

Abstract
Autophagy is important for the degradation of bulk cytoplasm, long-lived proteins, and entire organelles. In lower eukaryotes, autophagy functions as a cell death mechanism or as a stress response during development. However, autophagy’s significance in vertebrate development, and the role (if any) of vertebrate-specific factors in its regulation, remains unexplained. Through careful analysis of the current autophagy gene mutant mouse models, we propose that in mammals, autophagy may be involved in specific cytosolic rearrangements needed for proliferation, death, and differentiation during embryogenesis and postnatal development. Thus, autophagy is a process of cytosolic “renovation,” crucial in cell fate decisions.
Autophagy is a lysosomal pathway used by eukaryotes for degrading and recycling various cellular constituents, such as long-lived proteins and entire organelles. There are three main forms of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy (Klionsky, 2005; Massey et al., 2006). Microautophagy and the mammalian-specific chaperone-mediated autophagy directly involve the lysosome, which either engulfs small portions of cytosol or receives chaperone-associated cargoes, respectively. Macroautophagy is responsible for the turnover of organelles and portions of cytosol sequestered in a double-membrane-bound vesicle, the autophagosome. Autophagosomes originate from a precursor structure, the phagophore, which starts growing at both ends and finally closes and wraps the bulk cytoplasm and organelles (Figure 1A). Autophagosomes slide along cytoskeletal structures and fuse with lysosomes, forming a single large and membrane-surrounded vesicle called the autophagolysosome, where both their membrane and contents are degraded by lytic enzymes (reviewed in Xie and Klionsky, 2007).
Figure 1. Autophagosome Formation in Vertebrates.

(A) Autophagosomes (AΦ) originate from a precursor structure (phagophore), which grows, closes, and wraps the cytoplasmic components and organelles. Eventually, the autophagosome fuses with the lysosome (Ly), forming a structure termed autophagolysosome or autolysosome (AL). The contents of the autophagosome are then degraded by the lysosomal enzymes.
(B) Autophagosome nucleation is driven by phosphatidylinositol (PI) phosphorylation. This process (highlighted in blue) is mediated by a lipid kinase signaling complex (Beclin 1, Vps15, Vps34). Ambra1 promotes Beclin 1/Vps34 interaction, whereas Ulk1 is downstream of mTOR (which is inhibitory for autophagy) and is involved in autophagy induction (see text), although it is not a subunit of the Beclin 1/Vps34 complex. UVRAG and Bif-1 have been described as additional regulators of the Beclin 1/Vps34 complex.
(C) Autophagosome elongation is triggered by lipid modification of LC3 (by phosphatidylethanolamine, PE), as highlighted in yellow. This process is mediated, among others, by Atg7 (an E1-like ubiquitin conjugating enzyme), Atg3 (an E2-like ubiquitin conjugating enzyme), and Atg4C, to which LC3 is bound at first. Atg7 also acts in a ubiquitin-like conjugation system involving the E2-like ubiquitin conjugating enzyme Atg10 and Atg12/Atg5 which, at the end of the process, are transferred to Atg16L. The complex Atg12/Atg5/Atg16L mediates LC3-PE binding to the autophagosome membranes.
Most of the genes involved in macroautophagy (hereafter autophagy), the so-called Autophagy-related (Atg) genes, have been discovered in S. cerevisiae (Ohsumi, 2001). These include genes that regulate autophagosome formation, which requires two evolutionarily conserved ubiquitin-like conjugation systems—the Atg12-Atg5 and the Atg8(LC3)-PE (phosphatidylethanolamine) systems (Figure 1) (Ohsumi, 2001; Suzuki and Ohsumi, 2007)—as well as genes that function in other stages of autophagy. Atg genes were originally described in yeast, and in some cases their orthologs have been isolated and functionally characterized in mammals (Table 1) (Ferraro and Cecconi, 2007; Levine and Kroemer, 2008).
Table 1. Mouse Models of Autophagy Impairment with Clear Developmental Phenotypes.
| Molecule and Role in Autophagy | Mouse Model Allele Symbol and Type | Type of Mutation | Key Aspects of the Phenotype | Reference | |
|---|---|---|---|---|---|
| Ambra1: WD40 protein promotes Vps34 complex formation (autophagosome nucleation) | ambra1gt/gt (all tissues) | gene trap allele: Ambra1Gt(pGT1.8geo)1Fcec | gene trap insertion predicted to cause a 459 aa C-terminal truncation; fusion transcript, but no protein, detected | decreased autophagy, increased apoptosis and cell proliferation in embryonic brain; neural tube defects and embryonic lethality | Fimia et al., 2007 |
| Atg5: ubiquitin-like protein covalently modifies Atg12 and binds Atg16L to promote LC3-PE recruitment to autophagosome membranes (autophagosome elongation) | atg5-/- (all tissues) | knockout allele: Atg5tm1Nmz | neomycin-resistance (neo) gene replaces the transcription start site and first two exons | perinatal death due to nutrient and energy depletion; suckling defect; increased apoptosis in embryos | Kuma et al., 2004; Qu et al., 2007 |
| atg5-/flox (tissue-specific) | conditional allele: Atg5tm1Myok (floxed) | loxP sites flank exon 3 and neo cassette; Cre removes both exon 3 and neo | |||
| Atg5tm1Myok combined w/Tg(Nes-cre)1Kln transgenic (Cre driver) | Cre expressed in neuronal and glial precursors by e11.0 (Nestin promoter) | ubiquitin-positive inclusions in the brains of e15.5 embryos and postnatal mice, early onset neurodegeneration | Hara et al., 2006 | ||
| Atg5tm1Myok combined w/Tg(Zp3-cre)93Knw transgenic (Cre driver) | Cre expressed in female germline (Zp3 promoter) | null embryos derived from Atg5-deficient eggs fail to develop beyond the eight-cell stage; Atg5-deficient eggs fertilized by wild-type sperm are normal | Tsukamoto et al., 2008 | ||
| Atg5tm1Myok combined w/Cd19tm1(cre)Cgn transgenic (Cre driver) | Cre expressed throughout B lymphocyte development (Cd19 promoter) | the pro-B to pre-B cell transition is defective | Miller et al., 2008 | ||
| Atg7: E1 ligase-like protein activates LC3-I and Atg12 (autophagosome elongation) | atg7-/- (all tissues) | conditional allele: Atg7tm1.1Tchi | loxP sites flank exon 14 | excised during oogenesis, this allele results in a similar phenotype to Atg5tm1Myok/Tg(Zp3-cre)93Knw as above | Komatsu et al., 2005 |
| atg7-/flox (tissue-specific) | combined w/Tg(Nes-cre)1Wme transgenic (Cre/Flp) | this Nestin-Cre, in which Cre has a nuclear localization signal, drives variable/mosaic activity by e10.5, detectable in all adult tissues examined, including the germline | progressive deficits in motor function and death a few months after birth | Komatsu et al., 2006 | |
| combined w/Tg(Pcp2-cre)2Mpin transgenic (Cre/Flp) | Cre expressed in Purkinje cells starting at P6 and complete 2-3 weeks postpartum (L7/Pcp2 promoter) | degeneration of the axon terminals during postnatal brain development | Komatsu et al., 2007 | ||
| Beclin 1/Atg6: subunit of the Vps34 complex (autophagosome nucleation) | becn1-/- (all tissues) | knockout allele: Becn1tm1Blev | neo replaces exons 1 and 2 | abnormal ectodermal layer with reduced cavitation and early embryonic lethality | Qu et al., 2003; Qu et al., 2007 |
| knockout allele: Becn1tm1Htz | neo replaces exons 1 and 2 and 1.6 kb of upstream sequence | widespread cell death, reduced embryo size | Yue et al., 2003. |
Allele symbols are standard according to http://www.informatics.jax.org.
A core molecule in autophagy regulation is the kinase mammalian Target Of Rapamycin (mTOR). By sensing signals that monitor nutrient levels, mTOR can trigger protein translation by specific phosphorylation of the ribosomal protein S6 kinase (pS6K) (Blommaart et al., 1995). When nutrients are lacking, mTOR repression shifts cellular metabolism toward autophagy and recycling of cytosolic constituents (Figure 1), although the precise targets of mTOR in autophagy remain unidentified.
