

Abstract
Microarray-based comparative genomic hybridization has become a widespread method for the analysis of DNA copy number changes across the human genome. Initial methods for microarray construction using large-insert clones required the preparation of DNA from large-scale cultures. This rapidly became an expensive and time-consuming process when expanded to the number of clones needed for higher resolution arrays. To overcome this problem, several PCR-based strategies have been developed to enable array construction from small amounts of cloned DNA. Here, we describe the construction of microarrays composed of human-specific large-insert clones (40–200 kb) using a specific degenerate oligonucleotide PCR strategy. In addition, we also describe array hybridization using manual and automated procedures and methods for array analysis. The technology and protocols described in this article can easily be adapted for other species dependent on the availability of clone libraries. According to our protocols, the procedure will take approximately 3 days from labeling the DNA to scanning the hybridized slides.
INTRODUCTION
Since its introduction in 1992 (ref. 1), comparative genomic hybridization (CGH) has facilitated the analysis of genome-wide copy number changes particularly for tissues such as solid tumors, from which metaphase chromosomes are difficult to obtain. In this approach, normal metaphase chromosomes are used as the target for hybridization and, as a consequence, the resolution at which copy number changes can be detected is limited to approximately 3–5 Mb (see ref. 2). The replacement of metaphase chromosomes with DNA sequences arrayed onto glass slides has largely overcome this limitation. This methodology was first described in 1997 and termed matrix or array-CGH3,4.
In a typical array-CGH experiment, test and reference genomic DNAs are differentially labeled and then cohybridized to the microarray consisting of large-insert clones with known chromosomal location. After stringency washing and drying, the slide is scanned to measure the fluorescence intensity of each sequence on the array. The fluorescent ratio for the test and reference DNAs reported by each clone is normalized and the log 2-transformed ratio is then plotted against the position of the clone sequence along the chromosomes (Fig. 1). Gains or losses across the genome are identified by values increased or decreased from a 1:1 ratio (log 2 value of 0). In this approach, the resolution is dependent only on the insert size and the density of the clones used for array construction.
Figure 1.
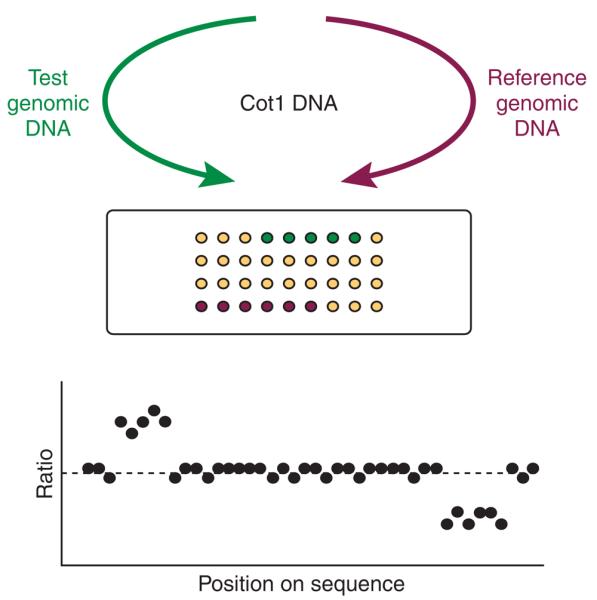
Principle of array-CGH. Genomic DNA from two cell populations is differentially labeled and hybridized to a microarray in the presence of Cot1 DNA to suppress repeat sequences. The fluorescent ratios on each array spot are calculated and normalized so that the median log 2 ratio is 0.
Array-CGH can detect chromosomal alterations associated with DNA copy number change but is insensitive to balanced rearrangements, such as inversions and reciprocal translocations. Furthermore, this methodology measures the relative DNA copy number of one test genome compared to a reference, but overlooks its absolute DNA ploidy state. This limitation can impair data interpretation, especially in aneuploid and polyploid tumor samples. In these cases, additional cytogenetic analyses—such as multicolor-fluorescence in situ hybridization5—are necessary to support and confirm CGH results.
