

Abstract
Objectives
The voltage-dependent L-type calcium channel α-subunit 1c (Cav1.2, CACNA1C) undergoes extensive mRNA splicing, leading to numerous isoforms with different functions. L-type calcium channel blockers are used in the treatment of hypertension and arrhythmias, but response varies between individuals. We have studied the interindividual variability in mRNA expression and splicing of CACNA1C, in 65 heart tissue samples, taken from heart transplant recipients.
Methods
Splice variants were measured quantitatively by polymerase chain reaction in 12 splicing loci of CACNA1C mRNA. To search for functional cis-acting polymorphisms, we determined allelic expression ratios for total CACNA1C mRNA and several splice variants using marker single nucleotide polymorphisms in exon 4 and exon 30.
Results
Total CACNA1C mRNA levels varied ∼50-fold. Substantial splicing occurred in six loci generating two or more splice variants, some with known functional differences. Splice patterns varied broadly between individuals. Two heart tissues expressed predominantly the dihydropyridine-sensitive smooth muscle isoform of CACNA1C (containing exon 8), rather than the cardiac isoform (containing exon 8a). Lack of significant allelic expression imbalance, observed with total mRNA and several splice variants, argued against CACNA1C polymorphisms as a cause of variability. Taken together, highly variable splicing can cause profound phenotypic variations of CACNA1C function, potentially associated with disease susceptibility and response to L-type calcium channel blockers.
Keywords: cis-acting polymorphism, L-type calcium channel α-subunit 1c, mRNA splicing
Introduction
Voltage-dependent calcium channels control the influx of calcium into the cells and play important roles in the regulation of skeletal and smooth muscle contraction, gene expression, neurotransmitter release, hormone secretion and activation of Ca2+-dependent ion channels [1-3]. Functional voltage-dependent calcium channels are complex proteins consisting of pore-forming subunit α1 and auxiliary subunits β/γ/α2/δ. The α1-subunit consists of four homologous repeats (I, II, III and IV), each composed of six transmembrane segments (S1—S6) connected by intracellular or extracellular linkers [4]. Among 10 voltage-dependent calcium channel α1-subunits cloned thus far, four belong to L-type (long-lasting) Ca2+ channels, denoted Cav1.1, Cav1.2, Cav1.3 and Cav1.4 [5,6]. Cav1.1 specifically expresses in the skeletal muscle, whereas Cav1.2 expresses in the cardiac and smooth muscle, brain and other tissues [6], regulating cardiac function and blood pressure [7,8]. Moreover, L-type voltage-dependent calcium channels are targets of Ca2+ channel blocking drugs widely used in the treatment of hypertension and cardiac arrhythmia [9]. Therapeutic responses to L-type calcium channel blockers are, however, highly variable while the underlying causes remain uncertain. The goal of this study is to determine whether variable expression and splicing of Cav1.2 play a role in drug response.
The Cav1.2 (α1c, CACNA1C) L-type Ca2+ channel is encoded by a complex gene; both genetic variations and alternative splicing of the primary transcripts are thought to contribute to interindividual variability. Human CACNA1C, spanning > 500 kb, maps to chromosome 12p11.2. While more than 200 single nucleotide polymorphisms (SNPs) have been identified in CACNA1C, the functional consequences are largely unknown, with the exception that rare mutations in exon 8a and exon 8 cause severe arrhythmia disorder [10]. Using transcript scanning, Tang et al. [11] have identified 19 of 55 exons undergoing alternative splicing, distributed over 12 splicing loci. Alternative splicing affects regions encoding the N-terminal, transmembrane segments, linkers between repeats I, II and III, linkers between S5 and S6 in repeats I and II, linker between S3 and S4 in repeats IV, as well as the C-terminal of CACNA1C. These variations, studied individually, affect CACNA1C function by altering electrophysiology properties [11-13], Ca2+ dependency [14], channel regulation [15], affinity to dihydropyridines (DHPs) [16-18] and loss of channel functions [11]. Taking into consideration all combinations of splice variants from the 12 loci, the number of possible splice variants is staggering and functional consequences are difficult to evaluate.
Individual genetic variability can arise from direct changes in protein structure, for example those caused by nonsynonymous SNPs, or from altered gene regulation and mRNA processing and translation. An important process in mRNA processing, mRNA splicing is observed in over 70% of genes, and can generate different functional isoforms that can vary during developmental stages or in different tissues, as shown for CACNA1C [11,19]. Moreover, defects in splicing are the basis for a number of diseases [20,21]. The central role of CACNA1C in maintaining blood pressure [8] and cardiac function [7], and as a target of Ca2+ channel blockers, raises the question whether genetic variability and variable splicing in CACNA1C contribute to the cardiac disease susceptibility or to the treatment response to Ca2+ channel blockers, such as verapamil and DHPs. The response to verapamil shows large interindividual variability that appears to be unrelated to drug serum levels [22]. Although several splice variants have been identified in CACNA1C with known functional consequences, the interindividual variability in splice variants has not been established. Moreover, it remains to be tested whether cis-acting polymorphisms that affect expression and splicing of CACNA1C exist.
In this study, we have developed a sensitive and quantitative method to measure all known splice variants of CACNA1C in human heart tissues (left ventricle), classified as either ischemic or nonischemic, from 65 different heart transplant recipients. This permits us to assess interindividual variability in these patients and compare it to a pooled sample obtained from five normal heart left ventricles. Furthermore, to investigate whether gene expression and variations in splicing are regulated by cis-acting or trans-acting elements, we determined allele-specific expression of total mRNA and of several splice variants derived from locus 10, using marker SNPs in exon 4 and exon 30, and SNaPshot analysis [a polymerase chain reaction (PCR)-based primer extension assay]. This approach permits the unambiguous detection of functional cis-acting polymorphisms affecting gene regulation and mRNA processing, including splicing [23-25]. The results failed to reveal any such functional cis-acting polymorphisms, but demonstrated large interindividual variability in CACNA1C splicing, likely caused by trans-acting factors. Several of these splice variants have previously been associated with altered channel functions.