The discovery of the molecular basis of autophagy has enabled the search for links with human pathological conditions. Genetic or pharmacological alteration in this process impairs cell survival rate or cell metabolism, thereby affecting tissue homeostasis (Levine and Kroemer, 2008; Mizushima et al., 2008). Many neurodegenerative conditions, for example, can be traced back to defective autophagy. The role of autophagy in Huntington’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, or Alzheimer’s disease may reside in the failure to clear aggregates of mutated toxic proteins (reviewed in Rubinsztein, 2006). Autophagy has also been identified as a crucial process in oncogenesis and cancer progression (Gozuacik and Kimchi, 2004; Jin and White, 2007; Levine and Kroemer, 2008; Mizushima et al., 2008). Several autophagy-related proteins have tumor suppressor activity (Beclin 1, Atg5, Bif-1, Atg4C, UVRAG), and some autophagy gene mutations, detected in humans or induced in cellular or animal models, can lead to an accumulation of DNA damage and genome instability (Mathew et al., 2007). In cancer progression, autophagy may, on the other hand, provide tumor cells with a survival strategy, thus suggesting a therapeutic use for autophagy downregulation in solid tumors (reviewed in Levine and Kroemer, 2008; Mizushima et al., 2008).
Further, antigen presentation, innate immune signaling, and pathogen degradation may all involve autophagosome recruitment and activity, and autophagy plays an important role in immunity and infectious diseases (Levine and Deretic, 2007).
After Christian de Duve’s first electron-microscopy-based description of autophagy (1963) and Per Seglen’s seminal work on the convergence between autophagic and endocytic pathways (De Duve, 1963; De Duve and Wattiaux, 1966; Gordon and Seglen, 1988), Yoshinori Ohsumi and Michael Thumm in the early 1990s independently began a detailed analysis of autophagy in yeast, going on to discover most of the Atg genes (Takeshige et al., 1992; Thumm et al., 1994; Tsukada and Ohsumi, 1993). Meanwhile, other studies focused on the peculiar morphology that some cells with autophagic activity show during embryogenesis in lower eukaryotes. In 1977, Jacques Beaulaton and Richard Lockshin elegantly demonstrated that in Lepidoptera the normal degeneration of intersegmental muscles, a crucial step in insect metamorphosis, was accompanied by the presence of autophagic vacuoles containing mitochondria (Beaulaton and Lockshin, 1977). Similar evidence was found in the regressing prothoracic and salivary glands of other insects, including Drosophila (Scharrer, 1966; Schin and Laufer, 1973; Schin and Clever, 1965). Increased autophagy in insect development was associated with type II cell death, a form of nonapoptotic programmed cell death involving excessive lysosomal activity, formation of a large number of autophagosomes, and presumed cell self-cannibalization (Baehrecke, 2003; Berry and Baehrecke, 2007). In only a few cases, most notably in the developing nervous system, was such a death pathway found in higher eukaryotes (Clarke, 1990; Hornung et al., 1989). These studies suggested that autophagy has an important role in development, although its specific functions remained unclear when using morphologic analysis alone.
The era of Atg gene discovery in lower eukaryotes was followed by in-depth investigation of the same process in vertebrates. In 1998, Beth Levine’s and Noboru Mizushima’s laboratories isolated, in mammals, Beclin 1, and Atg5 and Atg12, respectively (Liang et al., 1998; Mizushima et al., 1998b). Several mouse models for Atg gene inactivation revealed interesting embryonic phenotypes (Table 1, and listed in Table 1 of Levine and Kroemer, 2008). In theory, there could be a link between apoptosis and autophagy in these cases, suggesting that autophagy may trigger cell demise upon strong environmental or extrinsic stress during development, thereby matching the scenario believed to occur in lower eukaryotes. However, the mechanisms and functions of autophagy need further investigation. The question remains as to whether autophagy constitutes an understudied form of cell destruction or, alternatively or in parallel, whether it is a regulated process capable of rapidly changing the cell in response to environmental stimuli or intrinsic metabolic needs.
In order to function during embryogenesis (in proliferation, migration, communication, differentiation, or death), cells undergo phases of quiescence or enhanced metabolism. Thus, they require dynamic tools to suddenly modify their organelle or protein content so as to adapt rapidly and respond to adverse conditions. Since development time is so rapid, proteasome-mediated turnover of individual proteins is probably not sufficient to effect the necessary cellular architectural changes. Autophagy, which can mediate bulk protein and organelle turnover, could therefore help to profoundly renovate cells or modify their external appearance within a few hours. Therefore, a fuller knowledge of autophagy in development might not only reveal the evolutionary significance of this process, it may also explain its roles in human pathologies.
Mouse Models of Autophagy
To establish the role of autophagy in mammalian development or adult tissue homeostasis, various mouse and cellular models have been generated by using multiple genetic approaches, such as gene targeting, gene trapping, dysregulation in cell or tissue culture of embryonic origin, tissue chimerism, and tissue-specific conditional mutagenesis. The resulting developmental phenotypes and the genetic features of the most relevant mouse models are summarized in Table 1. Note that the complex phenotypes we include here may not be uniquely related to autophagy defects, but could also be due to the inactivation of nonautophagic functions of these genes.
Atg1/Ulk1
The mouse ortholog of yeast Atg1 is Ulk1/Unc51.1 (Figure 1), a serine/threonine kinase involved in the early steps of autophagosome generation (Tomoda et al., 1999). Atg1 forms a complex with other Atg regulatory proteins such as Atg13 and Atg17 (Kamada et al., 2000; Tsukada and Ohsumi, 1993). Under nutrient-rich conditions, i.e., upon autophagy inhibition, yeast Atg13 is hyperphosphorylated, blocking its association with Atg1. However, under nutrient starvation, i.e., when autophagy activation is required, Atg13 becomes partially dephosphorylated, with subsequent Atg1-Atg13 interaction and the generation of the autophagosome (Kamada et al., 2000). Ulk1 was the first autophagy-related gene to be manipulated in a mouse system, where it is expressed in granule cells in the cerebellar cortex. Retroviral infection of immature granule cells with a dominant-negative Ulk1 resulted in inhibition of neurite outgrowth in vitro and in vivo (Tomoda et al., 1999, 2004). Moreover, infected neurons failed to express any specific markers of neuronal differentiation, suggesting that neurodevelopment was arrested early (Tomoda et al., 1999). Ulk1 localizes to both axonal shafts and growth cones of extending axons and is essential for neurite extension and parallel fiber formation in cerebellar granule neurons (Tomoda et al., 2004). An autophagy-independent role for Ulk1 has been suggested in various species, such as control of axonal elongation and branching in C. elegans (Ogura et al., 1994) and mammals (Zhou et al., 2007). What seems to be misregulated by Ulk1 manipulation is either the establishment of a proper constellation of surface factors necessary for cell migration and adhesion, or normal cell metabolism, essential for an energy-consumptive cellular task such as axonal growth. Interestingly, Ulk1 was found to localize to autophagic phagophores under starvation conditions, interacting with the Atg17 ortholog FIP200 (Hara et al., 2008). FIP200 itself was relocalized upon starvation, switching from the cytoplasm to the phagophore (Hara et al., 2008). Given that FIP200 regulates diverse cellular processes such as cell size, proliferation, migration, and focal adhesion, the Ulk1-FIP200 interaction might have other vital functions aside from autophagy.
Beclin 1
The autophagy-related gene becn1 (encoding Beclin 1; coiled-coil, myosin-like BCL2-interacting protein) has been targeted and inactivated in mice (Qu et al., 2003; Yue et al., 2003). Beclin 1 is the mammalian ortholog of yeast Atg6, a component of the class III phosphatidylinositol-3-OH kinase (also known as Vps34) complex regulating autophagosome formation in mammals (Liang et al., 1999). It can inhibit tumorigenesis in vitro and in vivo and is expressed at decreased levels in several human cancers (Degenhardt et al., 2006; Liang et al., 1999; Mathew et al., 2007; Qu et al., 2003; Yue et al., 2003). Beclin 1 is also decreased in degenerating neurons in Alzheimer’s disease tissues and Beclin 1 heterozygous animals (becn1+/-) develop an Alzheimer’s-like pathology (Pickford et al., 2008). This protein interacts with some members of the Bcl-2 protein family through a BH3-like domain (see below; Maiuri et al., 2007a; Oberstein et al., 2007; Pattingre et al., 2005).