Array-CGH has been particularly useful for the analysis of copy number changes in tumors, where gains and losses can help identify genes involved in the development and pathogenesis of cancers6-8. More recently, this methodology has been applied to detect microduplications and microdeletions in patients with constitutional rearrangements9-16. In our laboratory, the analysis of 50 patients with learning disability and dysmorphic features revealed copy number changes in more than 20% of cases for whom previous screens by conventional cytogenetic methods had given negative results14. However, some of these copy number changes were found to be inherited from phenotypically normal parents. This improvement in diagnostic yield has led to the implementation of array-CGH as a new diagnostic tool in many clinical cytogenetic laboratories. Furthermore, array-CGH has been instrumental in the discovery that copy number differences are common between normal individuals and account for a significant proportion of variation in the human population17-19. More recently, array-CGH has also been applied successfully to study cross-species similarities and evolutionary breakpoints by hybridizing primate genomic DNA onto human-specific large-insert clone arrays20-22.
In this article, we describe the protocols we have designed for implementing array-CGH technology using spotted large-insert clone arrays. The sections “DNA probe labeling,” “array hybridization” and “data analysis” are also relevant for application to commercially produced large-insert clone arrays.
Array design
Large-insert clones, such as bacterial artificial chromosomes (BACs) and P1 artificial chromosomes (PACs) with an average size of about 100–150 kb, are commonly used for the construction of genomic DNA microarrays. Initially, arrays with a resolution of one large-insert clone per megabase across the human genome were used for copy number analyses in tumors and patients with constitutional rearrangements, for example refs. 6-8, 11, 14 and 15. However, high-resolution arrays covering specific chromosomes or even the entire genome with overlapping BAC and PAC clones are now more readily available23-26. A further increase in resolution can be achieved by replacing BAC and PAC clones with overlapping cosmid or fosmid clones (20–40 kb), which for array construction can be prepared in the same way as BAC and PAC clones.
Target DNA preparation
Initially, clone DNA extracted from large-scale bacterial cultures was used for direct spotting. However, this approach is time consuming and expensive when expanded to the number of clones required to construct an array with a resolution of 1 Mb (~3,500 clones) or even tiling path resolution (~30,000 clones). To overcome this problem, several approaches have been used to amplify the target DNA enzymatically. These strategies included ligation-mediated PCR27, rolling circle PCR using Phi29 (see ref. 28) and degenerate oligonucleotide PCR (DOP-PCR) using an aminemodified version of the standard DOP-PCR primer 6MW29.
We routinely use the protocol described by Fiegler et al.23,30. This approach is based on DOP-PCR for the amplification of the target DNA sequences (Fig. 2). DOP-PCR uses a mixture of primers, whereby each of the primers consists of a fixed 5′ sequence and six defined bases at the 3′ end flanking random hexanucleotide sequences. Used with a cycling protocol that incorporates a small number of initial low-temperature annealing cycles, DOP-PCR allows a general amplification of any target DNA31. However, the efficiency of the reaction relies on the frequency with which bases in the target sequence match the six 3′ bases of the primer. If the matches to the primer are infrequent in a particular target sequence, the DOP-PCR product will be a poor representation of the target. To reduce this problem, we amplify the target clones in three separate PCRs using three different DOP-PCR primers that have been designed to be efficient in amplifying human genomic DNA, but inefficient in the amplification of Escherichia coli DNA, which is a known contaminant of DNA preparations from clone cultures32. In this way, the generated PCR product contains higher proportions of clone insert sequences, thus increasing the capacity of the spotted product to hybridize with the DNA of interest. Furthermore, combining the three different DOP-PCR products before a secondary PCR with a 5′ amine-modified primer ensures a better representation of the original target sequence. The aminoprimer used in the final PCR is designed such that the 3′ end matches the 5′ end of the DOP-PCR primers to enable universal amplifications of the primary PCR products, whereas the amino modification allows covalent attachment of the PCR products to specially coated glass slides (CodeLink-activated slides, GE Healthcare). In our hands, the use of the three DOP-PCR primers, particularly in combination, resulted in a significant sixfold increase in hybridization sensitivity and reproducibility compared to arrays constructed using the standard DOP-PCR primer 6MW30.
Figure 2.

Schematic overview of target DNA preparation for array production (a) DOP-PCR: The variation of six consecutive bases within the primer (N) and the low annealing temperature during the first amplification cycles allow the primers to bind along the template DNA. For each primer (DOP1, DOP2 and DOP3), the amplification efficiency relies on the frequency with which bases in the target sequence match the six 3′ end bases of the primer. (b) Amino-linking PCR: Amplification of the target DNA by DOP-PCR is followed by a secondary PCR using a 5′ amine-modified primer. The 3′ end of the amino-linked primer matches the 5′ end of each of the three DOP-PCR primers (here only the amplification of DOP2-derived PCR products is shown as an example).