Materials and methods
Tissue samples
Sixty-five left ventricle heart tissues were obtained from patients undergoing heart transplantation at the Ohio State University Medical Center. The tissues from different regions (aorta, right and left ventricles, right and left atria) of the heart were dissected systematically, followed by a histology check-up to confirm the tissue type and ischemic or nonischemic conditions. Patient information is shown in Table 1. Genomic DNA and total RNA were prepared from these tissues as described previously [23,26]. Pooled total RNA from left ventricles of normal human adult hearts, from five donors who had died of nonheart diseases, was purchased from BioChain Institute, Inc. (Hayward, California, USA).
Table 1.
Characteristics of 65 subjects included in this study
| Ischemic | Nonischemic | Total | |
|---|---|---|---|
| Gender | |||
| Male | 32 | 14 | 46 |
| Female | 7 | 12 | 19 |
| Race | |||
| Caucasian | 36 | 20 | 56 |
| African American | 3 | 6 | 9 |
Quantitative analysis of mRNA expression in heart tissue
Complementary DNA (cDNA) was generated from 0.5 μg total RNA using oligo-dT, and in addition, using several CACNA1C gene-specific primers that target different exons to enhance cDNA yield from the large mRNA transcript and bypass partial degradation that may have occurred post mortem (Supplemental data Table 1). Total CACNA1C mRNA levels were measured by real-time PCR analysis as described in [23], using a set of primers that span exon 3 and exon 4 (E3F: TGACTATTTTTGCCAATTGTGTGG and E4R: GCGGAGGTAGGCATTGGG). To confirm the results, another set of primers spanning exon 5 and exon 6 was also designed (E5F: TGTCCGGAGTCCCAAGTCTC and E6R: CATGGCCTTGATGATGGAA). The sequence targeted by the primers does not contain any known SNPs and is not alternatively spliced. Standard curves were constructed using varying amounts of the respective amplicons. The precision of the analyses between different experiments was calculated as [100 — (standard deviation/mean)100)]%, and accuracy was calculated as [(Calculated log-amount)/(nominal log-amount added)]100%, as described in [27].
Quantitative analysis of splice variants
We used two methods for splice variant analysis. First, for splice variants with different sizes, we used PCR amplification of cDNA using fluorescently labeled primers. For each splice locus, a pair of PCR primers flanking the splice site was designed using the Primer Express Program (Applied Biosystems, Foster City, California, USA), with one primer labeled with fluorescent dye HEX or FAM (Sigma, St Louis, Missouri, USA) (Supplemental data Table 1). After initial denaturing at 95°C for 5 min, the PCR reactions were run for 30 cycles under the following conditions: 95°C for 30 s, 60°C for 1 min and 72°C for 1 min. Then the PCR amplification products were separated on an ABI 3730 DNA analyzer (Applied Biosystems Foster City, California, USA) and the data analyzed using Gene Mapper 3.0 software. Splice variants with different molecular weight yielded peaks with different retention times. The peak area for each splice variant is proportional to the amount of cDNA amplified, tested with the respective amplicons purified by gel electrophoresis. The minimum size difference clearly separable was 2 base pairs (bp) for PCR products ranging from 100 to 500 bp. Splice variants observed in each splice locus were confirmed by at least two sets of primers that give rise to differently sized PCR products. The optimal primer sets (Supplemental data Table 1) were selected for quantitative analysis. A second method using splice variant-specific primers (Supplemental data Table 2) as described in [28] was required for splice variants of identical size, or those not flanked by a common sequence. The amount of each splice variant was expressed as the percentage of total transcripts from each splice locus.
Quantitative analysis of allelic ratios in genomic DNA and mRNA using SnaPshot
The goal of these measurements is to detect differences in allelic DNA and mRNA ratios in samples heterozygous for a marker SNP in the transcribed region, as a measure of allelic expression imbalance (AEI) [23,24,26]. Briefly, a segment of DNA or cDNA (∼100 bp) surrounding the marker SNPs in exon 30 (rs216008) or exon 4 (rs1544514) was amplified by PCR using primers described in Supplemental data Table 2 (E30F/E30R and E4F/E4R). To test the allelic mRNA expression of splice variants near exon 30, the different splice variants were amplified using splice variant-specific primers (Supplemental data Table 2). The PCR products were then subjected to a primer extension assay (SNaPshot, Applied Biosystems) using extension primers (Supplemental data Table 2, E30-PEF or E4-PEF) designed to anneal to the amplified DNA adjacent to the SNP site. Allelic DNA ratios, normalized to 1, serve as internal control. Allelic mRNA ratios (after normalized by DNA ratios) deviating from 1, that is, AEI, indicate the presence of cis-acting factors/polymorphisms in CACNA1C that affect mRNA expression or splicing [23,24].
Statistics
Data are expressed as mean ± SD. Statistical analysis was performed using SPSS 13 program (Ohio State University, Columbus, Ohio, USA).
Results
CACNA1C expression in heart tissue
CACNA1C displayed robust but variable expression in heart tissues. Using β-actin as constitutive control for reverse transcription-polymerase chain reaction (RT-PCR), CACNA1C/β-actin ratios ranged from 2 to 100 [mean 45 ± 22 (SD)] (Fig. 1a). Using another set of primers spanning exon 5 and exon 6, we obtained similar results [CACNA1C/β-actin ratios ranged from 4 to 100, mean 34 ± 18 (SD)]. As RT-PCR cycle thresholds for β-actin varied in a relatively small range (PCR cycle thresholds from 20 to 24, equivalent to an 8-fold difference), this result reveals a high degree of interindividual variation of CACNA1C mRNA levels. The method was highly reproducible between experiments, with precision calculated to be 100.0 ± 0.3%, and accuracy 101 ± 1.3%. No difference exists between ischemic and nonischemic hearts (Fig. 1b, P > 0.05, t-test). No significant differences between sex and race (Caucasian and African American) were detectable [P > 0.05, one-way analysis of variance (ANOVA)].
Fig. 1.

The expression levels of CACNA1C mRNA in 65 heart tissues. (a) The distribution histogram of CACNA1C mRNA levels. After normalizing for β-actin expression, the level of CACNA1C mRNA in each sample was expressed as percentage of highest expression level among 65 samples. Each sample was measured three times. X-axis represents the relative amount of CACNA1C mRNA, while y-axis the number of samples. The average expression level among 65 samples is 45 ± 22 (SD). (b) CACNA1C mRNA levels in ischemic and nonischemic hearts. The average levels for ischemic and nonischemic groups are 42 ± 21 (n = 39) and 50 ± 23 (n = 26), respectively. Mean ± SD. Compared ischemic versus nonischemic groups, P > 0.05, t-test.