What is the effect of becn1 disruption in mouse? Embryos devoid of Beclin 1 (becn1-/-) exhibit a developmental delay culminating in a severely reduced size at embryonic day 7.5 (e7.5) (Table 1) (Yue et al., 2003). Although Beclin 1 expression was detected in the extraembryonic visceral endoderm, this structure forms properly, along with the head organization center, found in its anterior part (anterior visceral endoderm). Analysis of in vitro aggregates of becn1-/- inner-mass-derived embryonic stem cells (becn1-/- embryoid bodies) and of the mutant embryo itself produced some surprising evidence (Figure 2) (Yue et al., 2003): (1) becn1-/- cells failed to form expanded cystic embryoid bodies (hollow structures mimicking the blastocyst) due to a reduction in cell clearance at the core of the embryoid body (Figure 2A), and (2) widespread cell death was visible in becn1-/- embryos at e6.5 and e7.5 (Figure 2B). becn1 and the downstream autophagy gene atg5 (see below) are required for the proper generation of “eat-me” and “come-get-me” engulfment signals from the inner ectodermal apoptotic cells during cavitation of the embryoid body via a mechanism involving autophagy-dependent ATP energy homeostasis in dying cells (Qu et al., 2007). Similarly, becn1-/- e6.5 postimplantation embryos suffer from an accumulation of apoptotic cells and formation defects of their proamniotic cavity (Figure 2B). The cause of the phenotype of becn1-/- embryoid bodies is the failure of the dying cells to display the appropriate signals on their surfaces, which is necessary for their recognition. Indeed, in mammalian development the identity concept “autophagy = cell death,” often proposed in lower eukaryotes, does not seem to be universally applicable. However, becn1 deficiency may also result in early embryonic lethality through as yet unidentified autophagy-independent functions in mammalian cells that parallel its autophagy-independent roles in other membrane-trafficking pathways in yeast.
Figure 2. Developmental Phenotypes in Mice Lacking atg5, beclin 1, or ambra1.
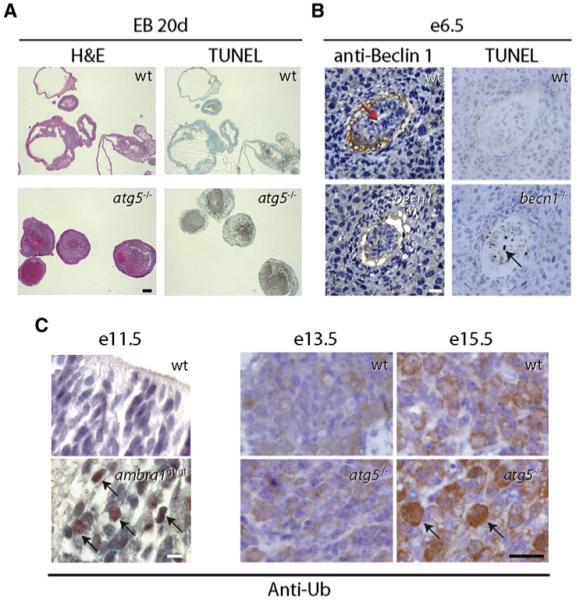
(A) Representative images showing lack of cavity formation and accumulation of TUNEL-positive cells at day 20 (20d) of development of wild-type (wt) and atg5-/- embryoid bodies (EBs) (excerpt from Qu et al., 2007). Similar findings were obtained with becn1-/- embryoid bodies. Scale bar, 200 μm. H&E, hematoxylin and eosin staining; TUNEL, terminal uridine deoxynucleotidyl transferase dUTP nick end labeling for detection of DNA fragmentation (a hallmark of apoptosis).
(B) Becn1-/- embryos at e6.5 fail to cavitate (left panels; red arrow labels proamniotic cavity in wt embryo; sections stained with anti-Beclin 1 antibody) and have increased numbers of TUNEL-positive cells (right panels; black arrow). Scale bar, 150 μm. (B.L., unpublished data.)
(C) Developing brains lacking ambra1 or atg5 stained positive for ubiquitin (black arrows) at e11.5 and e15.5, respectively (excerpts from Fimia et al., 2007 and Hara et al., 2006). Sections were stained with an antibody directed against ubiquitin (anti-Ub). Scale bars, 10 μm (e11.5) and 25 μm (e13.5/e15.5). Right subpanels reprinted by permission from Macmillan Publishers Ltd: Nature, Hara et al., 441, 885-889, copyright 2006.
Atg5 and Atg7
An important contribution to unraveling functions of autophagy in vertebrate development came from mutations targeting two fundamental genes in this process, atg5 and atg7 (Table 1) (Komatsu et al., 2005; Kuma et al., 2004). Atg5 and Atg7 function in autophagosome expansion and completion (Figure 1) (Mizushima et al., 1998a). Atg12, for which a targeted mouse model is not yet available, is a small factor that covalently binds to Atg5. The pathway of conjugation of Atg12 to Atg5 is similar to that of ubiquitination; Atg12 is first activated by Atg7 (which acts as an ubiquitin-activating enzyme E1) and then transferred to Atg10 (which has a similar function to E2) (Figure 1). A recent landmark study demonstrated a critical role for atg5 in early mammalian preimplantation development at the stage of transition from the maternal to zygotic gene program (Tsukamoto et al., 2008). Embryos derived from atg5-/- oocytes fertilized by atg5-/- sperm (and thereby lacking maternal rescue effects) fail to develop beyond the four- and eight-cell stages. Presumably this is because, without the autophagy machinery, maternal proteins and RNAs cannot be degraded in order to activate zygotic transcription and translation. However, in contrast to becn1-/- embryos, which die around day e7.5, atg5-/- mice derived from atg5+/- offspring are born but die perinatally. Kuma et al. (2004) observed that unsuckled atg5-/- mice die earlier than wild-type mice, whereas their survival can be slightly prolonged by forced feeding. They also noticed that plasma and tissue amino-acid concentrations in nonsuckling mutant mice are reduced relative to control mice a few hours after delivery, due to a severe depression of energy production that is reversible by forced feeding (Kuma et al., 2004). Since the induction of autophagy is immediate and transient in the perinatal time period, reaching maximal levels only 3 to 6 hr after birth and declining to basal levels within a day or two, Mizushima and collaborators postulated that the induction of autophagy following birth is required to provide energy before nursing (Kuma et al., 2004). In another study, examination of late embryonic tissues—such as the developing neuroretina and lung—in atg5-/- mice revealed the presence of an increased number of apoptotic corpses (Qu et al., 2007), thus supporting the involvement of autophagy in the removal of apoptotic corpses during development.
An analogous early postnatal death, accompanied by reduced pup size, has been described for atg7-/- mice (Komatsu et al., 2005). The reason for the reduced body size of atg7-/- neonates is unclear, but may be related to abnormal placental function or to inefficient reutilization of nutrients during embryogenesis. It is noteworthy that, in atg7-/- tissues, an accumulation of ubiquitin is detected on lipid droplet-like structures, membranous structures, and amorphous substances in the cytoplasm of neonatal hepatocytes (Komatsu et al., 2005). The increase in ubiquitin levels when autophagy is downregulated indicates (1) a role for autophagy in targeting ubiquitinated proteins for degradation, and/or (2) a possible role for the ubiquitin-proteasome system in compensating for the lack of protein turnover that may lead to accumulation of unfolded proteins during late development.