Array spotting
A variety of commercial and “in-house designed” robots is available for array spotting. These typically use glass capillaries, split metal pins or ceramic pins to spot the DNA products. Each robot will have specific requirements regarding spotting protocols, which are supplied by the manufacturer. In addition, spotting buffer compositions and concentrations are dependent on the chosen slide surface chemistry. We routinely use the commercially available Microgridder II (Biorobotics) and array our products onto Code-Link activated slides (GE Healthcare). These slides are coated with a hydrophilic polymer containing ester reactive groups and thus allow a covalent immobilization of amine-modified DNA. We found that the covalent attachment chemistry to CodeLink activated slides has superior performance characteristics compared to other surface chemistries. However, other slides commonly used for array production are GAPS II-coated slides (coated with amino-silane; Corning) and poly l-lysine- or chromium-coated microscope slides (e.g., http://www.nuncbrand.com), which all facilitate ionic interactions between the DNA and the glass surface. Finally, normal untreated glass slides can also be used if the DNA is silanized before spotting33. Depending on the number of clones to be spotted and the size and density of printed features, it is preferable to incorporate replicate spots of each clone either next to each other or randomly distributed across the slide. A random distribution of replicate clones on the array is preferable as this helps to minimize spatial hybridization artifacts.
DNA probe labeling by random priming
In array-CGH, test and reference genomic DNAs are differentially labeled typically by using the two fluorochromes Cy3 and Cy5. To obtain good labeling and thus good hybridization results, high quality genomic DNA is required (e.g., ratio 280/260>1.8). We find that the genomic DNA extraction kit from Qiagen (cat. no.13343) produces DNA suitable for use in array-CGH.
The labeling protocol described in this article has been established for an array 2 cm × 3 cm in size. This protocol can be simply scaled for use with arrays covering different areas. It is common practice to use a random-prime labeling approach to incorporate the Cy-dyes into the genomic DNA, as we and others have found labeling methods such as nick translation or direct labeling PCR produce inferior results (H.F. and R.R., personal communication).
Although Cy3 and Cy5 are the most commonly used fluorochromes for DNA labeling, both dyes are very sensitive to environmental conditions such as high ozone and humidity levels34. To limit this problem, antioxidants such as cysteamine (see MATERIALS) can be added to hybridization and washing solutions. In addition, owing to the different sizes of the Cy3 and Cy5 molecules, sequence-specific dye bias can occur during the DNA labeling reaction and introduce artifactual ratio changes. To avoid this bias, an indirect labeling approach has been developed using the incorporation of aminoallyl-modified dNTPs into both DNAs, followed by independent direct labeling with reactive fluorochromes (i.e., Bioprime Plus Array CGH Indirect Genomic Labeling System). Alternatively, the most common strategy to limit hybridization artifacts due to a labeling bias is to perform replicate dye-reversal hybridizations. This however increases the costs for the analysis of individual DNA samples. Clones/oligonucleotides can also be spotted in multiple copies onto an array such that intensities for one particular sequence can be compared and if necessary excluded from the analysis. Unfortunately, this might only be feasible when arrays are produced “in-house” and might not apply for commercially available arrays.
Array hybridization (manual)
After DNA labeling, both probes are co-precipitated and dissolved in hybridization buffer, which among other components contains 50% formamide, 10% dextran sulfate and 0.1% (v/v) Tween 20. Whereas formamide acts as a denaturing agent increasing the hybridization stringency, dextran sulfate helps accelerate the hybridization rate. Furthermore, Tween 20, a detergent, minimizes the occurrence of nonspecific fluorescence background. Hybridization to the array is then performed at 37 °C under a glass coverslip in the presence of Cot1 DNA. The large excess of Cot1 DNA suppresses the repeat sequences that are present in both target and probe DNA. Insufficient suppression of repeat sequences would result in unspecific hybridization and consequently noisier hybridization profiles and compressed copy number change ratio values.
After hybridization, slides are first washed in PBS/0.05% Tween 20 to remove excess hybridization buffer, followed by a stringency wash for 30 min in 50% formamide/2× SCC at 42 °C. This stringent wash is critical for the efficient elimination of nonspecific hybridization signals and must be monitored carefully. Finally, slides are washed again in PBS/0.05% Tween 20 before spin-drying and preparation for scanning. It is practical to process up to six slides per run using manual handling in this way.