Interindividual variation of CACNA1C splice variants
Tang et al. [11] had identified numerous splice variants of CACNA1C transcripts. To test whether the expression of these splice variants differ among individuals, we measured each known splice variant quantitatively in 65 human heart tissues. For most of the splice variants, we used PCR amplification of cDNA, employing fluorescently labeled primers for rapid detection by capillary electrophoresis. Figure 2 depicts separation and detection of 10 splice variants amplified from locus 10 (E30-E34) as an example. Of 12 splice variants known for this locus, 10 can be identified using this method, whereas two variants have identical size and electrophoretic retention. The size of each PCR product matches the expected size, which was confirmed by at least one other set of primers (data not shown). For splice variants that have the same molecular weight, for example, exon 8/8a, exon 21/22, exon 31/32, or those not flanked by a common sequence (exon 1a/1), we used real-time RT-PCR to quantitate each splice variant and calculated the fraction of total transcripts accounted for by each splice variant. The primers used to amplify different splice variants were selected to yield similar amplification efficiency (data not shown).
Fig. 2.

Separation of 10 splice variants derived from locus 10 (exon 30—exon 34) using fluorescently labeled primer polymerase chain reaction (PCR). Complementary DNA fragments spanning exon 30 and exon 34 were amplified with a pair of primers shown in Supplemental data Table 1, with one of them labeled with HEX fluorescence dye. The PCR products were run on ABI 3730 capillary electrophoresis DNA analyzer. The different sized PCR products were separated, and the splice variants were identified by their size.
Shown in Table 2, the major splice variant in loci 2, 4a, 5, 6, 8, 9a and 11, accounted for 90–100% of total transcripts at each locus. Some rare variants were below detection threshold in all or part of the samples tested. Therefore, interindividual variations of splice variants derived from these loci are not likely to affect CACNA1C function. In locus 12, E45* had been reported to be involved in oxygen sensing [29]. The splice variant containing E45* accounted for less than 9% of total transcripts in locus 12 and was detectable only in 13 out of 65 samples, six from the nonischemic group and seven from the ischemic group. No difference exists in the level of splice variant containing E45* between ischemic and nonischemic groups (P > 0.05, t-test). In contrast, two or more main splice variants coexisted in loci 1, 3, 4b, 7, 9b, 10a, 10b, showing differences between individuals (Table 2, Fig. 3). In loci 1, 4b, 9b and 10b, exon 1a/1, exon 8a/8, exon 21/22 and exon 31/32 are mutually exclusive exons, with the expression of the exon 1a, exon 8a, exon 21 and exon 32 predominant over the other (Table 2, Fig. 3). This result is consistent with the reference sequence of the cardiac form of CACNA1C (Cav2a), except for the predominant inclusion of exon 32 instead of exon 31 as reported [30,31].
Table 2.
Quantitative analysis of CACNA1C splice variants in human heart left ventricle (n = 65) using fluorescently labeled primers or real-time PCR (labeled with #)
| Locus | Splice variants | 65 samples measured individually |
Pooled sample from 65 heartsa |
Pooled sample from five normal heartsb |
Functional consequencea(ref) | |
|---|---|---|---|---|---|---|
| Mean ± SD (%) | Range % (min to max) | |||||
| 1# | E1a-E2 | 70 ± 7 | 47–87 | 67 | 71 | Activated by PKC [15] |
| E1-E2 | 30 ± 7 | 13–53 | 33 | 29 | Inhibited by PKC [15] | |
| 2 | E2-E3 | 99.9 ± 0.1 | 99–100 | 100 | 100 | |
| E2-E3 + 4nt | 0.04 ± 0.10 | 0–1 | 0 | 0 | Frame shift | |
| 3 | E6-E7 | 50 ± 3 | 40–59 | 52 | 53 | |
| E6-E7-12nt | 50 ± 3 | 41–60 | 48 | 47 | ||
| 4a | E7-E8/E8a-E9-E10 | 99 ± 1 | 91–100 | 95 | 96 | |
| E7-E9-E10 | 1 ± 1 | 0–9 | 3 | 3 | ||
| E7-E10 | 0 | 0 | 2 | 1 | ||
| 4b# | E7-E8 | 26 ± 12 | 13–91 | 25 | 15 | Smooth muscle isoform, higher affinity for DHP [18] |
| E7-E8a | 74 ± 12 | 9–87 | 75 | 85 | Cardiac isoform, lower affinity for DHP [18] |
|
| 5 | E9-E10 | 98 ± 1 | 94–99 | 97 | 96 | |
| E9-E9*-E10 | 2 ± 1 | 0.6–6 | 3 | 4 | ||
| 6 | E10-E11 | 99.0 ± 0.4 | 97–100 | 99 | 99 | |
| E10-E10*-E11 | 1.0 ± 0.5 | 0–3 | 1 | 1 | ||
| 7 | E14-E15 | 75 ± 4 | 60–83 | 75 | 78 | |
| E14-E15-73nt | 25 ± 4 | 17–40 | 25 | 22 | Frame shift, hemi-channel [11] | |
| 8 | E16-E17 | 99.0 ± 0.4 | 98–99 | 96 | 97 | |
| E16-E17 + 9nt | 1 ± 0.4 | 0.2–2.3 | 4 | 3 | ||
| E16-E17 + 12nt | 0.2 ± 0.2 | 0–1 | 0.2 | 0.2 | ||
| 9a | E20-E21/22-E23 | 99.0 ± 0.6 | 97–100 | 98 | 98 | |
| E20-E23 | 1.0 ± 0.6 | 0–2 | 2 | 2 | ||
| 9b# | E20-E21 | 91 ± 2 | 81–94 | 89 | 87 | |
| E20-E22 | 9 ± 2 | 6–19 | 11 | 13 | More sensitive to DHP [14] | |
| 10a | E30-E34 | 0.2 ± 0.4 | 0–2 | 0.3 | 0.2 | Nonfunctional channel [11] |
| E30-E33-E34 | 2 ± 1 | 1–7 | 1 | 0.6 | ||
| E30-E31-E32-E34 | 1 ± 0.2 | 0–1 | 0.5 | 0.5 | ||
| E30-E31-E32-6nt-E34 | 0.03 ± 0.06 | 0–0.3 | 0 | 0 | ||
| E30-E31-E32-E33-E34 | 1.0 ± 0.5 | 0.3–3 | 1.00 | 1 | ||
| E30-E31-E32-6nt-E33-E34 | 0.2 ± 0.2 | 0–1 | 0 | 0.2 | ||
| E30-E31/E32-E33-E34 | 48 ± 8 | 16–63 | 50 | 42 | Reference isoform [11] | |
| E30-E32-6nt-E34 | 3 ± 1 | 1–6 | 3 | 2.9 | A isoform, Change gating property [11] |
|
| E30-E32-6nt-E33-E34 | 15 ± 2 | 11–26 | 10 | 20 | B isoform, | |
| E30-E31/E32-E34 | 24 ± 6 | 9–41 | 34 | 32 | C isoform | |
| 10b# | E30-E31 | 13 ± 6 | 5–33 | 20 | 19 | Fetal isoform [19] |
| E30-E32 | 87 ± 6 | 67–95 | 80 | 81 | Adult isoform [19] | |
| 11 | E40-E41 | 96 ± 1 | 93–98 | 93 | 92 | |
| E40-E41 + 57nt | 4 ± 1 | 2–7 | 7 | 8 | Change gating property [13] | |
| 12 | E44-E46 | 99 ± 3 | 90–100 | 99 | 98 | |
| E44-E45*-E46 | 1 ± 3 | 0–9 | 1 | 2 | ||
| E44-E45-E46 | 0 | 0 | 0 | 0 | ||
| E44-E45*-187nt-E46 | 0 | 0 | 0 | 0 | ||
Data represent the percentage of each splice variant of the total transcripts in each locus. Data are the mean ± SD of 65 samples, each was measured twice. PKC, protein kinase C; DHP, dihydropyridine.
