb.175.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr AB, Ornitz DM, Sasisekharan R, Venkataraman G, Waksman G. Heparin-induced self-association of fibroblast growth factor-2. Evidence for two oligomerization processes. J Biol Chem. 1997;272:16382–16389. doi: 10.1074/jbc.272.26.16382. [DOI] [PubMed] [Google Scholar]
- Herr AB, Ballister ER, Bjorkman PJ. Bivalent binding of IgA1 to FcalphaRI suggests a mechanism for cytokine activation of IgA phagocytosis. J Mol Biol. 2003;327:645–657. doi: 10.1016/s0022-2836(03)00149-9. [DOI] [PubMed] [Google Scholar]
- Joseph JW, Kolodner R. Exonuclease VIII of Escherichia coli I. Purification and physical properties. J Biol Chem. 1983a;258:10411–10417. [PubMed] [Google Scholar]
- Joseph JW, Kolodner R. Exonuclease VIII of Escherichia coli. II Mechanism of action. J Biol Chem. 1983b;258:10418–10424. [PubMed] [Google Scholar]
- Kmiec E, Holloman WK. βprotein of bacteriophage λ promotes renaturation of DNA. J Biol Chem. 1981;256:12636–12639. [PubMed] [Google Scholar]
- Kolodner R, Hall SD, Luisi-Deluca C. Homologous pairing proteins encoded by the Escherichia coli recE and recT genes. Mol Microbiol. 1994;11:23–30. doi: 10.1111/j.1365-2958.1994.tb00286.x. [DOI] [PubMed] [Google Scholar]
- Kovall R, Matthews BW. Toroidal structure of lambda-exonuclease. Science. 1997;277:1824–1827. doi: 10.1126/science.277.5333.1824. [DOI] [PubMed] [Google Scholar]
- Kovall R, Matthews BW. Structural, functional, and evolutionary relationships between λ-exonuclease and the type II restriction endonucleases. Proc Natl Acad Sci USA. 1998;95:7893–7897. doi: 10.1073/pnas.95.14.7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovall RA, Matthews BW. Type II restriction endonucleases: structural, functional and evolutionary relationships. Curr Opin Chem Biol. 1999;3:578–583. doi: 10.1016/s1367-5931(99)00012-5. [DOI] [PubMed] [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophageλ. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Chang J, Joseph N, Ghirlando R, Rao DN, Yang W. MutH complexed with hemi- and unmethylated DNAs: coupling of base recognition and DNA cleavage. Mol Cell. 2005;20:155–166. doi: 10.1016/j.molcel.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Little JW. An exonuclease induced by bacteriophage lambda. II Nature of the enzymatic reaction. J Biol Chem. 1967;242:679–686. [PubMed] [Google Scholar]
- Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu Rev Biochem. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- Mitsis PG, Kwagh JG. Characterization of the interaction of lambda exonuclease with the ends of DNA. Nucleic Acids Res. 1999;27:3057–3063. doi: 10.1093/nar/27.15.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniyappa K, Radding CM. The homologous recombination system of phage λ. Pairing activities of β protein. J Biol Chem. 1986;261:7472–7478. [PubMed] [Google Scholar]
- Muyrers JPP, Zhang Y, Buchholz F, Stewart AF. RecE/RecT and Redα/Redβ initiate double-stranded break repair by specifically interacting with their respective partners. Genes & Devel. 2000;14:1971–1982. [PMC free article] [PubMed] [Google Scholar]
- Muyrers JPP, Zhang Y, Stewart AF. Techniques: recombinogenic engineering – new options for cloning and manipulating DNA. Trends Biochem Sci. 2001;26:325–331. doi: 10.1016/s0968-0004(00)01757-6. [DOI] [PubMed] [Google Scholar]
- Perkins TT, Dalal RV, Mitsis PG, Block SM. Sequence-dependent pausing of single lambda exonuclease molecules. Science. 2003;301:1914–1918. doi: 10.1126/science.1088047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteete AR. What makes the bacteriophage λ Red system useful for genetic engineering: molecular mechanism and biological function. FEMS Microbiol Lett. 2001;201:9–14. doi: 10.1111/j.1574-6968.2001.tb10725.x. [DOI] [PubMed] [Google Scholar]
- Radding CM, Rosenzweig J, Richards F, Cassuto E. Separation and characterization of exonuclease, β protein, and a complex of both. J Biol Chem. 1971;246:2510–2512. [Google Scholar]
- Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DG. Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature. 2004;432:187–193. doi: 10.1038/nature02988. [DOI] [PubMed] [Google Scholar]
- Stahl MM, Thomason L, Poteete AR, Tarkowski T, Kuzminov A, Stahl FW. Annealing vs. invasion in phage λ recombination. Genetics. 1997;147:961–977. doi: 10.1093/genetics/147.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Rutvisuttinut W, Scott W, Myers RS. The enzymatic basis of processivity in λ exonuclease. Nucleic Acids Res. 2003;31:1585–1596. doi: 10.1093/nar/gkg266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolun G, Myers RS. A real-time DNase assay (ReDA) based on PicoGreen fluorescence. Nucleic Acids Res. 2003;31:e111. doi: 10.1093/nar/gng111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- van Oijen AM, Glainey PC, Crampton DJ, Richardson CC, Ellenberger T, Xie XS. Single-molecule kinetics of λ exonuclease reveal base dependence and dynamic disorder. Science. 2003;301:1235–1238. doi: 10.1126/science.1084387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.