Array hybridization using an automated slide processor
To enable high-throughput processing of arrays, we use an automated workstation (Hs.4800, Tecan Inc.). This station enables one person to process easily 12 slides per unit per day, and can be configured with up to 4 different units each of which can be run independently and simultaneously.
The protocol we have developed is based on the manual procedure described above with two major changes: (i) the hybridization buffer contains only 5% dextran sulfate so that it is less viscous and thus compatible with injection into the hybridization chambers and with the mixing steps during the hybridization; (ii) the washing step with 50% formamide/2× SSC at 42 °C has been replaced by washes with 0.1× SSC at 54 °C to preserve the materials used in the construction of the hybridization chambers.
Data analysis
Array images for data analysis are commonly acquired using a commercial microarray scanner, which can be purchased from various companies such as Agilent Technologies, Perkin-Elmer, Tecan or Axon Instruments. Alternatively, image acquisition can be achieved by use of various CCD camera systems3,25.
There are several software solutions for image analysis available. Spot intensity extraction programs are usually supplied with commercially available scanners, but can also be purchased separately. Typical programs used are GenePix (Axon Instruments), ScanArray (Perkin-Elmer) or Agilent Feature extraction software (Agilent Technologies). Other programs—such as TIGR Spotfinder (http://www.tm4.org/spotfinder.html) or UCSF Spot (http://www.jainlab.org/)—are freely available and can be downloaded from the internet.
For further data analysis, the ratio values between the fluorescent intensities in both channels on every spot on the array are calculated (usually after the subtraction of local background fluorescence) and then normalized. We routinely normalize the array by dividing each individual ratio by the median ratio value of all autosomal clones. Normalization by subarray (or block normalization) can help correct spatial artifacts, such as fluorescence signal heterogeneity along the array or spatial biases introduced by laser misalignment during image acquisition. Furthermore, the analysis of tumor samples showing strong aneuploidy may require a normalization approach based on only chromosomal regions with modal copy number6. The normalized and log 2-transformed ratio for each clone is then plotted against its position along the genome. Although analysis can be performed using a spreadsheet program, custom scripts or macros and dedicated commercial programs are now available, for example, Bluefuse (Bluegnome, Ltd). Bluefuse provides a complete analysis package covering every step from spot intensity extraction to visualization, statistical analysis of the results and graphical display. In addition, free programs for visualization and/or statistical analysis, such as SPROC (http://www.jainlab.org) and a-CGH (http://bioconductor.org/packages/1.9/DNACopyNumber.html), can now be downloaded freely from the web. We have also recently developed a complete analysis pipeline for the automatic detection of constitutional copy number variation, using a whole-genome tiling path microarray composed of 26,574 large-insert clones23.
MATERIALS
REAGENTS
•2× TY medium, pH 7.4 (see REAGENT SETUP)
•Solution I (see REAGENT SETUP)
•Solution II (see REAGENT SETUP)
•Solution III (see REAGENT SETUP)
•Isopropanol
•70% ethanol
•100% ethanol
•80% ethanol
•T0.1E solution, pH 8 (see REAGENT SETUP)
•RNaseA (ICN Biochemicals, cat. no. 101076)
•DOP-PCR primers:
•DOP 1: 5′ CCGACTCGAGNNNNNNCTAGAA 3′
•DOP 2: 5′ CCGACTCGAGNNNNNNTAGGAG 3′
•DOP 3: 5′ CCGACTCGAGNNNNNNTTCTAG 3′
•TAPS (N-[tris(hydroxymethyl)methyl]-3-aminopropanesulfonic acid (Sigma-Aldrich, cat. no. T5130)