Same amounts of cDNA from 65 heart tissues were pooled, and splice variants measured twice and the average was shown.
Same amounts of total RNA from five normal heart left ventricles were pooled, then the cDNA synthesized and the splice variants measured. Data are the average of two cDNA preparations.
Fig. 3.

The splice patterns of CACNA1C mRNA from different loci in 65 heart tissues. The relative abundance of splice variants in each locus was expressed as percentage of total transcripts from that locus. Each horizontal bar represents the composition of CACNA1C mRNA splice variants in heart tissue from different individuals. Heart tissues were divided into ischemic and nonischemic groups. Each sample was measured twice, and the mean is shown.
The distribution of splice variants at each locus can vary considerably (Table 2, Fig. 3). Specifically, in locus 4b, two individuals expressed more exon 8 than exon 8a, different from the common expression pattern of the cardiac CACNA1C isoform (exon 8a) (Fig. 3c). Similarly, the minor variant ranged from 17 to 40% in locus 7 (Table 2, Fig. 3d). On the other hand, the two splice variants of locus 3 shared approximately equal amounts of total transcripts, and interindividual variation was small (range from 40 to 60%) (Table 2, Fig. 3b).
Splice locus 10 gives rise to 12 known splice variants [11]. Using the labeled primer method, we can identify 10 splice variants (Fig. 2, Table 2). Six variants that either exclude or include both exon 31 and exon 32 were predicted to produce nonfunctional proteins [11]. These accounted for ∼4% of total transcripts, ranging from 2 to 10% among different individuals (Fig. 4). Splice variants that contain a complete exon 31 or exon 32, and exon 33, represent the reference sequence or wild-type [11], which occupied 48% of the total transcripts, with considerable interindividual variability (Table 2, Fig. 4). The other variants, with six base pairs deleted from exon 32, or the entire exon 33 deleted, or both, are considered to have altered functions and are denoted as A, B and C form [11]. The combined A, B and C forms comprised 48% of total transcripts, ranging from 32 to 77% (Table 2, Fig. 4), implying interindividual variability in CACNA1C channel properties.
Fig. 4.
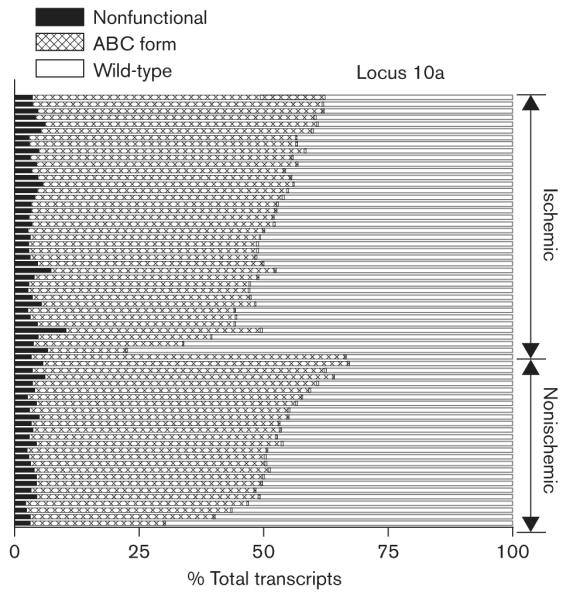
The splice patterns of CACNA1C mRNA from locus 10a (exon 30—exon 34) in 65 heart tissues. The amount of each splice variant was expressed as percentage of total transcripts from this locus. Wild-type, the reference sequence; nonfunctional, the splice variants that include or exclude both exon 31 and exon 32; ABC form, the splice variants that contain 6 bp deletion in exon 32 or delete whole exon 33, or both (see Table 2 for detail). Each horizontal bar represents the composition of CACNA1C mRNA splice variants in heart tissue from different individuals. Heart tissues were divided into ischemic and nonischemic groups. Each sample was measured twice and the mean is shown.
The splice patterns of ischemic and nonischemic hearts are similar in most of the loci, except for locus 4b, in which the two samples with predominant exon 8 instead of exon 8a were from ischemic hearts (both of them were from female Caucasians, Fig. 3c). Differences in splice variants in all loci between ischemic and nonischemic groups were, however, not significant, including locus 4b (P > 0.05, t-test). Similarly, the expression of splice variants in all loci did not vary with sex and race (P > 0.05, one-way ANOVA).
