

Summary
SRp38 is an atypical SR protein splicing regulator. To define the functions of SRp38 in vivo, we generated SRp38-null mice. The majority of homozygous mutants survived only until E15.5 and displayed multiple cardiac defects. Evaluation of gene expression profiles in the SRp38(−/−) embryonic heart revealed a defect in processing of the pre-mRNA encoding cardiac triadin, a protein that functions in regulation of Ca2+ release from the sarcoplasmic reticulum during excitation-contraction coupling. This defect resulted in significantly reduced levels of triadin, as well as of the interacting protein calsequestrin 2. Purified SRp38 was shown to bind specifically to the regulated exon, and to modulate triadin splicing in vitro. Extending these results, isolated SRp38(−/−) embryonic cardiomyocytes displayed defects in Ca2+ handling compared with wild-type controls. Taken together, our results demonstrate that SRp38 regulates cardiac-specific alternative splicing of triadin pre-mRNA and reflecting this is essential for proper Ca2+ handling during embryonic heart development.
Introduction
Alternative splicing of mRNA precursors plays a major role in expanding proteomic complexity and contributes significantly to cell- and tissue-specific gene expression according to cell type and developmental stage (Black, 2000; Blencowe, 2006). Splicing decisions that determine the expression patterns of different protein isoforms can have dramatic developmental consequences (Hammes et al., 2001), and defects in the splicing pathway have been shown to be associated with a variety of human diseases (Wang and Cooper, 2007). These observations indicate that understanding the mechanisms that control splice site selection is of vital importance.
Splicing is carried out in the spliceosome, a macromolecular complex containing five small nuclear ribonucleoprotein particles and a large number of auxiliary proteins (Jurica and Moore, 2003; Konarska and Query, 2005). Among the best-characterized non-snRNP proteins are the serine/arginine (SR)-rich family of splicing factors. SR proteins are highly conserved among animals and plants and play key roles in both constitutive and alternative splicing (Fu, 1995; Manley and Tacke, 1996; Graveley, 2000; Black, 2003). All SR proteins contain one or two RNP-type RNA-binding domains and an arginine-serine-rich domain. Typical SR proteins affect splicing in two distinguishable ways: First, SR proteins play essential but redundant roles in constitutive splicing, functioning as general splicing factors in a manner that involves stabilizing the binding of snRNPs to pre-mRNAs. Second, SR proteins bind in a sequence-specific manner to exonic splicing enhancers to facilitate recruitment of snRNPs to splice sites and thereby enhance exon inclusion. Beyond their role in splicing, the function of SR proteins has more recently been extended to mRNA export (Huang and Steitz, 2001), mRNA stability (Lemaire et al., 2002; Zhang and Krainer, 2004), genomic stability (Li and Manley, 2005) and translation (Sanford et al., 2004), indicating that SR proteins are involved in multiple cellular processes.
While SR proteins were discovered and characterized by biochemical methods, genetic approaches have been employed to address their physiological functions in living cells and organisms. Inactivation of ASF/SF2 in chicken DT40 cells led to general defects in RNA metabolism (Wang et al., 1996) and to apoptotic cell death (Li et al., 2005). Deletion of SRp20 in mice caused embryonic lethality at the blastocyst stage (Jumaa et al., 1999). Similar early lethal phenotypes were also observed in both SC35 (Wang et al., 2001) and ASF/SF2 (Xu et al., 2005) knockout mice. These experiments suggested that SR proteins perform fundamental functions crucial for cell viability. However, heart-specific knockouts of either SC35 (Ding et al., 2004) or ASF/SF2 (Xu et al., 2005) had little effect on cardiac development, instead resulting in cardiomyopathy in adult mice. Interestingly, only a specific set of alternative splicing events were affected in the ASF/SF2-ablated hearts; expression of most transcripts was unaltered (Xu et al., 2005). These findings point to the possibility that SR proteins may act as specific splicing regulators that play defined roles in specific cells and tissues.
SRp38 is an unusual member of the SR protein family. Although structurally similar to typical SR proteins, SRp38 is unable to activate splicing in standard in vitro assays, suggesting that it cannot function as a general splicing activator (Shin and Manley, 2002). Instead, SRp38 functions as a general splicing repressor, but only when activated by dephosphorylation (Shin and Manley, 2002). Another unusual property of SRp38 is that loss of SRp38 does not affect cell viability, although a prolonged G2/M phase and poor recovery following heat shock were observed in SRp38(−/−) DT40 cells (Shin et al., 2004). Despite its inactivity as a general splicing factor, recent experiments have shown that phosphorylated SRp38 can function as a sequence-dependent splicing activator(Feng et al., 2008).
In this report, we describe the generation and characterization of SRp38(−/−) mice. Consistent with the dispensability of SRp38 for cell viability, mice survive through early embryogenesis but die progressively from mid-gestation until birth with multiple cardiac defects. To investigate the possible molecular basis for this, microarray analysis of RNA isolated from mutant and wild-type embryonic ventricles was performed, but revealed only limited differences. Notably, however, the ratio of alternatively spliced isoforms of the mRNA encoding the cardiac protein triadin was significantly altered in mutant embryos, resulting in significantly reduced levels of triadin as well as of the interacting protein calsequestrin 2 (CSQ2). These proteins play a critical role in excitation-contraction (EC) coupling, functioning in regulation of Ca2+ release from the sarcoplasmic reticulum. By analyzing RNA binding by purified SRp38 and alternative splicing of triadin pre-mRNAs in cell extracts and stable transfection assays, we demonstrate that SRp38 directly modulates triadin alternative splicing. Importantly, using primary cultures of embryonic cardiomyocytes and Ca2+ imaging, we observed abnormal Ca2+ release from the sarcoplasmic reticulum in the SRp38(−/−) myocytes. The frequency of Ca2+ sparks was significantly increased, while the spark amplitude was reduced. We conclude that SRp38-regulated alternative splicing plays a critical role during embryonic heart development.
Results
Generation of SRp38-deficient mice
Our previous data has shown that SRp38 is not required for viability of DT40 cells (Shin et al., 2004). Given this, we reasoned that disruption of SRp38 in mice would not result in early embryonic lethality, and might give rise to informative phenotypes. We therefore set out to disrupt SRp38 in mice by a conventional gene targeting strategy (see Experimental Procedures). In brief, a gene targeting vector was constructed (Figure S1A, middle) and electroporated into E14 embryonic stem cells, and following homologous recombination a neoR cassette was introduced in an antisense orientation to disrupt the gene near the 5′ end (Figure S1A, bottom). Double-drug resistant clones were screened by Southern blotting using exon-specific and neo-specific probes as indicated in Figure S1A (data not shown). Two homologous recombinants were injected into C57BL/6J blastocysts to produce male chimeras that transmitted the mutated SRp38 allele through the germline to 50% of their agouti 129/C57BL/6 offspring. F1 SRp38(+/−) males and females were then interbred to produce F2 litters. Genotypes were confirmed by PCR analysis (Figure S1B) and Southern blot (Figure S1C).
The heterozygous mice appeared completely normal and fertile. However, no homozygous mutant mice were identified among 82 pups from heterozygous intercrosses, suggesting that a lack of SRp38 results in embryonic lethality. Examination of embryos at various times during gestation revealed that the majority of SRp38(−/−) embryos died before or around developmental day E15.5, with only ~5% (6 out of ~110) surviving until birth (Figure S1D). Genetic background had no apparent effect on the phenotypes of the SRp38(−/−) mice (data not shown).