•TAPS salt solution (see REAGENT SETUP)
•TAPS2 buffer (see REAGENT SETUP)
•Amino-linked primer:
•5′ GGAAACAGCCCGACTCGAG 3′
•Amplitaq polymerase, 5 U μl−1 (Roche, cat. no. N808-0145)
•dNTPs (Amersham Biosciences, cat. no. 27-2035-01)
•Amino-linking buffer (see REAGENT SETUP)
•4× microarray spotting buffer: 1 M sodium phosphate buffer, pH 8.5, 0.001% sarkosyl
•1% (w/v) ammonium hydroxide solution (prepared with HPLC water)
•0.1% (w/v) sodium dodecyl sulfate solution (prepared with HPLC water)
•HPCL water
•BioPrime Labeling Kit (Invitrogen, cat. no. 18094011)
•10× dNTP mix (1 mM dCTP, 2 mM dATP, 2 mM dGTP, 2 mM dTTP in TE buffer)
•1 mM Cy3-dCTP (NEN Life Science, cat. no. NEL 576)
•1 mM Cy5-dCTP (NEN Life Science, cat. no. NEL 577)
•Manual array hybridization buffer (see REAGENT SETUP)
•3 M NaAc, pH 5.2
•Human Cot1 DNA (Invitrogen, cat. no. 15279-101)
•20% formamide solution
•50% formamide/2× SSC
•PBS/0.05% Tween 20
•Automated array pre/hybridization buffer (see REAGENT SETUP)
•Herring sperm DNA (Sigma, cat. no. D7290)
•0.1× SSC
•PBS/0.05% Tween 20/2 mM cysteamine
•Cysteamine (Sigma, cat. no. M9768)
EQUIPMENT
•Microscope slide rack
•Microscope slide storage boxes
•Microcon YM30 columns (Millipore/Amicon, cat. no. 42411)
•1 ml deep-well microtiter plates (Beckman, cat. no. 267007)
•“U” bottom microtiter plate (Greiner, cat. no. 650161)
•PTC-225 Tetrad thermocyclers (MJ Research)
•96-well PCR plates
•2.5% agarose gels
•Multiscreen 0.2-μm filter plate (Millipore, MSGVN 2250)
•384-well plates (Genetix)
•CodeLink activated slides (GE Healthcare UK Limited).
•Humid chamber (NaCl saturated water in an air-tight container)
•Centrifuge (Sorvall RT 6000D or equivalent)
•Heat block
•37 °C incubator
•Coverslip (2.4 × 3.6 cm)
•Troughs
•Hybridization chambers (plastic slide mailers containing Whatman paper saturated with 20% formamide solution)
•Parafilm
•Eppendorf 5417R centrifuge
•Eppendorf 5415D centrifuge
•1.5 ml Eppendorf tubes
•2 ml Eppendorf tubes
•Hs.4800 automated slide processing station (Tecan Inc.) equipped with 51 × 20 mm hybridization chambers
REAGENT SETUP
2× TY medium, pH 7.4 (per liter) Dissolve 16 g tryptone, 10 g yeast extract and 5 g NaCl in 1 liter ddH2O.
Solution I 50 mM glucose, 5 mM Tris pH 8.0 and 10 mM EDTA; store at 4 °C.
Solution II 0.2 N NaOH and 1% SDS.
Solution III 3 M KOAc; store at 4 °C.
T0.1E solution pH 8 Add 10 ml of 1 M Tris/HCl pH 8 and 0.2 ml of 0.5 M EDTA pH 8 to 989.8 ml ddH2O.
TAPS salt solution 250 mM TAPS, 166 mM (NH4)2SO4 and 25 mM MgCl2. To prepare the solution, add 6.08 g TAPS pH 9.3, 2.20 g (NH4)2SO4 and 2.5 ml of a 1 M MgCl2 stock solution to 93.5 ml ddH2O and store at −20 °C CRITICAL STEP pH of 9.3 is crucial for the reaction to work; adjust pH with 5 M KOH.
TAPS2 buffer (final volume 1 ml) Combine 960 μl TAPS salt solution, 33 μl bovine serum albumin (5%) and 7 μl β-mercaptoethanol; UV-sterilize before use.
Amino-linking buffer 500 mM KCl, 25 mM MgCl2 and 50 mM Tris pH 8.5 CRITICAL STEP The amino-linking buffer should be made up from 1 M stock solutions before use (for 20 ml of amino-linking buffer, add 10 ml of 1 M KCl, 1 ml of 1 M Tris pH 8.5 and 0.5 ml of 1 M MgCl2 to 8.5 ml of ddH2O) and filter-sterilized (0.2 μm syringe filter). Storage at 4 or −20 °C is not recommended; however, the buffer can be stored for a maximum of 1 week at room temperature (approximately 22 °C).
Manual array hybridization buffer 50% deionized formamide, 10% dextran sulfate, 0.1% Tween 20, 2× SSC, 10 mM Tris pH 7.4 and 10 mM cysteamine. To prepare 30 ml of buffer, add 6 ml of 50% dextran sulfate, 300 μl of 1 M Tris pH 7.4, 30 μl of Tween 20, 3 ml of 20× SSC and 1.5 ml of 0.2 M cysteamine to 4.17 ml of HPLC-treated ddH2O. In a fume hood, mix 15 ml of the above mix with 15 ml of deionized formamide (keep buffer on ice).