The relative amounts of several splice variants correlated with total CACNA1C expression; for example, splice variants with exon 8a and exon 32 positively correlated with total CACNA1C expression (Pearson r 0.385, P < 0.01 and 0.346, P < 0.01 respectively), and nonfunctional variants in locus 10 negatively correlated with total CACNA1C mRNA expression (Pearson r -0.546, P < 0.01). The usage of exon 1a or exon 1, initiated from different promoters did not correlate with total mRNA levels (Pearson r 0.027, P > 0.05), indicating that the transcription efficiencies from the two promoters are similar.
To test whether the same extent of splicing events also occur in normal hearts, we obtained a pooled total RNA sample of normal human heart left ventricles from five donors. As shown in Table 2, the expression of splice variants in all loci of normal hearts did not differ from a pooled sample of all 65 tissues from heart failure subjects, nor did it differ from the average of 65 failing hearts measured separately (P > 0.05, ANOVA). In locus 4b, the expression of the cardiac form of CACNA1C (exon 8a) is slightly higher in normal hearts than failing hearts, but this difference did not reach statistical significance (P > 0.05, ANOVA). This again is accounted for by the two outlier subjects with high levels of the exon 8 variant, normally expressed in smooth muscle.
Allele-specific mRNA expression of CACNA1C and its splice variants
To test whether cis-acting polymorphisms affect CACNA1C mRNA expression, we measured allelic expression ratios, using a marker SNP in exon 30 (C > T, synonymous, rs216008). Differences in the allelic ratios of genomic DNA and mRNA (after conversion to cDNA) (AEI) indicate the presence of cis-acting factors [23,24]. Sixty-five heart tissue samples were genotyped for this SNP, yielding 43 CC homozygotes, 21 CT heterozygotes and one TT homozygote. Genotype distribution did not differ significantly from Hardy—Weinberg’s equilibrium (χ2 analysis, P > 0.05), and is consistent with published data (http://ncbi.nih.gov and http://www.hapmap.org/index.html.en). We used the 21 heterozygous samples for allele-specific expression analysis. It is shown in Fig. 5a and b that the mRNA ratio for C and T alleles did not differ significantly from that of DNA ratio in all samples tested (DNA ratio = 1.00 ± 0.04; RNA ratio = 1.00 ± 0.03, n = 21). Using another marker SNP located in the unspliced exon 4 (G > A, synonymous, rs1544514), we obtained similar results (DNA ratio = 1.00 ± 0.06; RNA ratio = 1.03 ± 0.04, n = 20). The SNP in exon 4 is separated from SNP in exon 30 by over 160 000 bp and is not in linkage disequilibrium, so that only four samples are heterozygous for both exon 4 and exon 30 SNPs. A total of 37 samples were thus heterozygous for either one or both of these two marker SNPs, none showing allelic mRNA expression imbalance. As the primers used to amplify the cDNA target sequences are within exon 30 or exon 4 (E30F/E30R and E4F/E4R, Table 1), which does not undergo alternative splicing, this result indicates an absence of AEI for total CACNA1C mRNA — and hence an apparent absence of cis-acting factors determining gene expression or mRNA processing.
Fig. 5.

(a) Tracing of fluorescence peaks measured with SNaPshot in genomic DNA and RNA from heart tissue of the same individual for marker single nucleotide polymorphism (SNP) in exon 30 (rs216008, C > T). (b) Allele-specific CACNA1C mRNA expression in heart tissues. Genomic DNA and complementary DNA (cDNA) were amplified with exon 30 internal primers (E30F and E30R, Supplemental data Table 1). Allelic mRNA and DNA ratios were measured in 21 heart samples heterozygous for exon 30 SNP using SNaPshot. The C/T ratio measured for DNA, assumed to be equally abundant, was set to 1. Ratios for mRNA (cDNA) were normalized to that of DNA. Data are mean ± SD, n = 6 determinations for each sample. After normalization, for all 21 samples, DNA ratio = 1.00 ± 0.04; RNA ratio = 1.00 ± 0.03. (c) CACNA1C mRNA levels in heart tissue samples with different exon 30 SNP genotypes. Mean ± SD.
CACNA1C mRNA expression measured by RT-PCR did not differ among different genotypes (rs216008, Fig. 5c, one-way ANOVA), consistent with a lack of significant cis-acting regulatory polymorphisms. Measuring allelic ratios and mRNA levels for all CACNA1C splice variants combined may, however, have obscured the influence of cis-acting polymorphisms on individual splice variants. Therefore, we measured AEI specifically for splice variants, providing a definitive measure of cis-acting variants affecting splicing. To screen for any cis-acting SNPs that regulate alternative splicing in locus 10, we used the exon 30 marker SNP, to measure allelic ratios for seven major splice variants (E30-E31, E30-E32, E30-E31-E33, E30-E32-E33, E30-E32-E34, E30-E32-6nt-E33, and E30-E32-6nt-E34) derived from this locus. cDNAs were amplified using splice variant specific primers (exon boundary primers, Supplemental data Table 2). For all variants tested, none showed RNA ratio for C and T alleles that differed significantly from that of genomic DNA ratios (all are visually and statistically similar to Fig. 5b, Supplemental data Figs 1 and 2). The absence of AEI argues against the presence of cis-acting polymorphisms affecting splicing in locus 10. For the other splice loci, suitable marker SNPs were not available.
In locus 4b (encompassing exon 8), several individuals displayed distinct expression patterns (Fig. 3c). To test whether any polymorphisms in an exonic or intronic region can determine the usage of exon 8a or exon 8, we sequenced exon 7, exon 8a, exon 8 and the surrounding intronic sequences (∼200 bp upstream and downstream of the exons) in eight samples that showed distinct splice patterns. We observed only one common SNP located 93 bp upstream of exon 8a (rs1008832, G/A), with reported 50% heterozygosity. This SNP, however, cannot account for the different splicing pattern, judged by the allele frequency and lack of association with splicing measured in the heart tissues. For example, two samples expressing predominantly the minor exon 8 splice variant were homozygous for AA and GG genotypes, respectively.
Correlation analysis between splice variants at different loci
Splicing events at one locus could affect the splicing at other loci. To test for this possibility, we performed correlation analyses between all splice loci across all 65 tissue samples. Splicing did not correlate significantly between any of the 12 loci, suggesting that each splice site operates independently. Hence, random combinations could give rise to countless isoforms in each tissue, some with altered functions.