SRp38(−/−) mice display multiple cardiac defects
We next examined morphological and histological properties of the SRp38(−/−) mice. The few mutant mice that survived to full term were growth retarded and did not live through the first day (Figure 1A). However, up until E12.5 SRp38(−/−) embryos showed no apparent morphological abnormalities (data not shown). From E13.5 to E16.5, the SRp38(−/−) embryos displayed noticeable edema along the back as well as slight growth retardation, indicative of severe cardiac defects (Figure 1B). Strikingly, despite the fact that SRp38 is widely expressed (Shin and Manley 2002), other tissues and organs appeared normal.
Figure 1. The majority of SRp38(−/−) mice die before birth and display severe edema.
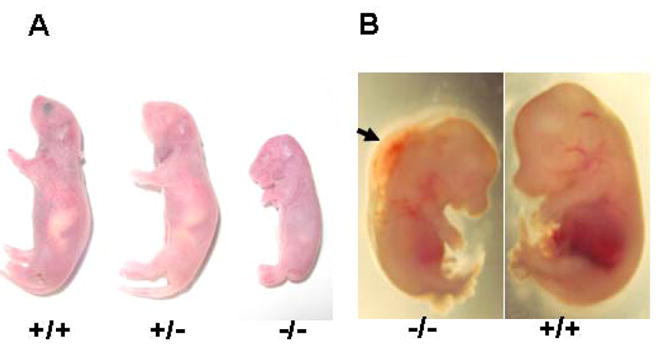
(A) Mutant mice that survive to term are severely growth retarded and die within the first day. (B) Severe edema is visible in the E14.5 SRp38(−/−) embryo. The arrow indicates edema along the back of the SRp38(−/−) embryo.
To examine the cardiac defects of the SRp38 null embryos, histological sections were generated from E14.5 and E16.5 wild-type and SRp38(−/−) embryos (Figures 2A–2F). HE staining (see Experimental Procedures) revealed severe morphological abnormalities, including atrial septal defects (ASD) and ventricular septal defects (VSD), in the E14.5 SRp38(−/−) heart (compare Figures 2A and 2B). High magnification further revealed that the myocardium was thin, undifferentiated and arranged in a disorganized manner, in contrast to that in wild-type hearts (compare Figures 2C and 2D). Moreover, an atrioventricular canal defect (AVCD) was detected in the E16.5 mutant embryos. Dilated right atria and a complete AVCD with a single atrioventricular junction were obvious (Figure 2F), while at this stage septation was complete in the wild-type control (Figure 2E). Consistent with these cardiac defects, severe degeneration were observed in the livers of many of the SRp38(−/−) embryos (Figures 2G and 2F). These data indicate that SRp38 is critical for normal cardiac development. TUNEL assays and immunostaining with phospho-histone-H3 ser 10 antibodies were also performed; however, no evidence of aberrant apoptosis or abnormal proliferation was observed in the mutant hearts at E14.5 (Figure S2).
Figure 2. SRp38(−/−) mice have multiple heart defects and severe liver damage.
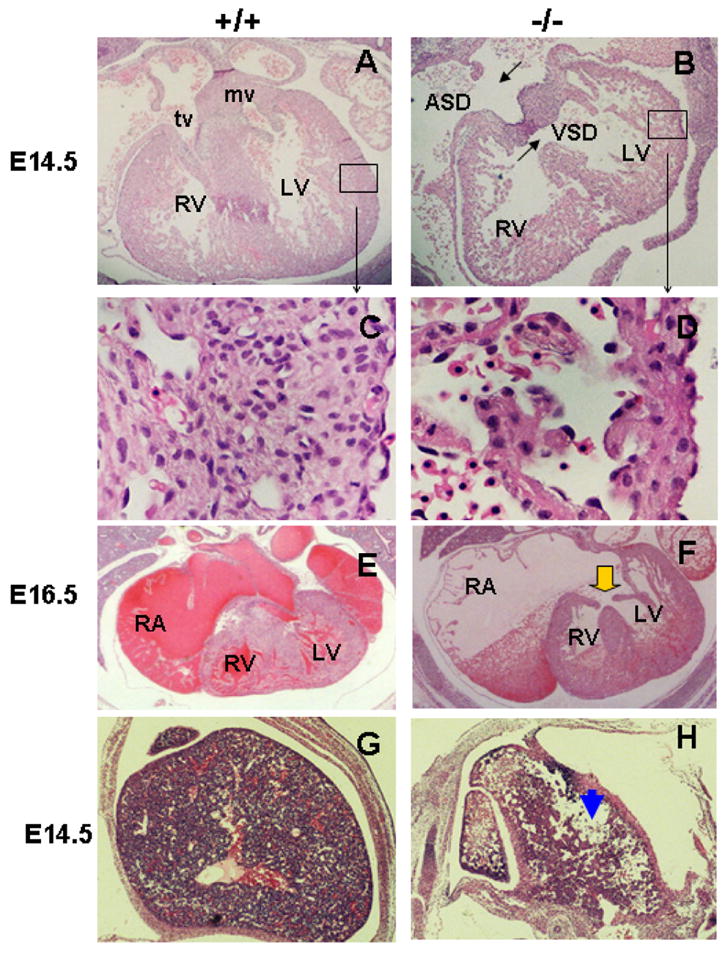
HE staining was performed on heart transverse sections of SRp38 wild-type (A,C,E) and mutant (B,D,F) embryos to examine cardiac morphology at E14.5 (A–D) and E16.5 (E–F). SRp38(−/−) hearts have atrial septal defects (ASD) and ventricular septal defects (VSD) at E14.5 (compare A and B). High-power view of the left ventricle (boxed area) shows disorganized cardiomyocytes and thin myocardium in the mutant as compared to the wild-type littermate (C). Atrioventricular canal defect (AVCD, arrow) and enlarged right atria (RA) are observed in the SRp38(−/−) heart at E16.5 (compare E and F). RV, right ventricle; LV, left ventricle; tv, tricuspid valve; mv, mitral valve. HE-stained transverse sections of the liver showed large amounts of tissue loss in the SRp38(−/−) embryos (H, arrowhead, compared to wild-type control G)
Cardiac triadin expression is deregulated in mice lacking SRp38
To identify genes regulated by SRp38 that might be responsible for the abnormalities in the SRp38(−/−) heart, we first performed microarray analysis using ventricular RNAs extracted from SRp38(−/−) and wild-type embryos at E13.5 and E14.5. Given that SRp38 functions as a general splicing repressor as well as a sequence-specific activator, we reasoned that loss of SRp38 might have a general effect on gene expression. We therefore analyzed an expression array designed to detect changes in total mRNA levels (See Experimental Procedures). While the majority of genes examined were found to be expressed similarly in SRp38(−/−) and wild-type ventricles, a number of differences were observed (Table S1). Among these were genes encoding enzymes involved in energy or hemoglobin metabolic pathways (see table S1, indicated by * after gene names, such as mino levulinate synthesis), which might have been affected by poor functioning of and/or circulation in the mutant hearts. Changes in other affected genes, listed in table S1 and indicated by # after gene names (such as fibulin5 and elastin), may reflect the underdeveloped myocardium (i.e., thin and non-laminated; see Figure 2D) observed in the mutant hearts, rather than direct effects of SRp38 loss. RT-PCR analysis of the downregulated transcripts showed no accumulation of alternative or aberrantly spliced RNAs (Figure S3), and none of these transcripts contain consensus SRp38 binding sites. We conclude that depletion of SRp38 did not invoke widespread defects in gene expression in cardiomyocytes. Similar observations were made previously with heart-specific ablation of SC35 (Ding et al., 2004) and ASF/SF2 (Xu et al., 2005).