Due to the perinatal lethality of atg5 and atg7 mutant mice, several tissue-specific conditional mouse models have been generated for these two genes (see Table 1). Mice deficient in atg5 or atg7 specifically in neural cells develop progressive deficits in motor function and die a few months after birth (Hara et al., 2006; Komatsu et al., 2006). In atg5-/- and atg7-/- neurons, as previously observed in the hepatocytes of atg7-/- mice (see above), autophagy inhibition is accompanied by an abnormal amount of ubiquitinated proteins that accumulate and progressively form aggregates and inclusions. Although no major morphological defects were reported during embryonic nervous system development, the increase in ubiquitinated proteins was detected from e15.5 onward in atg5-/- neurons (Figure 2) (Hara et al., 2006). Moreover, when atg7 is selectively inactivated in the Purkinje cell subpopulation of the cerebellum alone, degeneration of the axon terminals is observed (Table 1) (Komatsu et al., 2007). Consistent with the Ulk1-inactivation phenotype, axon terminals appear much more vulnerable than the dendrites to autophagy impairment. Importantly, the analysis of the phenotypes of mice lacking neuronal atg5 and atg7 highlighted that the physiological significance of autophagy can be divided into two categories. “Baseline (or basal) autophagy” prevents the accumulation of abnormal proteins during nervous system development, whereas “induced autophagy” helps the cell adapt to an adverse environment.
Atg5 has also been specifically knocked out in the heart and in immune cells. In contrast to its effect in adult cardiomyocytes, atg5 disruption during cardiogenesis does not induce cardiac hypertrophy or dysfunction (Nakai et al., 2007). Even though compensatory mechanisms may account for preventing the pathological consequences of autophagy inhibition during development, an important finding was observed in response to blood pressure overload in postnatal mice. This insult produced severe cardiac dysfunction after only one week, such as left ventricular dilatation and death, associated with enhanced protein synthesis and proteasome-dependent protein degradation (Nakai et al., 2007). In addition, inducible postnatal deletion of atg5 in cardiomyoctes results in the rapid onset of heart failure (Nakai et al., 2007), suggesting that autophagy is a beneficial pathway responsible for cardiomyocyte remodeling during cardiogenesis and essential for stress responses involved in cardiac homeostasis. In the immune system, lymphoid precursor cells devoid of atg5 were analyzed in chimeric mice by fetal liver reconstitution after irradiation (Pua et al., 2007). A general reduction of both thymocytes and B lymphocytes was observed, probably caused by both a lowered lymphoid precursor activity and the inability of these cells to undergo homeostatic proliferation (Pua et al., 2007). In addition, selective apoptosis impaired the total number of atg5-deficient CD8+ T cells (Pua et al., 2007). Finally, a specific defect was observed in B cell development, at the pro-B to pre-B cell transition, in mice with B lymphocyte-specific deletion of atg5 (Miller et al., 2008).
LC3 and Atg4
Another molecule essential for autophagy is Atg8, whose mammalian ortholog is Microtubule-Associate Protein 1 Light Chain 3 (MAP1LC3/LC3) (Kabeya et al., 2000). Atg4/autophagin (present in mouse in four different isoforms) cleaves LC3 into the cytosolic version LC3-I. LC3-I is activated by Atg7, transferred to Atg3, and then modified with a lipid attachment to become the membrane-bound form LC3-II (Figure 1) (Ichimura et al., 2000; Kim et al., 2002; Lang et al., 1998). This conversion is not only an essential step of the membrane rearrangement dynamics during the formation of autophagosomes, but it also represents a hallmark for detecting autophagy (Klionsky et al., 2008; Mizushima et al., 2004). Both Atg4/autophagin (i.e., the isoform most widely distributed in human tissues, Atg4C) and LC3 have been knocked out in mouse (Cann et al., 2008; Marino et al., 2007). Interestingly, mutant atg4C-/- mice develop normally (Marino et al., 2007) and LC3-deficient mice display no developmental abnormalities (Cann et al., 2008). Thus, neither protein seems to be necessary for induction of the required level of autophagy essential for the viability of neonatal mice. This is likely due to a functional redundancy in the Atg4 family of cysteine proteinases and the presence of at least two murine forms of LC3 (LC3α and LC3β) (Cann et al., 2008).
Ambra1
Another striking example of how autophagy may influence embryogenesis came from the gene trapping of activating molecule in Beclin 1-regulated autophagy (Ambra1) (Fimia et al., 2007). This molecule can bind to Beclin 1 upon autophagic stimuli, thereby promoting the interaction between Beclin 1 and its target kinase Vps34 (Figure 1). Ambra1 has no apparent orthologs in lower eukaryotes. This protein is crucial for nervous system development (Table 1) and is expressed from the first stages of neurulation with high specificity for the neural plate. Its loss of function leads to gross neural tube defects, such as exencephaly and spina bifida, which culminate in death at around e16.5. These findings open a new scenario for the putative tissue-specific regulation of autophagy. Specific regulation of autophagosome formation via Beclin 1 recruitment into the class III PI3K complex could conceivably take place in specific organs, due to the variety of signaling networks responsible for tissue differentiation during embryogenesis. In this case, the type of autophagy would again be basal autophagy, which is responsible for a proper recharge of cellular components, rather than induced autophagy, which responds to specific environmental conditions. Notably, ambra1 mutant neuroepithelium at e11.5 is highly enriched for ubiquitin (Figure 2), as described for the atg5 neuron-specific inactivation at e15.5 (see above).
Summary
The diverse phenotypes of different atg gene knockout mice described here may reflect intrinsic differences in the developmental effects of genes that function in different stages of autophagy, the presence of varying levels of autophagy deficiency with inactivation of different atg genes, autophagy-independent effects of specific genes, or other as yet unidentified factors. However, taken together, these cellular systems and mouse models suggest that autophagy is a finely regulated process that appears to be essential for development. Interestingly, the complex ontogenesis of the nervous system is particularly sensitive to autophagy dysregulation, as revealed by the neural tube defects and by the axonal growth and migration impairment upon inactivation or downregulation of proautophagic genes.
Autophagy and Cell Death
Crosstalk between Cell Death and Autophagy
In lower eukaryotes, there are many examples of autophagy in programmed cell death during embryogenesis, implying that autophagic cell death is important for development (reviewed in Baehrecke, 2003; Levine and Klionsky, 2004). All the mouse models described seem to indicate that mammalian cells undergoing autophagy are generally healthy; they activate the pathway as an adaptive response to extrinsic signals or they pursue baseline (or induced) self-eating to allow “house cleaning” and morphogenesis. The parallel with cell death, however, is not just a misinterpretation. Indeed, autophagy and apoptosis are strictly connected. Bcl-2 is an antiapoptotic factor regulating the mitochondrial response to cell death stimuli (for a recent review see Youle and Strasser, 2008). Such Bcl-2 activity is controlled by a specific pool of this molecule residing on the mitochondrial membrane. By binding several proapoptotic or antiapoptotic Bcl-2 family members (all containing a BH3 domain), Bcl-2 fine-tunes mitochondrial membrane dynamics and triggers the release of cytochrome c and other apoptosis effectors from mitochondria. Beclin 1 was first described as a Bcl-2-interacting cellular protein that plays a role in antiviral host defense (Liang et al., 1998) and has since been shown to also contain a BH3 domain (reviewed in Levine et al., 2008). This interaction has subsequently been linked to autophagy inhibition (Maiuri et al., 2007a, 2007b; Pattingre et al., 2005; Wei et al., 2008), and the relative amounts of Beclin 1 and Bcl-2 (and also other Bcl-2 family members, e.g., Bcl-XL) seem regulate the transition from cell homeostasis to cell death. A nonmitochondrial pool of Bcl-2like molecules, namely those residing in the endoplasmic reticulum, inhibit Beclin 1 function in autophagosome formation, and Bcl-2/Beclin 1 complexes in the endoplasmic reticulum are disrupted by JNK1-mediated Bcl-2 phosphorylation (Akao et al., 1994; Krajewski et al., 1993; Wei et al., 2008). Thus, the cell’s decision concerning death or survival is determined by the physiological levels of autophagy, as partly determined by the relative amounts, subcellular localization, or phosphorylation status of Bcl-2-like family members. Accordingly, the absence of autophagy genes increases cell death during nutrient deprivation and other forms of cellular stress.