Discussion
In this study, we have employed quantitative methods to measure the expression level of CACNA1C mRNA, and specifically of all known mRNA splice variants, in 65 human heart tissues. The results reveal extensive interindividual variations in mRNA levels and splice patterns of CACNA1C transcripts. Importantly, several splice variants that predominate in some individuals have clearly distinct channel functions. In comparison with a pooled sample obtained from five normal hearts, we did not detect any significant differences in the splice patterns at all loci between failing hearts and normal hearts, indicating that the formation of splice variants is not unique to failing heart. Moreover, splice patterns did not differ between ischemic and nonischemic hearts. In a separate study on brain autopsy tissues, the same splice variants were also detectable, with similar interindividual variability (Wang et al., unpublished), suggesting that factors other than ischemic conditions play a role in regulating splicing. The present study is the first to report the full extent of splicing variability of CACNA1C mRNA in human heart tissue. Understanding the relevance of expression and splice patterns in disease and drug response will require further study.
Are variable mRNA levels caused by cis-acting genetic polymorphisms in CACNA1C?
Employing a definitive test for the presence of factors acting in cis on CACNA1C expression and mRNA processing, we measured allele-specific expression of CACNA1C mRNA, using two indicator SNPs in exons that are not subject to splicing. Any AEI, that is one allele expresses more than the other (relative to the number of genomic DNA alleles present), indicates the presence of cis-acting factors [23-25]. The lack of detectable AEI in a total of 37 tissues heterozygous for either of the two indicator SNPs argues against the presence of any cis-acting polymorphism affecting mRNA expression. This was surprising in view of the complex nature of the CACNA1C gene, which spans over 500 kb. Previous studies had detected abundant cis-acting polymorphisms affecting gene regulation and mRNA processing in a large portion of genes [23,24,32,33]. It is, however, possible that the indicator SNPs failed to reveal AEI because we measured average allelic ratios across all splice variants.
Factors contributing to variable mRNA levels
CACNA1C mRNA levels are surprisingly variable among different individuals (range from 2 to 100). To confirm this result, we repeated the experiments using another set of primers spanning different exons, obtaining similar results (mRNA levels range from 4 to 100). The interindividual variability in mRNA levels appears not to be caused by the quality of the total RNA, because in another study, we also measured the expression of more than 10 other genes in these 65 samples, with expression levels for all genes ranging maximally ∼10-fold. Therefore, the variable expression of CACNA1C appears to be characteristics for this gene. This was supported by separate studies in 28 human autopsy brain samples, in which we observed ∼40-fold differences in CACNA1C expression (Wang et al., unpublished). Both primers for real-time PCR were designed to amplify the regions unaffected by alternative splicing serving as common exons for all splice variants. Therefore, the mRNA levels represent a mixture of all splice variants. It is possible that extensive splicing of CACNA1C is linked to variable mRNA levels, as transcription by RNA polymerase II and pre-mRNA processing are coordinate events within the cell nucleus [34]. Our study showed that the relative amounts of several splice variants correlated with total CACNA1C expression, consistent with this notion. On the other hand, alternative splicing can reduce mRNA levels by yielding isoforms that are degraded by nonsense-mediated mRNA decay or other mechanisms [35,36]. The latter is expected to yield a negative correlation between an unstable isoform and total mRNA, as observed for splice variants inclusion or exclusion both exon 31 and exon 32 in locus 10a (nonfunctional isoforms). This suggests that the relative formation of these variants is considerably greater than indicated by measuring mature mRNA, resulting from rapid decay during mRNA processing.
Is variable splicing a result of cis-acting polymorphisms in CACNA1C?
Pre-mRNA splicing is a complex process that requires introns to have a conserved 5′ splice site, a branch point sequence followed by a polypyrimidine tract (Py tract) and a 3′ splice site. The recognition and binding of 5′ splice site by small nuclear ribonucleprotein U1 initiate the sequential assembly of the spliceosome, which contains ∼300 components, resulting in efficient and precise splicing. Alternative splicing is regulated by multiple factors, acting through both cis-acting and trans-acting pathways [37,38]. Cis-acting elements include the DNA sequences required for efficient splicing, that is, 5′ splice site, 3′ splice site, branch sites and Py tract, as well as intronic or exonic splicing enhancer and silencer. Mutations in all these sequences can cause either exon skipping or alternative splicing. It has proven difficult to identify cis-acting polymorphisms that affect splicing, as they can be located not just in the splice regions, but anywhere in large introns.
We introduce here a novel approach to tackle this problem, using allelic expression ratios specific for each splice variant. The presence of AEI would be a strong indicator that formation of this splice variant depends on a cis-acting polymorphism (or alternatively epigenetic allele-specific events). One limitation to this approach is the need for a frequent marker polymorphism in the splice region. Splice locus 10 does contain a frequent SNP in exon 30, which we selected for AEI analysis of seven splice variants derived from the exon 30 to exon 34 region. The results showed no AEI of CACNA1C for these seven splice variants from locus 10, indicating that interindividual variability in splicing was likely not caused by cis-acting polymorphisms. Similarly, sequencing of the region surrounding exon 7, exon 8a and exon 8 in several samples showing distinct splice pattern in locus 4b, did not reveal any polymorphism that could account for different splicing. A common SNP located 93 bp upstream of exon 8a is unlikely to be responsible, because the genotype of this SNP was randomly distributed between eight samples with different splice patterns. Moreover, any responsible SNP is likely to be uncommon, because the smooth muscle splice pattern was prevalent in only two out of 65 heart tissues tested. Therefore, the expression of cardiac or smooth muscle form of CACNA1C in heart tissue is not likely to be determined by SNPs in exon 7, exon 8a, exon 8 and surrounding intronic regions (∼200 bp). We cannot, however, exclude at present the possibility that SNPs located at more distant intronic enhancer or silencer sites affect splicing.
The major trans-acting factors that regulate alternative splicing are members of a family of arginine/serine-rich protein (SR proteins), modulated by their concentration and their phosphorylation state [39]. The latter indicates that alternative splicing can be regulated by signal transduction pathways [40]. It remains to be determined whether the interindividual differences in mRNA levels and splicing of CACNA1C are related to the pathophysiology of the harvested heart tissues from patients requiring transplants. mRNA acquired from heart tissues of five donors who had died of other causes, however, failed to show significant differences in splice patterns.