The above transcripts seem unlikely to have been directly affected by the loss of SRp38 but rather the expression changes observed likely reflect the poor functioning of the mutant hearts. Especially intriguing however were differences in accumulation of mRNAs encoding distinct isoforms of the cardiac protein triadin. Triadin pre-mRNA has previously been shown to generate distinct mRNA isoforms by alternative splicing (Hong et al., 2001; also see Figure 3A). The major protein isoform, triadin 1, has been shown to play an important role in Ca2+ handling during EC coupling (Kirchhefer et al., 2001; Terentyev et al., 2005).
Figure 3. Altered triadin pre-mRNA splicing patterns and significantly reduced triadin and CSQ2 protein levels are observed in the SRp38(.

−/−) hearts. (A) Triadin gene expression is regulated by alternative RNA splicing. Exons 1–8 are shared by all three isoforms, while exon 9, exon 10 and exon 11 are specific to triadin 1, triadin 2 and triaidn 3 mRNAs, respectively. Note that exons and introns are not shown to scale. Primers used for real-time RT-PCR analysis were also indicated (The forward primer was designed to span the junction of exons 7–8 in order to detect only spliced products). (B) Comparison of the levels of triadin transcript in SRp38(−/−) and SRp38(+/+) hearts at E13.5 and E14.5. Ventricular RNA was extracted from SRp38(−/−) and SRp38(+/+) embryonic hearts and used for real-time RT-PCR analysis. Experiments were repeated at least three times, relative amounts of transcripts after normalization with beta-actin mRNA levels were plotted as mean ± SD for each sample. (C) Total triadin mRNA levels (triadin 1 plus triadin 2) were reduced in SRp38(−/−) hearts. Real-time RT-PCR was performed using primers targeted against constitutively spliced exons of triadin pre-mRNA and analyzed as described in (A). (D) CSQ2 mRNA levels in SRp38(−/−) and SRp38(+/+) hearts. Real-time RT-PCR was performed and analyzed as described in (A). (E) Decreased levels of triadin and CSQ2 proteins in the SRp38(−/−) heart. Ventricle extracts were prepared from E14.5 embryos, resolved by SDS-PAGE and analyzed with indicated antibodies.
To confirm the difference in triadin mRNA accumulation suggested by the microarray analysis, we performed real-time RT-PCR with the ventricular RNAs to quantitate levels of triadin mRNA isoforms. Consistent with the microarray data, a significant decrease in triadin 1 and an increase in triadin 2 mRNA levels was observed in the E13.5 SRp38(−/−) heart (Figure 3B, compare lanes 1 with 2, and 5 with 6), suggesting that SRp38 promotes accumulation of triadin 1 over triadin 2 mRNA. We also detected an increase in a third isoform, triadin 3, although the level was low (data not shown). At E14.5, triadin 1 and triadin 2 mRNA levels in the mutant heart did not change significantly relative to expression levels at E13.5 (Figure 3B, compare lanes 1 and 3, 5 and 7). In contrast, in the wild-type heart, triadin 1 mRNA levels at E14.5 were significantly increased relative to levels at E13.5 (compare lanes 2 and 4). Triadin 1 mRNA levels at E14.5 were 4–5 fold higher in wild-type compared to mutant hearts (compare lanes 3 and 4), while levels of triadin 2 mRNA were comparable (lanes 7 and 8). Aberrantly spliced triadin transcripts were not detected (Figure S4A). In summary, the ratio of triadin 1 to triadin 2 mRNA was significantly reduced in SRp38 mutant hearts at both E13.5 and E14.5 (Figure S4B), as were total triadin mRNA levels (Figure 3C),
To investigate whether the changes in triadin mRNA we observed were specific, we examined by real-time RT PCR the levels of transcripts encoding other sarcoplasmic reticulum membrane-related proteins in the E13.5 and E14.5 mutant and wild-type ventricles. With one exception, all transcripts tested, which included those encoding the ryanodine receptors (RyR2), junctin, SERCA 2a and PLB (Figure S5 and data not shown), were unchanged between wild-type and mutant hearts. However, CSQ2 mRNA levels were slightly decreased in the mutant at E13.5, and were significantly reduced at E14.5 (Figure 3D, lanes 1–4).
We next wished to determine whether the changes in triadin and CSQ2 mRNA levels were reflected at the protein level. To this end, protein extracts were prepared from E14.5 ventricles and used for Western blot analysis (Figure 3E). The triadin antibody used was capable of detecting both triadin 1 and triadin 2 isoforms (see Experimental Procedures). Consistent with the real-time RT PCR data, a striking decrease in total triadin protein levels was observed in the mutant ventricles, and CSQ2 protein levels were also greatly reduced. As expected, SRp38 was not detected in the mutant hearts. Levels of SERCA 2a were unchanged between wild-type and mutant hearts.
SRp38 specifically binds to triadin 1-specific exon 9 RNA in vitro
The above data suggests that SRp38 regulates expression of triadin and/or CSQ2 in developing cardiomyocytes. Because the change in mRNA expression of triadin isoforms was observed earlier than the decreased CSQ2 mRNA levels, and because the changes in triadin mRNAs were consistent with a defect in alternative splicing, we examined in more detail possible regulation of triadin pre-mRNA splicing by SRp38. The triadin gene contains 11 exons; alternative splicing leads to three isoforms that differ only in which of the last three exons is included or excluded in the mature RNA. In triadin 1, exon 9 (E9) is included; in triadin 2, exon 10 (E10) is included; and in triadin 3, exon 11 (E11) is included (Hong et al., 2001).
We first examined the sequences of the three exons for the presence of potential SRp38 binding sites. Strikingly, we found two purine-rich motifs, separated by nine nucleotides, in triadin 1-specific E9; each constitutes an excellent match to the SRp38 consensus sequence (AAAGACAAA) (Figures 4A and 4B; Shin and Manley 2002). Neither E10 nor E11 contained a match to the SRp38 consensus sequence (data not shown). To determine whether SRp38 binds E9 specifically, we prepared 32P-labelled E9 RNA containing the putative SRp38 binding sites (60 nucleotides in length) and compared binding in gel shift assays with E10 RNA (prepared in a similar fashion) (Figure 4C). The SRp38 derivative used contains only the RBD, but displays the same RNA binding specificity as the full-length GST-SRp38 protein (data not shown). The results indicate that SRp38 indeed binds to E9 RNA in a concentration-dependent manner (lanes 1–4), but not detectably to E10 RNA (lanes 5–8). SRp38 bound to E9 RNA with an affinity comparable to its affinity for the GluR-B flop exon (Feng et al., 2008; Figure S6A). In addition, two other SR proteins, ASF/SF2 and SC35, did not display significant affinity for E9 RNA (Figure S6B).