By contrast, excessive, nonphysiological levels of autophagy trigger autophagy-gene-dependent cell death (reviewed in Maiuri et al., 2007b). Cells from Bax/Bak double knockout mice resist apoptosis, but still undergo a nonapoptotic death after exposure to death-inducing stimuli (Lum et al., 2005). Electron microscopic and biochemical studies reveal that death in these cells is associated with autophagy, and can be blocked by knockdown of Beclin 1 and Atg5 (Shimizu et al., 2004). This suggests, in principle, that the Bcl-2 family of proteins not only regulates apoptosis, but also controls nonapoptotic programmed cell death that depends on autophagy genes. Such an enhancement of autophagy upon apoptosis inhibition has also been found in cells where mitochondrial cell death is impaired by downregulation of the apoptosome, a multimolecular complex that regulates caspase activation in the presence of cytosolic cytochrome c (structure and mechanisms reviewed by Riedl and Salvesen, 2007; Schafer and Kornbluth, 2006). When, upon exposure to specific apoptotic stimuli, the equilibrium of Bcl-2 family members is prone to trigger cell death, cytochrome c is released by mitochondria; it then binds the apoptotic protease activating factor Apaf1 and induces conformational changes of its structure. This change induces Apaf1 binding with the initiator caspase, caspase-9 (Casp9), which in turn can cleave and activate caspase-3 (Casp3). When death is induced and Casp3 is inhibited by broad-range or specific drugs (e.g., zVAD-fmk or qVDoph), autophagy occurs (Colell et al., 2007). This phenomenon has been associated with a transient decrease in mitochondrial mass. The specific autophagic degradation of damaged mitochondria that have lost their membrane potential after cytochrome c release could then spare a few intact mitochondria, which enable cell survival. In another experimental approach, the apoptosome was genetically inactivated by Apaf1 deletion in proneural cells (Ferraro et al., 2008). In this case, autophagy may also rescue the cell, by helping produce essential metabolites for ATP generation. More recently, the upstream regulator of apoptosis p53 has also been associated with autophagy regulation. By experiments of deletion, depletion, or inhibition, it has been shown that cytosolic p53 (and not nuclear p53) downregulates autophagy (Tasdemir et al., 2008).
Two other factors play a clear role in autophagy-apoptosis crosstalk. Damage-regulated autophagy modulator (DRAM) is a p53 target that modulates autophagy (Crighton et al., 2006). More specifically, DRAM is a lysosomal protein required for a p53-positive effect on autophagy induction and is essential for p53-mediated apoptosis. The contradictory results regarding p53’s role in autophagy may be due to the two functions of p53 itself, as both nuclear regulator of gene activity and cytosolic rheostat for various protein targets. Additionally, Atg5 has a role in apoptosis, resembling the proapoptotic factor Bid. A truncated form of Atg5, generated by calpain protease activity during the execution of programmed cell death, translocates to the mitochondria and accelerates its membrane permeability transition, thereby leading to the release of cytochrome c and the enhancement of the apoptosome-dependent cell death (Yousefi et al., 2006). A pioneer study by Baherecke and Lenardo (Yu et al., 2004) postulated that caspase-8 (Casp8), the initiator caspase in the extrinsic death receptor pathway, can also regulate autophagy. When Casp8 expression was reduced in cells exposed to apoptosis-inducing stimuli, cell death surprisingly increased, showing features of autophagy.
Autophagy May Allow Cell Survival in Cell Death Mutants
Developmental studies confirm the occurrence of autophagy in cell death mutants. Mutant mouse models for most proapoptotic and antiapoptotic genes have been generated (for review articles, see Joza et al., 2002; Ranger et al., 2001; Schafer and Kornbluth, 2006). Among these, Apaf1 is one of the most relevant (Cecconi et al., 1998; Yoshida et al., 1998). Two aspects of the Apaf1 knockout phenotype are particularly noteworthy: the nervous system hyperplasia and the persistence of interdigital webs. Loss of interdigital cells in the mouse embryo, a paradigm of cell death during development, is known to include an apoptotic mechanism. However, a necrotic-like morphology has been described in the persistent interdigital webs in the Apaf1 mutant limb buds (Cecconi et al., 1998; Chautan et al., 1999; Yoshida et al., 1998). Web persistence can also be found in wild-type limb explants subjected to apoptosis induction upon zVAD-fmk-mediated caspase inhibition; similar explants from the Apaf1-/- embryos showed mottled chromatin condensation, nuclear membrane detachment and rupture, dilated mitochondria and cytoplasmic vacuoles, and sometimes external plasma membrane rupture (Chautan et al., 1999). The sum effect of this cellular response is a slow cavitation of the web, followed by its final disappearance. The observed cell morphology could theoretically be due to a death mediated by excessive autophagy, similar to what has been postulated in many circumstances for lower eukaryote embryogenesis (type II cell death). Bax/Bak double knockouts also exhibit interdigital web survival (Lindsten et al., 2000). In contrast with the Apaf1 phenotype, this effect in the Bax/Bak mutant is irreversible. In the mitochondrial death pathway, Bax and Bak are defined as the gateway of apoptosis, with these two factors playing a role upstream of mitochondrial activation (Wei et al., 2001). Bax/Bak mutant cells (in which the switch to apoptosis has been impaired) exposed to death-inducing stimuli do not activate mitochondria at all; they not only survive, but are healthy. In Apaf1-/- mice, vice versa, Bax/Bak and other Bcl-2 family members enable cytochrome c release in response to developmental signals from surrounding tissues; the mitochondria start their depolarization program and autophagy sustains cell survival until metabolites for energy production are totally consumed and secondary cellular destruction takes place. This represents an unsuccessful attempt at survival rather than a death mechanism. In the nervous system, something very similar has been observed. Cerebral cortices in forebrain overgrowth (fog) mice, which show reduced Apaf1 mRNA levels, give rise to ectopic brain masses during development (Honarpour et al., 2001). These masses undergo intensive autophagy in the postnatal phases of neurodevelopment (Moreno et al., 2006).
With respect to the autophagy-deficient phenotypes, we have already described what happens in atg5 or becn1 mutant embryoid bodies or atg5 mutant retina (Qu et al., 2007). Autophagy mediated by these two factors in dying cells is crucial for the removal of dead corpses by the surrounding nonprofessional phagocytes (see Figure 2). More recently, inhibition of autophagy has also been shown to impair cell corpse removal in the developing avian retina (Mellén et al., 2008). Therefore, what may at first sight be interpreted as excessive cell death is, in fact, a defect in cell signaling and cell corpse removal. Also in ambra1 mutant brains, apparent excessive apoptosis is observed. Many TUNEL-positive cells accumulate in the caudal or rostral parts of the forming neural tube, which is evident from e9.0 onward (Fimia et al., 2007). Although it cannot be ruled out that defective generation of engulfment signals contributes to the enhanced cell death in this case, significantly, the onset of the phenotype (hyperplasia of the neuroepithelium) occurs much earlier, at around e7.0, when Apaf1 is not yet expressed in the embryo and, consequently, the apoptosome cannot mediate mitochondria-dependent programmed cell death. Excessive cell death in ambra1 mutant mice might be explained by the following alternative reasons: (1) autophagy defects in some subpopulations of neuronal precursors might induce surrounding tissue to undergo waves of apoptosis; (2) proneurons defective in autophagy might undergo apoptotic cell death as described for some cell culture systems; (3) supernumerary cells might be killed by programmed cell death, which actively controls cell number in neurodevelopment. Additional studies should elucidate the correct hypothesis and whether it is related to the autophagic functions of Ambra1.
In light of recent evidence for an active role of basal autophagy in neurodevelopment, some “apoptotic phenotypes” are worth reevaluating: one example could be the c-Jun-NH(2)-terminal kinase 1 and 2 double knockout embryo (Kuan et al., 1999; Sabapathy et al., 1999). Embryos lacking both kinases (JNK1 and JNK2) die at around e11.0 and display an open neural tube (exencephaly) at the hindbrain level with reduced apoptosis in the hindbrain neuroepithelium at e9.25. In contrast, a dramatic increase in cell death was observed one day later, at e10.5, in both the hindbrain and forebrain. Which kind of cell death is reduced and which kind is enhanced in this embryo? Could two different cell death processes be impaired in the mutant? Normal e9.25 fetuses contain a high proportion of apoptotic cells in the hindbrain region, concentrated mostly at the edges of the folding neural tube. This evidence has been reported by counting condensed nuclei in the area and by TUNEL staining (Kuan et al., 1999; Sabapathy et al., 1999). There are two possibilities: (1) these cells lack basal autophagy, i.e., do not expose the appropriate come-get-me and eat-me signals and are not properly phagocytosed; (2) they undergo type II nonapoptotic, autophagic cell death in absence of the JNK proteins. The increase in apoptosis is observed in these knockout models at e10.5 within the open neural tube and at morphogenetic apoptosis sites in the forebrain. In this case, deregulated cell death might be a consequence of either defective proapoptotic or proautophagic signals.