Functional consequences of the formation of different splice variants
Several splice variants of CACNA1C have been reported to differ functionally from the main isoform expressed in the heart. Exon 1a precedes exon 1 and is separated by large intron of ∼80 kb [15]. Exon 1a and exon 1 are alternative initial exons of human CACNA1C, regulated by different promoters [15]. L-type Ca2+ channels containing exon 1a or exon 1 yielding a long or short N-terminal domain, respectively, were designated long NT (N-terminal)-α1c and short NT-α1c. The stimulation of protein kinase C enhances activation of long NT-α1c channels but inhibits short NT-α1c channels [15]. While both exon 1a and exon 1 splice variants were detectable in all heart tissues analyzed, the expression patterns varied between individuals (Fig. 3a), suggesting interindividual variability in protein kinase C-mediated CACNA1C regulation.
Mutually exclusive exons E8a/8 encode the IS6 segment of CACNA1C, which is part of the DHP binding site. Ca2+ channels containing E8, previously denoted as a smooth muscle form (α1c-b), display 10-fold greater affinity for DHP channel blockers than channels containing E8a (cardiac form, α1c-a) [18]. The expression pattern of exon 8/8a varied dramatically among different individuals, ranging from 9 to 87% for exon 8a usage. Two heart tissues contained predominantly the smooth muscle-specific form, raising the question as to the pharmacological response to DHPs in individuals with this expression pattern. Interestingly, both these two samples were from ischemic hearts. As it occurred only in two out of 39 ischemic hearts, the relationship between ischemic and the expression of smooth muscle isoform of CACNA1C remains to be investigated. Similarly, channels containing exon 21 or exon 22 have different sensitivity to voltage-dependent DHP inhibition [14]. Our results showed predominant expression of exon 21 (∼90%), the cardiac form of CACNA1C (α1c-a), with relatively smaller interindividual variability (Fig. 3e).
A splice variant in locus 7 (E14-E15-73nt) with 73 base pair deletion in exon 15 results in a shift in the open reading frame and generation of a stop codon several base pairs downstream. The relative abundance of this interesting variant ranged from 10 to 40%. It presumably encodes an inactive hemi-channel containing only repeat I and repeat II without S6. Individuals with a higher portion of the 73 nt deletion of exon 15 might express less functional channels from their mRNA. Moreover, an inactive hemi-channel may act in a dominant-negative fashion by interfering with functional channel expression, channel property or interaction with β-subunit, as reported for a similar hemi-channel of Cav2.2 [41].
Exon 45* has been reported to be the oxygen sensor for CACNA1C in cell culture [29]. The splice variant containing E45* was, however, rare in failing heart samples (detectable only in a portion of samples) and normal hearts (Table 2), consistent with a previous report [11]. This result indicates that in adult hearts, oxygen sensing might not be critical for regulating CACNA1C function. In support of this notion, the expression of the E45*-containing variant did not differ between ischemic and nonischemic heart tissues.
Our analysis did not directly address the question which combinations of splice events at the various loci occur in parallel. This would reduce the number of possible CACNA1C splice variants expressed in heart tissues. We did not, however, observe any significant correlations between the extent of splicing at the various loci, suggesting that each splice event is independent, and that a large number of overall combined variants are present. A complete analysis of all CACNA1C variants at the protein level would be desirable; however, this exceeds the scope of this study.
In summary, the results presented here have revealed a large degree of interindividual variability in CACNA1C mRNA levels and splice variants, likely to mediate substantial phenotypic variation of the L-type CACNA1C channel. Furthermore, allele-specific expression results indicate that interindividual splice patterns result largely from trans-acting factors, rather than from cis-acting polymorphisms in CACNA1C. Interindividual variability in CACNA1C splicing is likely to play a role in disease or drug response and adverse effects, or both. As the interindividual variability does not appear to arise from genetic variants of CACNA1C, and cardiac tissue is not commonly available for analysis, we must now search for suitable biomarkers in accessible tissues where the pathophysiological and pharmacological implications of variable CACNA1C functions can be analyzed in clinical trials.
Acknowledgments
Sponsorship: This work was supported by NIH research grant HL74730.
References
- 1.Bonci A, Grillner P, Mercuri NB, Bernardi G. L-type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. J Neurosci. 1998;18:6693–6703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel—calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 3.Striessnig J. Pharmacology, structure and function of cardiac L-type Ca(2 +) channels. Cell Physiol Biochem. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- 4.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 5.Birnbaumer L, Campbell KP, Catterall WA, Harpold MM, Hofmann F, Home WA, et al. The naming of voltage-gated calcium channels. Neuron. 1994;13:505–506. doi: 10.1016/0896-6273(94)90021-3. [DOI] [PubMed] [Google Scholar]
- 6.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, et al. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 7.Seisenberger C, Specht V, Welling A, Platzer J, Pfeifer A, Kuhbandner S, et al. Functional embryonic cardiomyocytes after disruption of the L-type alpha1C (Cav1.2) calcium channel gene in the mouse. J Biol Chem. 2000;275:39193–39199. doi: 10.1074/jbc.M006467200. [DOI] [PubMed] [Google Scholar]
- 8.Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegnener JW, et al. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22:6027–6034. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grossman E, Messerli FH. Calcium antagonists. Prog Cardiovasc Dis. 2004;47:34–57. doi: 10.1016/j.pcad.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang ZZ, Liang MC, Lu S, Yu D, Yu CY, Yue DT, Soong TW, et al. Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem. 2004;279:44335–44343. doi: 10.1074/jbc.M407023200. [DOI] [PubMed] [Google Scholar]