Figure 4. SRp38 specifically binds to triadin 1-specific exon 9 RNA.
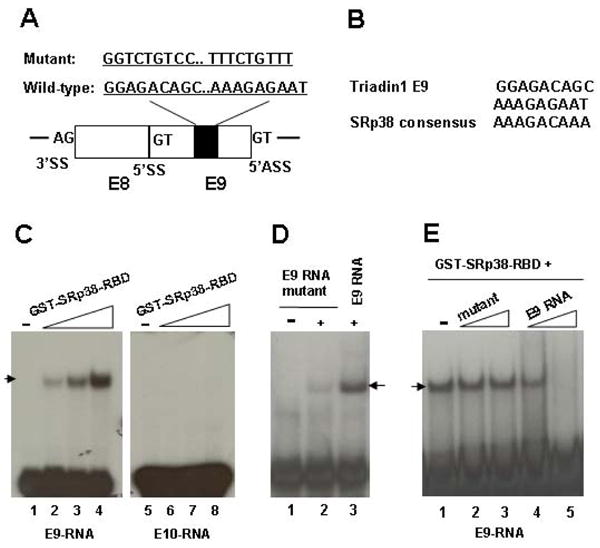
(A) Schematic diagram of triadin constitutive exon 8 (E8) and 5′ alternative exon 9 (E9), and auxiliary sequences. Filled boxes denote two putative SRp38 binding sites identified in E9. SS, splice site; ASS, alternative splice site. (B) Comparison of the two putative SRp38 binding sites within E9 with the selected SRp38 consensus sequence. (C) SRp38 binds to triadin1 specific E9 RNA but not to triadin 2-specific E10 RNA. In vitro transcribed triadin 1-specific E9 or triadin 2-specific E10 RNA were used in a gel shift assay with increasing amounts of GST-tagged SRp38 RBD as indicated. (D) SRp38 only weakly binds to E9 mutant RNA. Two SRp38 binding sites were replaced with the indicated nucleotides in E9 RNA. Gel shift assays were performed as in (C). (E) Competition assay with 32 P-labeled E9 RNA and increasing amounts of unlabeled RNAs as competitors. Gel shift assays were performed as in (C). Each competitor was used at 2 or 6-fold molar excess over the probe. RNA-protein complex was indicated by arrows.
We next wished to confirm that the putative SRp38 binding sites in E9 were responsible for the observed SRp38 binding. To this end, gel shift assays were performed with an E9-RNA in which the two motifs were mutated (Figure 4A; see also Experimental Procedures). SRp38 bound the mutant RNA very weakly, as compared to the binding to wild-type E9 RNA (Figure 4D, compare lanes 2 and 3). We also performed a competition assay with increasing amounts of unlabelled RNAs as competitors. As shown in Figure 4E, the mutant RNA was ineffective as a competitor while the wild-type RNA eliminated binding (compare lanes 2, 3 and 4, 5). Together, this data indicates that SRp38 specifically binds to the E9 RNA and that binding is dependent on SRp38 binding sites in the RNA.
SRp38 activates splicing in vitro of pre-mRNA containing triadin exon 9 in a sequence-dependent manner
We next asked if SRp38 could activate splicing of an E9-containing substrate. To this end, we utilized a strategy we established previously to measure SRp38-dependent splicing activation (Feng et al., 2008). Specifically, we constructed chimeric β-globin RNA substrates in which triadin E9 containing the SRp38 binding motif (wild type or mutant) and E10 were used to replace sequences in the downstream β-globin exon (β-E9, β-E9 mutant and β-E10; see Feng et al., 2008 and Experimental Procedures). Splicing of these RNAs was assayed either in complete HeLa nuclear extract (NE) or in a splicing-competent extract lacking all SR proteins including SRp38 (S100 + NF 40–60; see Feng et al., 2008 and Experimental Procedures) plus purified GST-SRp38 (Figure 5A). Significantly, the β-E9 RNA was efficiently spliced in a SRp38-dependent manner (lanes 1–4). Splicing of the β-E9 mutant and β-E10 substrates, on the other hand, was not activated at all by SRp38 (lanes 5–8 and 9–12, respectively. Note that splicing of the β-E10 RNA was stalled after the first step, even in NE, reflecting the activity of second-step inhibitory element in E10 (Y.F. and J.L.M., unpublished data).) To provide additional evidence that SRp38-dependent splicing was due to SRp38 binding to the E9 RNA, we performed competition assays with increasing amounts of non-labeled RNAs. Splicing of the β-E9 was completely inhibited by a 9-fold molar excess of the wild-type E9 RNA (Figure 5B, lane 3), while the same amount of the mutant RNA showed at most only a slight inhibition (Figure 5B, lanes 4–5).
Figure 5. SRp38 specifically activates splicing of a substrate containing triadin 1-specific exon 9 RNA.

(A) SRp38 activates splicing of a substrate containing triadin 1-specific E9. In vitro splicing was performed in Hela S100 extract using E9, E9 mutant or E10 containing substrates (β-E9, β-E9 mutant or β-E10) with indicated additions. SRp38 splicing activity in S100 requires a nuclear fraction (NF40–60) prepared by ammonium sulfate precipitation. (B) Splicing of β-E9 in vitro in the presence of unlabelled competitors. Competitors were used at 3 or 9-fold molar excess over the pre-mRNA.
SRp38 promotes inclusion of exon 9 in triadin pre-mRNA splicing in DT 40 cells
We next wished to ask whether SRp38 favors inclusion of exon 9 in triadin pre-mRNA splicing in an in vivo setting. To this end, we constructed a triadin mini-gene plasmid in which a truncated triadin cassette containing exons 8–11 was preceded by the chicken β-actin promoter and followed by an SV40 poly(A) site (Figure 6A). This plasmid was stably transfected into chicken DT40 cells with the genetic background SRp38(+/+) or SRp38(−/−). Stably transfected colonies were isolated and total RNAs were extracted and analyzed first by RT-PCR in the presence of 32P UTP. As shown in Figure 6B, while triadin 2 mRNA levels were comparable in the two cell types, there was significantly less of the triadin 1 variant in the SRp38(−/−) cells, suggesting that SRp38 promotes inclusion of triadin 1 specific exon 9. Actin mRNA levels were equivalent in the two cell lines (Figure 6B). To extend the above results, we analyzed RNA isolated from five different colonies derived from each cell line by real-time RT-PCR, and compared the ratio of triadin 1 to triadin 2 mRNA levels. Consistent with the above RT-PCR results, a significant difference was observed between SRp38(+/+) and SRp38(−/−) cells, such that there was an near five-fold increase in the ratio of triadin 1 to triadin 2 mRNA in the presence of SRp38 (Figure 6C). Thus, SRp38 is capable of regulating alternative splicing of triadin pre-mRNA in vivo as well as in vitro. To provide additional evidence that the effects on triadin pre-mRNA splicing were specific, we constructed and analyzed cell lines expressing a human β-globin minigene. Levels of β-globin mRNA were equivalent between SRp38(+/+) and SRp38(−/−) cells (Figure 6D).
Figure 6. SRp38 enhances inclusion of triadin exon 9 in stably transfected DT40 cells.