Although JNK proteins have been shown to regulate both autophagy and apoptosis by a Casp8-dependent mechanism (Yu et al., 2004), the mechanistic basis for the phenotype of JNK1/JNK2 double knockout embryos remains unclear. Interestingly, JNK1 appears to be essential for mediation of Bcl-2 phosphorylation, disruption of the Bcl-2/Beclin 1 complex, and autophagy activation during starvation (Wei et al., 2008). Given the parallels between the phenotypes of the JNK1/JNK2 and Ambra1 mutant mice, it is tempting to speculate that JNK proteins also regulate autophagy induction during neurodevelopment through effects on the Beclin 1/Ambra1 autophagyinducing complex.
An important open question is whether the excessive developmental cell death observed in some of the mouse mutants here described is a consequence or a secondary effect of defective autophagy in vivo. Further studies about the roles of such processes in the delicate balance of cell fate in development need to be performed for an unequivocal answer.
Autophagy and Cell Proliferation
In several adult mouse models of autophagy impairment, a significant dysregulation of the rate of cellular proliferation has been observed. However, during development, the only example to date of such alteration is represented by the ambra1 mutant embryos, in which alterations in cell cycle progression were detected at the beginning of neurulation and in ambra1-deficient mouse embryonic fibroblasts (Fimia et al., 2007). Ambra1 is mainly expressed in the neural plate at e7.0, the earliest stage for detecting phenotypic abnormalities in the mutant embryos. Although a complete uncoupling of the cell cycle defect from the autophagy deficiency cannot be excluded, a link between these two processes can be postulated. First, basal autophagy may be relevant for recycling proteins essential in negative cell cycle regulation; upon decrease in autophagy, those proteins would not be available, inducing the cell cycle to speed up. Second, when basal autophagy has been impaired, proneural cells may not process, rearrange, or expose their own suitable molecules for correct integration with the surrounding tissue. As a consequence, without a clear commitment, and being prone to division, they may continue to proliferate. Any disturbance of the neural precursor cells at such an early stage may first have consequences for proliferation and, subsequently, for cell migration and differentiation of the neuroepithelium. As described above, alteration of the cell death program in neural precursors induces a high rate of cell proliferation, resulting in a hyperplasic brain, the formation of discrete ectopic brain masses, or both. Also, defects in the cytoskeleton of proneural cells may lead to defects in neural tube closure by impairing cell cycle regulation. The findings from David Rubinzstein’s laboratory are of particular interest in this context (Ravikumar et al., 2005). A mechanistic link has been hypothesized between dynein mutations and inclusion formation in certain motor neuron diseases: dynein mutations may impair autophagosome-lysosome fusion and the clearance of aggregate-prone proteins, making neurons more susceptible to proapoptotic insults. This hypothesis is based on the fact that autophagosomes are transported along the neural axons by the dynein motor complex on the microtubuli to perinuclear regions where lysosomes are more abundant (Jahreiss et al., 2008). A crucial role for the cytoskeleton in autophagosome flux has long been postulated (Fass et al., 2006; Kochl et al., 2006) and autophagy is impaired during mitosis (Eskelinen et al., 2002). A defect in autophagosome formation could, conversely, potentially result in dysregulated microtubular dynamics, which are a determinant in cell cycle regulation.
Clearer effects of autophagy dysregulation on cell cycle are evident in the homeostasis of adult tissues. Here, when autophagy genes are deleted or downregulated, such as in hemizygous conditions, a high incidence of tumors is observed in a wide range of target tissues. Beclin 1, UVRAG, Atg4C, or Atg5 complete or partial inactivation in certain cancers highlights each of these molecules’ potential in tumor suppression, strongly suggesting that autophagy’s role in cancer has hitherto been underestimated. Becn1+/- mice are in fact viable, but they develop a wide range of spontaneous malignancies with increased frequency (Liang et al., 1999; Qu et al., 2003; Yue et al., 2003). Atg4C-/- mice show a higher incidence of chemically induced fibrosarcomas as compared with their wild-type littermates (Marino et al., 2007). The viable Bif-1-/- mice, which do not exhibit major abnormalities, show increased frequency of spontaneous lymphomas and solid tumors (Takahashi et al., 2007). Bif-1 therefore joins the putative UVRAG-Beclin 1 complex as a potential activator of autophagy (Figure 1) that can also be considered a bona fide tumor suppressor (along with UVRAG) (Liang et al., 2006). Further studies involving mice lacking other components of the Atg4 proteinase family or the Beclin 1 proautophagic complex, and exhibiting mutations in the atg genes involved in autophagosome elongation and completion, will help define the role of each family member in vivo in both normal and pathological conditions, including cancer. Hence there is an urgent need for understanding the mechanisms whereby autophagy crosstalks with cell cycle regulation.
Autophagy and Differentiation
In principle, alterations in autophagy may influence cell fate during mammalian development in several ways. First, autophagy may, analogously to lower eukaryotes, destroy mammalian cells by reducing the size of the cytosol, impairing protein synthesis, engulfing energy-producing organelles, or stopping cell metabolism. Second, autophagy may guarantee cell survival under dramatic conditions of mechanical or biochemical stress either within the cell or when induced by the surrounding environment. Third, autophagy may help the cell to rapidly and efficiently change its cytosolic composition, accelerating routine protein and organelle turnover and modifying the cell’s appearance in terms of exposed receptors, transcriptional factors present in the nucleus, and cytoskeletal dynamics. Of these three possibilities, the latter is the most relevant for influencing cell-autonomous and/or regulated differentiation.
In at least three cases, the role of autophagy in this regard is supported by experimental evidence. In the case of becn1-/- mutant embryos, visceral endoderm development occurs, although cells seem to have an abnormal morphology and development is blocked after visceral endoderm formation (Yue et al., 2003). Both the anterior-posterior and the dorso-ventral axes of the mammalian embryo are established by a complex set of interactions between embryonic (epiblast) and extraembryonic tissues (which include the visceral endoderm among others). At e5.0, at least two signaling centers are necessary for the establishment of axes, one located in the anterior visceral endoderm and the other in the epiblast (the node). The epiblast induces the anterior visceral endoderm to form from the distal-most visceral endoderm of the embryo (Rodriguez et al., 2005). This signaling center expresses a unique set of molecular markers (i.e., Hex, Lhx1, Cer1, and Lefty1) and is required for specifying anterior neural identity. Later the node is induced in the epiblast and is characterized by the expression of Nodal and Wnt3, which in turn specify posterior cell fate. Accumulating evidence indicates that reciprocal interactions between the epiblast and visceral endoderm are essential for establishing both these signaling centers (reviewed in Takaoka et al., 2007). During the few days (e5.0-e7.0) of mouse development immediately after implantation, the gastrulation and differentiation fate of the embryo is therefore decided, and visceral endoderm, characterized by early expression of Beclin 1, is responsible for a related set of events. The fact that visceral endoderm cannot go on to develop in becn1-/- mutant embryos may therefore derive from a lack of signaling in the proper areas. In this specific stage of embryogenesis, the autophagosomes may selectively destroy subcellular structures or clusters of proteins, allowing a fast replacement by the protein synthesis apparatus that involves CAP-dependent and -independent translation or translational control of mature mRNAs.
Similarly, the brain patterning of Ambra1 mutant embryos is dysregulated during neural tube differentiation at e10.5 (Fimia et al., 2007), with the morphogen sonic hedgehog (Shh) diffused in the area of the neural tube floor plate rather than concentrated in the notochord, a bona fide signaling center in neurogenesis (Figure 3) (Marti and Bovolenta, 2002; Wilson and Maden, 2005). In normal development, the ventral-most part of the undifferentiated neural tube (where Ambra1 is highly expressed) may be responsible for producing specific receptors, transferring them onto the membrane, and then exposing them. The spread of Shh in the neural target field is influenced by the expression of extracellular and transmembrane proteins, which can bind to it and either restrict its diffusion or alter the rate of Shh degradation (reviewed in Dessaud et al., 2008). Again, autophagosome formation, mediated by Ambra1-dependent constitution of the Beclin 1/Vps34 multimolecular kinase complex, might regulate the proper receptor/ligand interaction, inducing a fast cellular makeover in neural tube areas subjected to strong cytosolic and nuclear rearrangements.