- 12.Soldatov NM, Oz M, O’Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40–42 of the human alpha1C subunit gene. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- 13.Soldatov NM, Zuhlke RD, Bouron A, Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in alpha1C subunit in the kinetics and Ca2 + dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 14.Soldatov NM, Bouron A, Reuter H. Different voltage-dependent inhibition by dihydropyridines of human Ca2+ channel splice variants. J Biol Chem. 1995;270:10540–10543. doi: 10.1074/jbc.270.18.10540. [DOI] [PubMed] [Google Scholar]
- 15.Blumenstein Y, Kanevsky N, Sahar G, Barzilai R, Ivanina T, Dascal N. A novel long N-terminal isoform of human L-type Ca2+ channel is up-regulated by protein kinase C. J Biol Chem. 2002;277:3419–3423. doi: 10.1074/jbc.C100642200. [DOI] [PubMed] [Google Scholar]
- 16.Liao P, Yu D, Lu S, Tang Z, Liang MC, Zeng S, et al. Smooth muscle-selective alternatively spliced exon generates functional variation in Cav1.2 calcium channels. J Biol Chem. 2004;279:50329–50335. doi: 10.1074/jbc.M409436200. [DOI] [PubMed] [Google Scholar]
- 17.Lacinova L, Klugbauer N, Hofmann F. State- and isoform-dependent interaction of isradipine with the alpha1C L-type calcium channel. Pflugers Arch. 2000;440:50–60. doi: 10.1007/s004249900244. [DOI] [PubMed] [Google Scholar]
- 18.Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F. Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ Res. 1997;81:526–532. doi: 10.1161/01.res.81.4.526. [DOI] [PubMed] [Google Scholar]
- 19.Diebold RJ, Koch WJ, Ellinor PT, Wang JJ, Muthuchamy M, Wieczorek DF, Schwartz A. Mutually exclusive exon splicing of the cardiac calcium channel alpha 1 subunit gene generates developmentally regulated isoforms in the rat heart. Proc Natl Acad Sci USA. 1992;89:1497–1501. doi: 10.1073/pnas.89.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000;25:106–110. doi: 10.1016/s0968-0004(00)01549-8. [DOI] [PubMed] [Google Scholar]
- 21.Caceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18:186–193. doi: 10.1016/s0168-9525(01)02626-9. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen BN, Parker RB, Noujedehi M, Sullivan JM, Johnson JA. Effects of COER-verapamil on circadian pattern of forearm vascular resistance and blood pressure. J Clin Pharmacol. 2000;40:1480–1487. [PubMed] [Google Scholar]
- 23.Wang D, Johnson AD, Papp AC, Kroetz DL, Sadee W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C > T affects mRNA stability. Pharmacogenet Genomics. 2005;15:693–704. [PubMed] [Google Scholar]
- 24.Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem. 2005;280:32618–32624. doi: 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- 25.Johnson AD, Wang D, Sadee W. Polymorphisms affecting gene regulation and mRNA processing: broad implications for pharmacogenetics. Pharmacol Ther. 2005;106:19–38. doi: 10.1016/j.pharmthera.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Pinsonneault J, Nielsen CU, Sadee W. Genetic variants of the human H + /dipeptide transporter PEPT2: analysis of haplotype functions. J Pharmacol Exp Ther. 2004;311:1088–1096. doi: 10.1124/jpet.104.073098. [DOI] [PubMed] [Google Scholar]
- 27.Owen A, Goldring C, Morgan P, Chadwick D, Park BK, Pirmohamed M. Relationship between the C3435T and G2677T(A) polymorphisms in the ABCB1 gene and P-glycoprotein expression in human liver. Br J Clin Pharmacol. 2005;59:365–370. doi: 10.1111/j.1365-2125.2005.02229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zygalaki E, Stathopoulou A, Kroupis C, Kaklamanis L, Kyriakides Z, Kremastinos D, Lianidou ES. Real-time reverse transcription-PCR quantification of vascular endothelial growth factor splice variants. Clin Chem. 2005;51:1518–1520. doi: 10.1373/clinchem.2004.046987. [DOI] [PubMed] [Google Scholar]
- 29.Fearon IM, Varadi G, Koch S, Isaacsohn I, Ball SG, Peers C. Splice variants reveal the region involved in oxygen sensing by recombinant human L-type Ca(2 + ) channels. Circ Res. 2000;87:537–539. doi: 10.1161/01.res.87.7.537. [DOI] [PubMed] [Google Scholar]
- 30.Biel M, Ruth P, Bosse E, Hullin R, Stuhmer W, Flockerzi V, Hofmann F. Primary structure and functional expression of a high voltage activated calcium channel from rabbit lung. FEBS Lett. 1990;269:409–412. doi: 10.1016/0014-5793(90)81205-3. [DOI] [PubMed] [Google Scholar]
- 31.Koch WJ, Ellinor PT, Schwartz A. cDNA cloning of a dihydropyridine-sensitive calcium channel from rat aorta. Evidence for the existence of alternatively spliced forms. J Biol Chem. 1990;265:17786–17791. [PubMed] [Google Scholar]
- 32.Bray NJ, Buckland PR, Owen MJ, O’Donovan MC. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum Genet. 2003;113:149–153. doi: 10.1007/s00439-003-0956-y. [DOI] [PubMed] [Google Scholar]
- 33.Yan H, Yuan W, Velculescu VE, Vogelstein B, Kinzler KW. Allelic variation in human gene expression. Science. 2002;297:1143. doi: 10.1126/science.1072545. [DOI] [PubMed] [Google Scholar]
- 34.Cramer P, Srebrow A, Kadener S, Werbajh S, de la Mata M, Melen G, et al. Coordination between transcription and pre-mRNA processing. FEBS Lett. 2001;498:179–182. doi: 10.1016/s0014-5793(01)02485-1. [DOI] [PubMed] [Google Scholar]
- 35.Hillman RT, Green RE, Brenner SE. An unappreciated role for RNA surveillance. Genome Biol. 2004;5:R8. doi: 10.1186/gb-2004-5-2-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baek D, Green P. Sequence conservation, relative isoform frequencies, and nonsense-mediated decay in evolutionarily conserved alternative splicing. Proc Natl Acad Sci USA. 2005;102:12813–12818. doi: 10.1073/pnas.0506139102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nissim-Rafinia M, Kerem B. Splicing regulation as a potential genetic modifier. Trends Genet. 2002;18:123–127. doi: 10.1016/s0168-9525(01)02619-1. [DOI] [PubMed] [Google Scholar]
- 38.Hastings ML, Krainer AR. Pre-mRNA splicing in the new millennium. Curr Opin Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 39.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamm S. Signals and their transduction pathways regulating alternative splicing: a new dimension of the human genome. Hum Mol Genet. 2002;11:2409–2416. doi: 10.1093/hmg/11.20.2409. [DOI] [PubMed] [Google Scholar]
- 41.Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, Dolphin AC. Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructs. J Neurosci. 2001;21:8495–8504. doi: 10.1523/JNEUROSCI.21-21-08495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]