(A) Diagram of reporter plasmid containing the indicated triadin genomic sequences, procedure for stable transfection of the reporter plasmid into SRp38(+/+) and SRp38(−/−) DT40 cell lines, and the two alternatively spliced triadin products. The forward primer was designed to span the junction of exons 7–8 in order to detect only spliced products. (B) SRp38 depletion decreases the level of triadin 1 mRNA in SRp38(−/−) DT 40 cells. RNA was extracted from SRp38(+/+) and SRp38(−/−) cells and 32P RT-PCR was performed. RNA was resolved on a 6% denaturing PAGE and triadin 1 and triadin 2 products were shown. Actin mRNA levels were used as a loading control. (C) The ratio of triadin 1 to triadin 2 mRNA levels is greatly reduced in SRp38(−/−) DT40 cells. RNA was extracted from SRp38(+/+) and SRp38(−/−) DT40 cell clones stably expressing the triadin reporter construct. Real-time RT-PCR analysis was performed as described in Figure 4A. Comparison was made between five independent SRp38(+/+) and five independent SRp38(−/−) DT40 cell clones. (D) Basal splicing activity in SRp38(+/+) and SRp38(−/−) DT40 cells. A plasmid containing the human β-globin gene was constructed and stably transfected into DT 40 cells following the same strategy employed in (A). RNA was extracted and 32P RT-PCR was performed.
Ca2+ handling is affected in isolated SRp38(−/−) cardiomyocytes
We next wished to investigate whether the reduced levels of triadin and/or CSQ2 might affect Ca2+ homeostasis in SRp38(−/−) embryonic cardiomyocytes. To this end, we developed an approach using primary cultures of cardiomyocytes isolated from wild-type and SRp38(−/−) embryos at E14.5. The myocytes were spherical in appearance immediately after dissociation, and become flattened and spread out after overnight incubation in culture. Wild-type and mutant cardiomyocytes appeared morphologically identical (Figure 7A). Western blots indicated that triadin and CSQ2 levels were as expected reduced in the SRp38(−/−) cultured cardiomyocytes (Figure S7). We measured intracellular cytosolic Ca2+ in these cells using fluo-4 and laser scanning confocal microscopy (see Experimental Procedures).
Figure 7. The incidence of spontaneous Ca2+ sparks is increased in SRp38(−/−) myocytes.

(A) Confocal images of cardiomyocytes isolated from embryos at E14.5. (B) Representative line-scan images of Ca2+ sparks in myocytes incubated in Tyrode’s solution containing 5 mM Ca2+. (C) Spark frequency was compared between SRp38(+/+) and SRp38(−/−) cardiomyocytes. Comparison of distribution of Ca2+ sparks between SRp38(+/+) and SRp38(−/−) cardiomyocytes was made by plotting number of sparks events against (D) spark amplitude (ΔF/F), (E) full duration at half-maximal amplitude (FDHM; ms), or (F) full width at half-maximal amplitude (FWHM; μm), respectively. SparkMaster was used to detect and analyze spark parameters. Data analysis was performed on 75 sparks from wild-type preparations and 198 sparks from multiple SRp38(−/−) cardiomyocyte preparations.
Both the SRp38(−/−) and wild-type myocytes displayed spontaneous contractile activity and alterations in Flou-4 fluorescence. While the pattern and frequency of spontaneous contractions was variable in isolated myocytes, the amplitude of Ca2+ transients (expressed as an elevation above diastolic levels; F/Fo) was not significantly different in the mutant myocytes (data not shown). Ryanodine (1 uM for 3 mins) did not abolish spontaneous Ca2+ transients, but led to a reduction in the amplitude of the Ca2+ transients and there was not a much difference between two groups (data not shown). This data supports the view that fetal cardiomyocyte contraction is mainly regulated by Ca2+ influx from extracellular sources rather than Ca2+ release from the sarcoplasmic reticulum (Artman et al., 2000).
We next examined whether the properties of Ca2+ sparks, the elementary Ca2+ release event from the sarcoplasmic reticulum (Cheng et al., 1993), might be influenced in SRp38(−/−) myocytes. To this end, we incubated wild-type and mutant cardiomyocytes with 5 mM Ca2+ to augment sarcoplasmic reticulum Ca2+ loading (Seki et al., 2003). Strikingly, 198 spark events were detected from 50 mutant myocytes isolated from 5 embryos, while only 75 sparks were observed from 51 wild-type myocytes from 7 embryos (Figure 7B). We then analyzed the frequency and amplitude of the sparks using SparkMaster (see Experimental Procedures). As shown in Figure 7C, spark frequency was significantly increased in the SRp38(−/−) myocytes as compared to the wild-type myocytes. The SRp38(−/−) myocytes also displayed a significantly smaller amplitude (1.72 ± 0.03, n=198, p< 0.01) compared to the wild-type cells (2.87 ± 0.20, n=75) (Figure 7D). Additionally, the width of Ca2+ sparks (FWHM) was altered in the SRp38(−/−) myocytes, decreasing from 3.24 ± 0.21 μm (n=75) to 2.48 ± 0.08 μm (n=198, p< 0.05) (Figure 7E), although no significant differences were detected in the duration of sparks (FDHM) between the two groups of cells (Figure 7F). The data indicates that there is greater occurrence of Ca2+ sparks in the SRp38(−/−) myocytes, reflecting a specific role of SRp38 in regulating Ca2+ release from the sarcoplasmic reticulum in embryonic cardiomyocytes.
Discussion
During mouse development, the heart undergoes dramatic morphological changes and critical modifications in gene expression. Here we have provided evidence that the splicing factor SRp38 is critically involved in cardiac development and that its loss leads to multiple cardiac defects, changes in triadin pre-mRNA splicing, and altered intracellular Ca2+ handling in isolated cardiomyocytes. Below we discuss how SRp38 functions as an important regulator of alternative splicing during heart development, and how loss of SRp38 leads to changes in triadin pre-mRNA splicing and altered Ca2+ handling in embryonic cardiomyocytes.
Given that SR proteins are essential splicing factors, inactivation of specific SR protein is expected to result in multiple general defects in RNA metabolism and therefore cell death or early embryonic lethality. This has been demonstrated by loss of ASF/SF2 in chicken DT40 cells (Wang et al., 1996) and gene-targeting deletion of SRp20 (Jumaa et al., 1999) and SC35 (Wang et al., 2001) in mice. These findings also exemplify the difficulties in analysis of splicing defects caused by ablation of SR proteins in vivo. However, by application of conditional gene-targeting approaches, Fu and colleagues demonstrated connections between the heart-specific inactivation of ASF/SF2, defects in several alternative splicing events, and a severe defect in the function of adult cardiac muscles (Xu et al., 2005). For example, one of the affected transcripts, the Ca2+/calmodulin-dependent kinase IIδ (CaMKIIδ) pre-mRNA, showed a defect in the postnatal splicing switch in ASF/SF2(−/−) hearts; this defect results in production of a kinase that is mistargeted to sarcolemmal membranes, causing severe EC coupling defects (Xu et al., 2005). However, it remains to be determined whether the misregulated CaMKIIδ pre-mRNA is a direct target of ASF/SF2, and how ASF/SF2 regulates its splicing. Especially given the recently identified roles for ASF/SF2 in mRNA export, genomic instability and translation (see Introduction), it is possible that ASF/SF2 affects expression of a factor(s) that in turn modulate(s) the splicing transition of the CaMKIIδ pre-mRNA during postnatal heart remodeling.