Figure 3. Autophagy-Dependent Regulation of Shh Signaling.
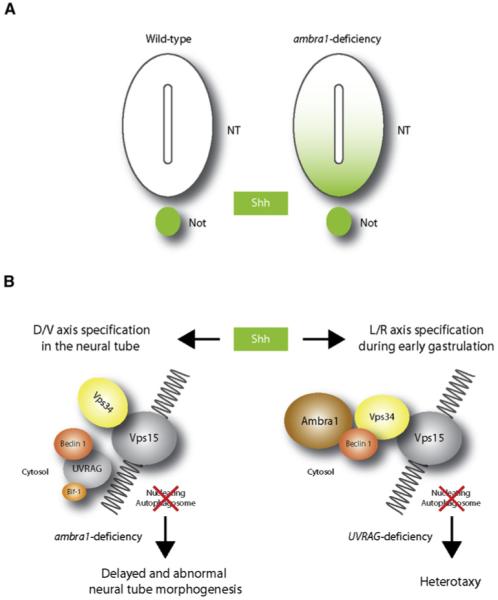
The morphogen sonic hedgehog (Shh) is involved in several aspects of vertebrate development. The model shown is speculative, and autophagy-independent effects may also explain the phenotypes of ambra1, becn1, and UVRAG targeted mutant mice. (A) In wild-type embryos at e9.5, Shh expression is retained within the notochord (Not). At the same developmental stage, ambra1-deficient embryos show Shh expression diffused in the ventral half of the neural tube (NT) and in the floor plate. (B) In the absence of Ambra1, the interaction between Beclin 1 and Vps34 is impaired and autophagy cannot proceed. In addition to the control of Shh signaling during dorso-ventral (D/V) differentiation of the neural tube, autophagy may be involved in regulating the left-right (L/R) axis. In fact, in wild-type embryos, Shh also signals the establishment of the left-right axis in the node cells (see text). In UVRAG mutant humans and Drosophila, the left-right axis symmetry is disturbed, leading to heterotaxy. This could be a consequence of incorrect Shh distribution within the node.
Autophagy genes could also play a role in the specification of the left-right axis. In vertebrates, left-right symmetry is determined in part by a flow of Shh in the left cells of the node during gastrulation, inducing left-gene expression in the left part of the primitive streak. Ciliary movements of node cells have been linked to this asymmetric concentration of Shh (Hirokawa et al., 2006). Interestingly, the factor UVRAG (another component of the dynamic Beclin 1/Vps34 complex) (Liang et al., 2006) is mutated in a human case of abnormal left-right axis formation, resulting in heterotaxy and multiple malformations (Iida et al., 2000). Since UVRAG is also expressed during embryonic life in presomitic stages, this phenotype could also be related to an impairment of Shh distribution in the node, caused by a decreased rate of autophagosome formation and a defective cell makeover in those cells (Figure 3). Notably, a similar phenotype in Drosophila has been identified, upon mutation of an UVRAG ortholog gene (reported in He and Overdahl, 2007). Another relevant role of Shh during embryogenesis is the establishment of the antero-posterior axis of the limb (Laufer et al., 1994). Interestingly, the first analysis of atg5 embryonic expression, performed in 1998 in Peter Gruss’ laboratory, showed high levels of atg5 (originally termed paddy) mRNA in the zone of polarizing activity (ZPA), the area of the limb bud responsible for antero-posterior axis formation (Pires da Silva and Gruss, 1998).
Conclusion
The importance of autophagy during development has been highlighted by a number of approaches in lower eukaryotes, from yeast to C. elegans, Drosophila and D. discoideum. In these organisms, autophagy is a relevant stress response, and the developmental stages involving highly stressful conditions, such as yeast sporulation or insect metamorphosis, activate autophagic pathways. Autophagy as a cell death mechanism, characterized by lysosomal activation and cell vacuolization, has also been widely investigated. However, only in the last decade have extensive studies on this process been performed in vertebrates, thanks to the elucidation of the major molecular mechanisms of autophagy. Given the phenotypes observed in lower eukaryotes, it seemed at one point that autophagy might be a stress response or a death mechanism in mammals. Instead, new findings in becn1, ambra1, atg5, and atg7, and other atg gene inactivation during development now indicate that autophagy is an important prosurvival mechanism and may act in renovating the cell cytosol by degrading bulk cytoplasm, specific long-lived proteins, or damaged mitochondria. Furthermore, autophagy may help the cell signal its condition to the surroundings or rapidly modify its protein content to better respond to extrinsic stimuli. Notably, the definition of autophagy as merely a programmed cell death pathway has also been recently called into question in Drosophila (Akdemir et al., 2006; Martin and Baehrecke, 2004; Juhász et al., 2007). Histolysis of the salivary gland, a classical example of autophagy-dependent cell death in Drosophila, requires Dark and Dronc, two orthologs of vertebrate Apaf1 and Casp9. This implies that autophagy per se is not the “lethal event” mediating histolysis of this organ. Instead, the induction of autophagy may lie upstream of, or parallel to, the apoptosome.
Another ground-breaking finding is that the nervous system is a preferential target for uncovering autophagy abnormalities, perhaps due to the existence of a neurospecific autophagy apparatus in the developing central nervous system. However, another cause may be the high sensitivity of the neural precursor cells to any changes in their protein or organelle content prior to (or at the beginning of) their complex differentiation.
Studying autophagy in mouse models during development may also have important consequences for medicine. Neural tube defects, such as spina bifida or exencephaly, are common congenital malformations leading to infant mortality or severe disability. Despite the multifactorial etiology of neural tube defects, the discovery of an orchestrated control of both apoptotic cell death and basal autophagy for regulating the correct morphogenesis and cell number in the developing central nervous system may reveal novel target factors for diagnostic and therapeutic purposes. Similarly, there is a need to further evaluate the contribution in humans of autophagy dysregulation to early developmental defects, e.g., in preimplantantion development abnormalities and in proamniotic cavity formation, where murine Atg5 and Beclin 1, respectively, are important factors.
The landmark discovery of the roles that autophagy plays in development may have implications for cancer. In light of autophagy’s role in tumorigenesis, it may be possible to pinpoint single components of the autophagy pathway that characterize, when mutated, the embryonic origin of various tumors. Admittedly, much work needs to be done in defining more fully the mechanisms that autophagy may use in cell cycle regulation and tumor suppression. One thing remains clear, however: the putative pharmacological modulation of genes in these processes could represent a novel strategy for treating a wide range of tumors.
ACKNOWLEDGMENTS
Work from the authors’ laboratories is partially supported by grants from Fondazione Telethon, Compagnia di San Paolo, AIRC, the Italian Ministry of University and Research (MUR), and the Italian Ministry of Health to F.C., and from the National Institutes of Health, American Cancer Society, and Howard Hughes Medical Institute to B.L. F.C. is an Associate Telethon Scientist. We thank Martin W. Bennet for the valuable editorial work.