Automated array pre/hybridization buffer 50% deionized formamide, 5% dextran sulfate, 0.1% Tween 20, 2× SSC, 10 mM Tris pH 7.4 and 10 mM cysteamine. To prepare 30 ml of buffer, add 3 ml of 50% dextran sulfate, 300 ml of 1 M Tris pH 7.4, 30 ml of Tween 20, 3 ml of 20× SSC and 1.5 ml of 0.2 M cysteamine to 7.17 ml of HPLC-treated ddH2O. In a fume hood, mix 15 ml of the above mix with 15 ml of deionized formamide (keep buffer on ice).
PROCEDURE
DNA preparation of large-insert clones
1| For each clone, set up 500 μl bacterial cultures in 2× TY medium with the appropriate antibiotic in a separate well of a 1 ml 96-deep-well microtiter plate. Grow at 260 r.p.m. for 18 h at 37 °C.
2| Transfer 250 μl of the culture into a “U” bottom microtiter plate and pellet the cells by centrifugation in a Sorvall RT 6,000D centrifuge at 977g for 4 min (PAC and BAC clones) or 2 min (cosmids and fosmids). Discard the supernatant.
CRITICAL STEP Until otherwise stated, all centrifugation steps throughout the procedure are performed at room temperature.
3| Add 25 μl of solution I to the wells and mix by carefully tapping the side of the plate.
4| Add 25 μl of solution II, mix as before and leave at room temperature for 5 min until the solution clears.
5| Add 25 μl of cold solution III, mix as before and leave at room temperature for 5 min.
6| After incubation, transfer the entirety of each sample to a 0.2-μm filter plate and filter the sample into a “U” bottom microtiter plate containing 100 μl of isopropanol per well by spinning the plate in a Sorvall RT 6,000D centrifuge at 977g for 2 min at 20 °C.
7| Remove the filter and incubate the samples for 30 min at room temperature before spinning the samples at 1,600g for 20 min at 20 °C.
8| Remove the supernatant and dry the precipitated DNA briefly before washing with 100 μl of 70% ethanol.
9| Centrifuge in a Sorvall RT 6,000D centrifuge at 1,600g for a further 10 min at 20 °C, remove the supernatant, dry the pellet and resuspend the DNA in 5 μl of T0.1E (pH 8) with RNaseA (10 μl of 1 mg ml−1 RNaseA per 1 ml T0.1E). Incubate at 4 °C overnight.
10| Dilute the DNA with sterile distilled water to a final concentration of 1 ng μl−1.
CRITICAL STEP The final concentration of DNA per well is approximately 200 ng per 5 μl after resuspending the DNA in 5 μl of T0.1E (pH 8) with RNaseA (10 μl of 1 mg ml−1 RNaseA per 1 ml T0.1E). We dilute the DNA by adding 195 μl of distilled water and check the concentration of a small number of wells across the plate to confirm the concentration.
PAUSE POINT The DNA solution can be stored at −20 °C until required. Although we cannot comment on the maximum storage time, we are still successfully using DNA prepared a number of years ago.
Array production
11| Set up three separate DOP-PCRs (one for each DOP primer) for each clone in 96-well plates in a final reaction volume of 50 μl, as shown in the table below.
CRITICAL STEP Make sure that you are working in a sterile environment, preferably in a PCR hood, using only reagents that have been sterilized before use. We routinely UV-sterilize water, plates/tubes, TAPS2 buffer, W1 and pipettes for 30 min before use. In addition, always include negative controls in your PCR setup for each of the primers used by replacing the template with the equivalent amount of water.
| Component | Stock concentration | Amount (μl) |
|---|---|---|
| TAPS2-buffer | 5 | |
| DOP primer | 20 μM | 5 |
| dNTPs | 2.5 mM each | 4 |
| Polyoxyethelene ether W1 | 1 % | 2.5 |
| AmpliTaq polymerase | 0.5 | |
| H2O | 28 | |
| Clone template DNA | 1 ng μl−1 | 5 |
12| Place the PCRs in a thermocycler and use the following program.