In the present study, we provided considerable evidence that SRp38 directly regulates alternative splicing of cardiac triadin pre-mRNA. Our initial experiments dealing with SRp38 indicated that the protein, when dephosphorylated, functions as a general splicing repressor, without apparent sequence specificity (Shin and Manley, 2002; Shin et al., 2004). Although these experiments provided no evidence that SRp38 could, like other SR proteins, function as a splicing activator, they did identify, by SELEX, a specific high-affinity binding site for SRp38, and we speculated that under some conditions SRp38 might indeed be capable functioning as a splicing activator. Indeed, Feng et al. (2008) showed recently that phosphorylated SRp38 is in fact a sequence-specific activator, both in in vitro and in vivo assays, and can regulate alternative splicing of the GluR-B pre-mRNA (Komatsu et al., 1999) in a sequence-specific manner. The RNA binding and splicing data with triadin pre-mRNA presented here is essentially identical, and indicates that triadin is indeed a direct SRp38 target. However, triadin exon 9 alternative splicing may be coupled with altered polyadenylation (see Figure 3A), which raises the possibility that SRp38 may play a more complex role in triadin pre-mRNA processing.
In our experiments, we observed reduced triadin 1 mRNA levels, but increased levels of triadin 2 mRNA in the absence of SRp38. However, the switch in triadin splicing led to a reduction in total levels of triadin protein, rather than accumulation of triadin 2. Given that no function has been attributed to triadin 2, it is possible that it is inactive and degraded, at least at this developmental stage. In addition to these changes, we also observed down-regulation of CSQ2 mRNA and protein expression, but not of other sarcoplasmic reticulum-related proteins. Related observations have been made when triadin 1 and CSQ2 levels were altered in transgenic mice: overexpression of triadin1 was reported to down-regulate both RyR2 and junctin (Kirchhefer et al., 2001), while inactivation of CSQ2 essentially eliminated expression of triadin and junctin (Knollmann et al., 2006). Together with our data, these findings suggest that triadin and CSQ2 associate not only with each other, but also with other proteins involved in Ca2+ handling, including RyR2 and junctin. Changes in expression of any of these proteins may significantly alter expression of the others, indicating that expression of these Ca2+ homeostatic proteins is tightly coordinated during cardiac development.
Consistent with the observed reduction in triadin and CSQ2 protein levels, our data demonstrated that intracellular Ca2+ handling was altered in isolated SRp38(−/−) cardiomyocytes. Specifically, Ca2+ sparks occurred with greater frequency but lower amplitude in SRp38(−/−) cardiomyocytes than in wild-type cells. It is likely that this phenotype is caused by reductions in triadin or CSQ2 alone or in combination with each other. Several reports have established the importance of triadin 1 in controlling Ca2+ handling in adult cardiomyocytes, although its precise role appears complex. For example, adenoviral-mediated overexpression of triadin 1 in cultured rat cardiomyocytes led to an increased frequency of spontaneous Ca2+ sparks, accompanied by decreased amplitudes (Terentyev et al., 2005), while transgenic overexpression of triadin 1 in the mouse was reported to have almost the opposite effect, increased Ca2+ spark amplitude but without changes in spark frequency (Kirchhefer et al., 2001). Results obtained with ablation of CSQ2 (Knollmann et al., 2006) also indicate that normal Ca2+ release relies on an intricate balance among the Ca2+ homeostatic proteins, including RyR2, triadin, CSQ2 and junctin. Due to differences in the sarcoplasmic reticulum in embryonic vs. adult cardiomyocytes (Itzhaki et al., 2006), it is possible that the Ca2+ handling defects we observed may involve additional mechanisms than reduced levels of triadin and/or CSQ2. Re-expressing one or both of these proteins will be an intriguing topic for future studies that focus on mechanisms of Ca2+ handling in embryonic cardiomyocytes by RyR2 and regulatory proteins such as triadin.
A number of cardiac-related defects were observed in the SRp38(−/−) embryos. Several of these have been observed in many unrelated knockout mice, and may reflect general or secondary effects (Conway et al., 2003). For example, hepatic necrosis has been observed in many mice with severe cardiac defects (Shou et al., 1998; Araki et al., 2004), as heart failure can cause secondary congestive liver disease. Similar liver damage was present in SRp38(−/−) embryos, which, in combination with the observed cardiac defects, almost certainly contributes to the observed embryonic lethality and growth retardation. The cardiac defects observed in the SRp38(−/−) mice are unlikely related to the changes in expression of triadin and/or CSQ2, as loss of triadin (Shen et al., 2007) or CSQ2 (Knollmann et al., 2006) has little effect on cardiac development. Therefore the various cardiac abnormalities on the one hand and the triadin splicing defects and concomitant altered Ca2+ handling on the other likely reflect independent effects of SRp38 inactivation. This suggests that SRp38 plays a broad role in regulating alternative splicing during cardiac development. While future studies will be required to elucidate the molecular bases for these effects, it is remarkable that the requirement for SRp38 during embryonic development seems to be limited to the heart. Given the broad tissue distribution of SRp38, at least in adults (Shin and Manley 2008), it is likely that SRp38 plays additional important roles later in development.
In summary, the loss of splicing factor SRp38 leads to multiple cardiac defects in mice. SRp38 specifically regulates alternative splicing of triadin pre-mRNA, as loss of SRp38 caused a switch in the triadin isoform pattern. SRp38 inactivation also diminished CSQ2 expression and altered intracellular Ca2+ cycling in embryonic cardiomyocytes. By understanding how SRp38 modulates cardiac triadin expression, we have moved closer towards elucidating the role of alternative splicing as a genetic modifier during heart development.
Experimental Procedures
Construction of the targeting vector and generation of SRp38 knockout mice
A 5.5-kb fragment upstream of the SRp38 exon 1 and a 3.5-kb fragment downstream of exon 2 were amplified by PCR and sequentially subcloned into the pPNT targeting vector (Qu et al., 2003). Linearized vector was electroporated into E14 embryonic stem cells. Southern blotting identified 20 out of 200 G418 and gancyclovir double-resistant clones that had undergone homologous recombination. Two ES clones were injected into C57BL/6 blastocysts. Six chimeric male mice with >90% ES cell contribution to coat color were bred with C57BL/6 females to produce F1 heterozygotes, which were interbred to produce F2 progeny. A genomic PCR assay to detect the wild-type allele (330 bp) or the mutant SRp38 allele (240 bp) was designed using a common intron primer (5′ GCTGGAATGGTGT CAGCACAGCG 3′) and the SRp38 wild-type exon2 primer (5′ TCCAAGAATCATATCCAGTGGCT 3′) or the neoR gene primer (5′ CTACCCGGTAGAATTGACCTGCA 3′).
Histological analysis
Embryos generated by timed matings were isolated, fixed in 10% buffered formalin and then embedded in paraffin and sectioned. The sections were stained with hematoxylin and eosin (HE) and analyzed by microscopy. Freshly dissected whole embryos were also observed and photographed by using a dissecting microscope.