REFERENCES
- Akao Y, Otsuki Y, Kataoka S, Ito Y, Tsujimoto Y. Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res. 1994;54:2468–2471. [PubMed] [Google Scholar]
- Akdemir F, Farkas R, Chen P, Juhasz G, Medved’ova L, Sass M, Wang L, Wang X, Chittaranjan S, Gorski SM, et al. Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development. 2006;133:1457–1465. doi: 10.1242/dev.02332. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH. Autophagic programmed cell death in Drosophila. Cell Death Differ. 2003;10:940–945. doi: 10.1038/sj.cdd.4401280. [DOI] [PubMed] [Google Scholar]
- Beaulaton J, Lockshin RA. Ultrastructural study of the normal degeneration of the intersegmental muscles of Anthereae polyphemus and Manduca sexta (Insecta, Lepidoptera) with particular reference of cellular autophagy. J. Morphol. 1977;154:39–57. doi: 10.1002/jmor.1051540104. [DOI] [PubMed] [Google Scholar]
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, Meijer AJ. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995;270:2320–2326. doi: 10.1074/jbc.270.5.2320. [DOI] [PubMed] [Google Scholar]
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev. Dyn. 2008;237:187–195. doi: 10.1002/dvdy.21392. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Chautan M, Chazal G, Cecconi F, Gruss P, Golstein P. Interdigital cell death can occur through a necrotic and caspase-independent pathway. Curr. Biol. 1999;9:967–970. doi: 10.1016/s0960-9822(99)80425-4. [DOI] [PubMed] [Google Scholar]
- Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, Fitzgerald P, Guio-Carrion A, Waterhouse NJ, Li CW, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983–997. doi: 10.1016/j.cell.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–134. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- De Duve C. The lysosome. Sci. Am. 1963;208:64–72. doi: 10.1038/scientificamerican0563-64. [DOI] [PubMed] [Google Scholar]
- De Duve C, Wattiaux R. Functions of lysosomes. Annu. Rev. Physiol. 1966;28:435–492. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–2503. doi: 10.1242/dev.009324. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM, Lucocq JM. Inhibition of autophagy in mitotic animal cells. Traffic. 2002;3:878–893. doi: 10.1034/j.1600-0854.2002.31204.x. [DOI] [PubMed] [Google Scholar]
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch. Biochem. Biophys. 2007;462:210–219. doi: 10.1016/j.abb.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Ferraro E, Pulicati A, Cencioni MT, Cozzolino M, Navoni F, di Martino S, Nardacci R, Carrì MT, Cecconi F. Apoptosome-deficient cells lose cytochrome c through proteasomal degradation but survive by autophagy-dependent glycolysis. Mol. Biol. Cell. 2008;19:3576–3588. doi: 10.1091/mbc.E07-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem. Biophys. Res. Commun. 1988;151:40–47. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Overdahl A. 2007 Keystone Symposium on autophagy in health and disease. Autophagy. 2007;3:527–536. doi: 10.4161/auto.4595. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Tanaka Y, Okada Y, Takeda S. Nodal flow and the generation of left-right asymmetry. Cell. 2006;125:33–45. doi: 10.1016/j.cell.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Honarpour N, Gilbert SL, Lahn BT, Wang X, Herz J. Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc. Natl. Acad. Sci. USA. 2001;98:9683–9687. doi: 10.1073/pnas.171283198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung JP, Koppel H, Clarke PG. Endocytosis and autophagy in dying neurons: an ultrastructural study in chick embryos. J. Comp. Neurol. 1989;283:425–437. doi: 10.1002/cne.902830310. [DOI] [PubMed] [Google Scholar]
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Iida A, Emi M, Matsuoka R, Hiratsuka E, Okui K, Ohashi H, Inazawa J, Fukushima Y, Imai T, Nakamura Y. Identification of a gene disrupted by inv(11)(q13.5;q25) in a patient with left-right axis malformation. Hum. Genet. 2000;106:277–287. doi: 10.1007/s004390051038. [DOI] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC. The Itinerary of Autophagosomes: From Peripheral Formation to Kiss-and-Run Fusion with Lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Kroemer G, Penninger JM. Genetic analysis of the mammalian cell death machinery. Trends Genet. 2002;18:142–149. doi: 10.1016/s0168-9525(01)02618-x. [DOI] [PubMed] [Google Scholar]
- Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000;150:1507–1513. doi: 10.1083/jcb.150.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Huang WP, Stromhaug PE, Klionsky DJ. Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J. Biol. Chem. 2002;277:763–773. doi: 10.1074/jbc.M109134200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J. Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993;53:4701–4714. [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lang T, Schaeffeler E, Bernreuther D, Bredschneider M, Wolf DH, Thumm M. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998;17:3597–3607. doi: 10.1093/emboj/17.13.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer E, Nelson CE, Johnson RL, Morgan BA, Tabin C. Sonic hedgehog and Fgf-4 act through a signaling cascade and feedback loop to integrate growth and patterning of the developing limb bud. Cell. 1994;79:993–1003. doi: 10.1016/0092-8674(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007a;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007b;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- Marti E, Bovolenta P. Sonic hedgehog in CNS development: one signal, multiple outputs. Trends Neurosci. 2002;25:89–96. doi: 10.1016/s0166-2236(02)02062-3. [DOI] [PubMed] [Google Scholar]
- Martin DN, Baehrecke EH. Caspases function in autophagic programmed cell death in Drosophila. Development. 2004;131:275–284. doi: 10.1242/dev.00933. [DOI] [PubMed] [Google Scholar]
- Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellén MA, de la Rosa EJ, Boya P. The autophagic machinery is necessary for removal of cell corpses from the developing retinal neuroepithelium. Cell Death Differ. 2008;15:1279–1290. doi: 10.1038/cdd.2008.40. [DOI] [PubMed] [Google Scholar]
- Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK, Mizushima N, Iwasaki A, He YW, Swat W, Virgin HW., 4th The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. doi: 10.4161/auto.5474. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998a;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998b;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Imbroglini V, Ferraro E, Bernardi C, Romagnoli A, Berrebi AS, Cecconi F. Apoptosome impairment during development results in activation of an autophagy program in cerebral cortex. Apoptosis. 2006;11:1595–1602. doi: 10.1007/s10495-006-9081-4. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XLBeclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Müller F, Ohshima Y. Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 1994;8:2389–2400. doi: 10.1101/gad.8.20.2389. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Mizushima N, et al. The autophagy protein Beclin 1 is reduced in early Alzheimer’s Disease and regulates Ab accumulation in vivo. J. Clin. Invest. 2008;118:2015–2018. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires da Silva A, Gruss P. Gene trap insertion into a novel gene expressed during mouse limb development. Dev. Dyn. 1998;212:318–325. doi: 10.1002/(SICI)1097-0177(199806)212:2<318::AID-AJA16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- Ranger AM, Malynn BA, Korsmeyer SJ. Mouse models of cell death. Nat. Genet. 2001;28:113–118. doi: 10.1038/88815. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, Brown SD, Rubinsztein DC. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005;37:771–776. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- Rodriguez TA, Srinivas S, Clements MP, Smith JC, Beddington RS. Induction and migration of the anterior visceral endoderm is regulated by the extra-embryonic ectoderm. Development. 2005;132:2513–2520. doi: 10.1242/dev.01847. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech. Dev. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Kornbluth S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev. Cell. 2006;10:549–561. doi: 10.1016/j.devcel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Scharrer B. Ultrastructural study of the regressing prothoracic glands of blattarian insects. Z. Zellforsch. Mikrosk. Anat. 1966;69:1–21. doi: 10.1007/BF00406264. [DOI] [PubMed] [Google Scholar]
- Schin KS, Clever U. Lysosomal and free acid phosphatase in salivary glands of chironomus tentans. Science. 1965;150:1053–1055. doi: 10.1126/science.150.3699.1053. [DOI] [PubMed] [Google Scholar]
- Schin K, Laufer H. Studies of programmed salivary gland regression during larval-pupal transformation in Chironomus thummi. I. Acid hydrolase activity. Exp. Cell Res. 1973;82:335–340. doi: 10.1016/0014-4827(73)90350-9. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007;581:2156–2161. doi: 10.1016/j.febslet.2007.01.096. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka K, Yamamoto M, Hamada H. Origin of body axes in the mouse embryo. Curr. Opin. Genet. Dev. 2007;17:344–350. doi: 10.1016/j.gde.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Takeshige K, Baba M, Tsuboi S, Noda T, Ohsumi Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992;119:301–311. doi: 10.1083/jcb.119.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavahery-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al. Autophagy regulation by cytoplasmic p53. Nat. Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349:275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–846. doi: 10.1016/s0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–120. doi: 10.1126/science.1154822. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autopahgy. Mol. Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Maden M. The mechanisms of dorsoventral patterning in the vertebrate neural tube. Dev. Biol. 2005;282:1–13. doi: 10.1016/j.ydbio.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Babu JR, da Silva S, Shu Q, Graef IA, Oliver T, Tomoda T, Tani T, Wooten MW, Wang F. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc. Natl. Acad. Sci. USA. 2007;104:5842–5847. doi: 10.1073/pnas.0701402104. [DOI] [PMC free article] [PubMed] [Google Scholar]