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 94 °C, 3 min | — | — |
| 2–11 | 94 °C, 1.5 min | 30 °C, 2.5 min | 72 °C, 3 min, ramp at 0.1 °C s−1 to 72 °C) |
| 12–41 | 94 °C, 1 min | 62 °C, 1.5 min | 72 °C, 2 min |
| 42 | — | — | 72 °C, 8 min |
13| Run 5 μl of each PCR product on a 2.5% agarose gel (200 V, 30 min).
CRITICAL STEP The average size of the product ranges from 0.2 to 2 kb. Ensure you have no signals in the negative controls.
PAUSE POINT PCR products can be stored at −20 °C until required. Although we cannot comment on the maximum storage time, we are still successfully using PCR products produced a number of years ago.
TROUBLESHOOTING
14| Combine equal amounts of the three different DOP-PCR products from each corresponding clone into a fresh 96-well plate.
15| Set up amino-linking PCRs in 96-well plates in a final reaction volume of 90 μl, using the combined PCR products as a template.
CRITICAL STEP Do not forget to include negative controls (water in place of template) in your experiment.
| Component | Stock concentration | Amount (μl) |
|---|---|---|
| Amino-linking buffer | 9 | |
| dNTPs | 2.5 mM each | 9 |
| Aminoprimer | 200 ng μl−1 | 4.5 |
| AmpliTaq polymerase | 0.9 | |
| H2O | 63.6 | |
| Template (combined DOP-PCR product) | 3 |
16| Place the PCRs in a thermocycler and amplify the template using the program shown below.
| Cycle number | Denature | Anneal | Extend |
|---|---|---|---|
| 1 | 95 °C, 10 min | — | — |
| 2–36 | 95 °C, 1 min | 60 °C, 1.5 min | 72 °C, 7 min |
| 37 | — | — | 72 °C, 10 min |
17| Run 2 μl of the PCR products on a 2.5% agarose gel (200 V, 30 min).
CRITICAL STEP Ensure you have no signals in the negative controls.
PAUSE POINT PCR products can be stored at −20 °C until required. Although we cannot comment on the maximum storage time, we are still using PCR products produced a number of years ago.
TROUBLESHOOTING
18| Transfer 88 μl of amino-linked DOP-PCR product mix into a Millipore Multiscreen 0.2-μm filter plate and add 29 μl 4× microarray spotting buffer.
CRITICAL STEP This spotting buffer works specifically with CodeLink activated slides. Spotting buffers vary and need to be selected according to the slide surface chemistry as well as for the arrayer used for spotting the DNA.
19| Filter the samples by centrifugation for 10 min at 600g (in a Sorvall RT 6,000D) and transfer into Genetix 384-well plates.
PAUSE POINT PCR products can be stored at −20 °C until required. Although we cannot comment on the maximum storage time, we are still using PCR products produced a number of years ago.
20| Array DNA elements onto CodeLink activated slides at 20–25 °C and 40–50% relative humidity according to the arrayer manufacturer's instructions.
CRITICAL STEP Arraying sessions can last up to 3 days without loss of binding efficiency of the DNA to the slides.
21| Transfer slides containing arrayed elements into a microscope slide rack, place in a humid chamber containing saturated NaCl solution, do not immerse the slides in this solution and incubate for 24–72 h at room temperature.
CRITICAL STEP Humidity levels of approximately 70–80% are used to drive the covalent reaction between reactive groups on the slide and the 5′ amino groups on the DNA elements.
22| Remove the slides from humid chamber, block reactive groups by immersing slides in a 1% solution of ammonium hydroxide and incubate for 5 min with gentle shaking.
23| Transfer slides into a solution of 0.1% sodium dodecyl sulfate and incubate for 5 min with gentle shaking.
24| Briefly rinse slides in HPLC water at room temperature and then place in 95 °C HPLC water for 2 min to denature the bound DNA elements.
CRITICAL STEP 1.25 liter of HPLC water can be boiled in a microwave and will remain between 95 and 100 °C during the 2 min denaturation step carried out on the bench.
25| Transfer slides to ice-cold HPLC water for 1 min and then briefly rinse twice in room temperature HPLC water.
26| Dry the slides by spinning in a centrifuge for 10 min at 156g.
27| Place slides in a storage box and store at room temperature in a cool, dry place until use.
PAUSE POINT Slides can be stored in the dark under dehumidified atmosphere for at least 3 months without loss of performance in hybridizations.