Microarray analysis and real-time RT-PCR and 32P RT-PCR
Ventricle total RNA extracted at E13.5 and E14.5 was used for microarray analysis. Briefly, double-stranded cDNA was synthesized from 10 μg of total Trizol-extracted RNA by using the Microarray cDNA Synthesis Kit (Roche). In vitro transcription by using double-stranded cDNA as a template in the presence of biotinylated UTP and CTP was carried out using an ENZO BioArray HighYield RNA Transcript Labeling Kit (ENZO Biochem). Biotin-labeled cRNA was purified, fragmented and used for hybridization on a Mouse Genome 430A 2.0 Array (Affymetrix) in the Columbia University Cancer Center microarray core facility. Data were analyzed with the Affymetrix GeneChip Expression Analysis Software.
Real-time RT-PCR reactions were carried out with total RNA extracted from the ventricle at the indicated stages. RT reactions were performed using the Transcriptor Reverse Transcriptase (Roche), and real-time PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems) and performed on Applied Biosystems 7300 Real Time PCR machine per manufacturer instructions. PCR results were quantified using ABI software (SDS); delta Rn values were exported to and analyzed using Microsoft Excel. Gene-specific primer sets (5′-3′) include: For triadin 1, triadin-F (GATGATGGCAAAAGAGGACAA) and triadin-R1 (GGAGAAAGTGAAACCAACAGC); For triadin 2, triadin-F (GATGATGGCAAAAGAGGACAA) and triadin-R2 (TTAAATCCCCATGGACAAACA); For triadin 3, triadin-F (GATGATGGCAAAAGAGGACAA) and Triadin-R3 (TGTGGGAGAAAACACCACAA); For total triadin mRNA levels, triadin-F (GATGATGGCAAAAGAGGACAA) and triadin-R (TGCACAGCTGGCATCTCTT); For calsequestrin2 (CSQ2), CSQ2-F (TCAAAGACCCACCCTACGTC) and CSQ2-R (GGGTCAATCCACAAGATGCT); For Junctin, Junctin-F (GCAGGAGGATAGAGGGAAGG) and Junctin-R (TTCCTCGCCTGTCTTTGTCT); For cardiac ryanodine receptor (RyR2), RyR2-F (AGCCCTCACGACTAAAGCAA) and RyR2-R (CCACCCAGACATTAGCTGGT); For SERCA-2a, SERCA-2a-F (AAGCTATGGGAGTGGTGGTG) and SERCA-2a-R (GCAATGCAAATGAGGGAGAT); For phospholamban (Pln), Pln-F (TACCTCACTCGCTCGGCTAT) and Pln-R (GATGCAGATCAGCAGCAGAC); For beta-actin, actin-F: ATGAGCTGCCTGACGGCCAGGTCATC and actin-R: TGGTACCACCAGACAGCACTGTGTTG. PCR utilizing [α 32p] UTP, was performed according a protocol previously described (Kashima et al., 2007).
Western blotting
For Western blotting analysis, frozen mouse ventricles were homogenized and lysed as described (Li and Minden, 2003). Samples were collected, resolved on a SDS-PAGE and analyzed using indicated antibodies. Primary antibodies employed include mouse anti-tubulin (Hybridoma bank), anti- CSQ2 (Affinity Bioreagents), mouse anti-triadin and anti-SERCA2a (Santa Cruz), and lab-made rabbit anti-SRp38 (Shin and Manley, 2002).
Plasmid constructions, gel-shift assay, in vitro splicing, cell culture and transfection
Plasmids (β-E9 and β-E10) used to produce substrates for in vitro splicing were constructed by replacement of sequences between Acc I site and BamH I site in the second exon with the indicated sequences. The β-E9 mutant was produced by subcloning two mutant oligos containing nucleotides (GGTCTGTCC..TTTCTGTTT) in place of two SRp38 putative binding sites (GGAGACAGC..AAAGAGAAT). The forward primer was GGTCTGTCCAGGAGGTCCT TTCTGTTTAATGGGCAAGAAGCAGATGCAGTCAACTGAAAA, and the reverse primer was TTTTCAGTTGACTGCATCTGCTTCTTGCCCATTAAACAGAAAGGACCTCCTGGA CAGACC. For in vitro gel-shift assays, we constructed plasmids by inserting the indicated sequences into pBluescript SK(+) plasmid. Gel-shift assays and in vitro splicing were performed as previously described (Feng et al., 2008). The triadin truncated reporter minigene was constructed as described previously (Feng et al., 2008). DT40 cells of different backgrounds including wild-type or SRp38(−/−) were maintained essentially as described previously(Shin et al., 2004). Transfection of the triadin truncated reporter minigene into DT40 cells was also performed as described previously (Feng et al., 2008)
Isolation of embryonic cardiomyocytes and primary cell culture
Embryos were removed from pregnant female mice at 14.5 days. Cardiac myocytes were isolated from embryonic hearts as previously described (An et al., 1996; Liu et al., 1999) with slight modifications. In brief, the dissected ventricles were incubated in Ca2+- free isolation buffer (in mM: 116 NaCl, 20 HEPES, 1.0 NaH2PO4, 5.5 Glucose, 5 KCl, and 0.8 MgSO4, pH 7.35) containing 0.4 mg/mL collagenase type II (Worthington) for 40 min at 37° C and then rinsed with the same buffer without enzyme. The ventricles were dissociated by pipetting up and down several times, and the isolated cells were placed on collagen-coated glass coverslips and incubated in DMEM (GIBCO) plus 10% FBS and 10 μg/mL gentamicin at 37° C for 18 to 24 hours before recording.
Confocal imaging of Ca2+ transients and Ca2+ sparks in embryonic ventricular myocytes
Ventricular myocytes were incubated with fluo-4 AM (5 μM; Molecular Probes) for 10 min. Cells were subsequently washed in Tyrode’s solution containing (in mM): 137 NaCl, 4 KCl, 1 MgCl2, 1.2 NaH2 PO4, 1.8 CaCl2, 10 glucose, 10 HEPES (pH 7.4) for 20 min to allow de-esterification. Confocal fluorescence imaging was performed with a confocal scanning laser microcopy (Zeiss 510). The line scan mode was used for quantitative analysis of Ca2+ transients and Ca2+ sparks. Confocal imaging of Ca2+ transients was performed with the myocytes in Tyrode’s solution containing 1.8 mM Ca2+, while Ca2+ sparks were recorded using 5 mM extracellular Ca2+ (Seki et al., 2003). Ca2+ sparks were detected and analyzed using SparkMaster (Picht et al., 2007). Analysis included spark frequency (sparks × s−1 × (100 μm)−1), amplitude (ΔF/F), full duration at half-maximal amplitude (FDHM; ms), and full width at half-maximal amplitude (FWHM; μm).
Statistics
Quantitative data were expressed as means ± SE. The paired or unpaired Student’s t-test was applied, and significance was defined at p < 0.05.
Acknowledgments
We are grateful to A. Beg, L. Yamasaki and A. Minden for advice and assistance with the generation of SRp38 (−/−) mice. We thank C. Yang at the Rockefeller ES cell facility for help with the ES cell culture and injection. We thank Microarray core facility at the Columbia University Cancer Center for help with microarray analysis. We also thank I. Boluk for help preparing the manuscript and members of the Manley laboratory for helpful discussions and comments. This work was supported by NIH grant NIH GM48259 to JLM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An RH, Davies MP, Doevendans PA, Kubalak SW, Bangalore R, Chien KR, Kass RS. Developmental changes in beta-adrenergic modulation of L-type Ca2+ channels in embryonic mouse heart. Circ Res. 1996;78:371–378. doi: 10.1161/01.res.78.3.371. [DOI] [PubMed] [Google Scholar]
- Artman M, Henry G, Coetzee WA. Cellular basis for age-related differences in cardiac excitation-contraction coupling. Prog Pediatr Cardiol. 2000;11:185–194. doi: 10.1016/s1058-9813(00)00049-7. [DOI] [PubMed] [Google Scholar]
- Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. doi: 10.1016/j.cell.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Kruzynska-Frejtag A, Kneer PL, Machnicki M, Koushik SV. What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis. 2003;35:1–21. doi: 10.1002/gene.10152. [DOI] [PubMed] [Google Scholar]
- Ding JH, Xu X, Yang D, Chu PH, Dalton ND, Ye Z, Yeakley JM, Cheng H, Xiao RP, Ross J, et al. Dilated cardiomyopathy caused by tissue-specific ablation of SC35 in the heart. Embo J. 2004;23:885–896. doi: 10.1038/sj.emboj.7600054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Chen M, Manley JL. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat Struct Mol Biol. 2008;15:1040–1048. doi: 10.1038/nsmb.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD. The superfamily of arginine/serine-rich splicing factors. Rna. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. Sorting out the complexity of SR protein functions. Rna. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes A, Guo JK, Lutsch G, Leheste JR, Landrock D, Ziegler U, Gubler MC, Schedl A. Two splice variants of the Wilms’ tumor 1 gene have distinct functions during sex determination and nephron formation. Cell. 2001;106:319–329. doi: 10.1016/s0092-8674(01)00453-6. [DOI] [PubMed] [Google Scholar]
- Hong CS, Ji JH, Kim JP, Jung DH, Kim DH. Molecular cloning and characterization of mouse cardiac triadin isoforms. Gene. 2001;278:193–199. doi: 10.1016/s0378-1119(01)00718-1. [DOI] [PubMed] [Google Scholar]
- Huang Y, Steitz JA. Splicing factors SRp20 and 9G8 promote the nucleocytoplasmic export of mRNA. Mol Cell. 2001;7:899–905. doi: 10.1016/s1097-2765(01)00233-7. [DOI] [PubMed] [Google Scholar]
- Itzhaki I, Schiller J, Beyar R, Satin J, Gepstein L. Calcium handling in embryonic stem cell-derived cardiac myocytes: of mice and men. Ann N Y Acad Sci. 2006;1080:207–215. doi: 10.1196/annals.1380.017. [DOI] [PubMed] [Google Scholar]
- Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol. 1999;9:899–902. doi: 10.1016/s0960-9822(99)80394-7. [DOI] [PubMed] [Google Scholar]
- Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2007;104:3426–3431. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhefer U, Neumann J, Baba HA, Begrow F, Kobayashi YM, Reinke U, Schmitz W, Jones LR. Cardiac hypertrophy and impaired relaxation in transgenic mice overexpressing triadin 1. J Biol Chem. 2001;276:4142–4149. doi: 10.1074/jbc.M006443200. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116:2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kominami E, Arahata K, Tsukahara T. Cloning and characterization of two neural-salient serine/arginine-rich (NSSR) proteins involved in the regulation of alternative splicing in neurones. Genes Cells. 1999;4:593–606. doi: 10.1046/j.1365-2443.1999.00286.x. [DOI] [PubMed] [Google Scholar]
- Konarska MM, Query CC. Insights into the mechanisms of splicing: more lessons from the ribosome. Genes Dev. 2005;19:2255–2260. doi: 10.1101/gad.1363105. [DOI] [PubMed] [Google Scholar]
- Lemaire R, Prasad J, Kashima T, Gustafson J, Manley JL, Lafyatis R. Stability of a PKCI-1-related mRNA is controlled by the splicing factor ASF/SF2: a novel function for SR proteins. Genes Dev. 2002;16:594–607. doi: 10.1101/gad.939502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005;122:365–378. doi: 10.1016/j.cell.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Li X, Minden A. Targeted disruption of the gene for the PAK5 kinase in mice. Mol Cell Biol. 2003;23:7134–7142. doi: 10.1128/MCB.23.20.7134-7142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Manley JL. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev. 2005;19:2705–2714. doi: 10.1101/gad.1359305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yasui K, Arai A, Kamiya K, Cheng J, Kodama I, Toyama J. beta-adrenergic modulation of L-type Ca2+-channel currents in early-stage embryonic mouse heart. Am J Physiol. 1999;276:H608–613. doi: 10.1152/ajpheart.1999.276.2.H608. [DOI] [PubMed] [Google Scholar]
- Manley JL, Tacke R. SR proteins and splicing control. Genes Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–1081. doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- Qu J, Li X, Novitch BG, Zheng Y, Kohn M, Xie JM, Kozinn S, Bronson R, Beg AA, Minden A. PAK4 kinase is essential for embryonic viability and for proper neuronal development. Mol Cell Biol. 2003;23:7122–7133. doi: 10.1128/MCB.23.20.7122-7133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki S, Nagashima M, Yamada Y, Tsutsuura M, Kobayashi T, Namiki A, Tohse N. Fetal and postnatal development of Ca2+ transients and Ca2+ sparks in rat cardiomyocytes. Cardiovasc Res. 2003;58:535–548. doi: 10.1016/s0008-6363(03)00255-4. [DOI] [PubMed] [Google Scholar]
- Shen X, Franzini-Armstrong C, Lopez JR, Jones LR, Kobayashi YM, Wang Y, Kerrick WG, Caswell AH, Potter JD, Miller T, et al. Triadins modulate intracellular Ca(2+) homeostasis but are not essential for excitation-contraction coupling in skeletal muscle. J Biol Chem. 2007;282:37864–37874. doi: 10.1074/jbc.M705702200. [DOI] [PubMed] [Google Scholar]
- Shin C, Feng Y, Manley JL. Dephosphorylated SRp38 acts as a splicing repressor in response to heat shock. Nature. 2004;427:553–558. doi: 10.1038/nature02288. [DOI] [PubMed] [Google Scholar]
- Shin C, Manley JL. The SR protein SRp38 represses splicing in M phase cells. Cell. 2002;111:407–417. doi: 10.1016/s0092-8674(02)01038-3. [DOI] [PubMed] [Google Scholar]
- Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC, Gyorke S. Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res. 2005;96:651–658. doi: 10.1161/01.RES.0000160609.98948.25. [DOI] [PubMed] [Google Scholar]
- Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- Wang HY, Xu X, Ding JH, Bermingham JR, Jr, Fu XD. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell. 2001;7:331–342. doi: 10.1016/s1097-2765(01)00181-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Takagaki Y, Manley JL. Targeted disruption of an essential vertebrate gene: ASF/SF2 is required for cell viability. Genes Dev. 1996;10:2588–2599. doi: 10.1101/gad.10.20.2588. [DOI] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR, Jr, Ye Z, Liu F, et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell. 2004;16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]