DNA probe labeling by random priming and removal of unincorporated nucleotides (protocol established for an array size of 2 × 3 cm)
28| Mix 0.15 μg test and 0.15 μg reference genomic DNA in separate reactions each with 60 μl of 2.5 random primers solution supplied in the BioPrime Labeling Kit and add water to a final volume of 130.5 μl.
29| Denature the solution in a heat block for 10 min at 100 °C and immediately cool on ice. Then add the following reagents on ice to each of the reactions:
10× dNTP mix, 15.0 μl
Cy3- or Cy5-labeled dCTP, 1.5 μl
Klenow fragment (40 U ml−1), 3.0 μl
30| Incubate the reaction at 37 °C overnight in the dark and stop by adding 15 μl of stop buffer supplied in the kit.
31| To remove unincorporated labeled nucleotides using Microcon YM30 columns place filter into Eppendorf tubes provided by the suppliers.
32| Pipette labeled test and reference DNA together on top of the filter. If both labeling reactions together exceed a total volume of 300 μl, then we recommend using two separate filters, one for each reaction.
33| Spin for 5 min at 12,000g using an Eppendorf 5415D centrifuge. The labeled probe should be contained in the filter (filter should still be moist) and unincorporated dyes should be in flow-through.
34| Discard flow-through, put filter back onto Eppendorf tube and add 300 μl of HPLC water.
35| Spin for 5 min at 12,000g.
36| Discard flow-through (flow-through should be less colored than before; filter should still be moist).
37| Add 100 μl of HPLC water and place filter upside-down onto a fresh Eppendorf tube provided by the suppliers.
38| Spin for 2 min at 400g.
39| Run 3 μl of the collected sample (approximately 120 μl total volume) on a 2.5% agarose gel.
CRITICAL STEP You should see a smear with the bulk fragments below 500 bp.
TROUBLESHOOTING
Array hybridization
40| Array hybridization can be carried out manually (option A) or automatically (option B). We generally run 6 and 12 arrays, respectively, per setup. Both protocols are optimized for an array size of 2 × 3 cm: the amounts detailed below can be scaled up/down for larger/smaller arrays.
(A) Manual array hybridization
(i) Add the following solutions to a 1.5 ml Eppendorf tube.
| Component | Amount (μl) |
|---|---|
| Cy3 and Cy5 labeled and cleaned test and reference DNAs from Step 39 | ~160 (entire cleaned labeling reaction) |
| Human Cot1 DNA (1 μg μl−1) | 135 |
| 3 M NaAc pH 5.2 | 35 |
| 100% EtOH (cold) | 1,000 |
(ii) Mix the samples and precipitate at −20 °C overnight or for 30 min at −70 °C.
PAUSE POINT Precipitated DNA samples can be stored at −20 °C for several days until required.
(iii) Spin the precipitated DNA for 30 min at maximum speed, at 4 °C in a refrigerated benchtop centrifuge (i.e., Eppendorf 5417R centrifuge).
(iv) Wash the pellets with 500 μl of 80% EtOH and re-spin at maximum speed for 5 min.
(v) Remove the supernatant and re-spin the samples at maximum speed for 1 min.
Take off the supernatant with a small tip to prevent accidentally discarding the DNA pellet. Repeat this process until the pellet is dry.
(vi) Resuspend DNA pellet in 35 μl hybridization buffer. Denature the sample for 10 min at 70 °C.
(vii) Incubate for 60 min in a 37 °C heat block to suppress repetitive sequences present in the labeled DNA.
CRITICAL STEP Fluorescent dyes are light sensitive: for long incubation periods, keep the labeled samples in the dark.
(viii) Apply 30 μl of the hybridization solution onto the array and cover with a coverslip (2.4 × 3.6 cm). Place the slide into a slide mailer humidified with 20% formamide/2× SSC, seal with parafilm and incubate at 37 °C for 24–48 h.
(ix) After incubation, remove the coverslip by placing the slide into a tall glass trough containing PBS/0.05% Tween 20 to wash off any excess hybridization solution.
(x) Transfer the slide into a new glass trough and wash in PBS/0.05% Tween 20 for 10 min at room temperature (shaking at maximum speed).
(xi) Place the slide into a preheated 50% formamide/2× SSC solution and incubate for 30 min at 42 °C (shaking).
(xii) Transfer the slide into fresh PBS/0.05% Tween 20 and wash again for 10 min at room temperature (shaking at maximum speed).
(xiii) Dry the slide by spinning at 150g for 1 min.
PAUSE POINT Store in a light-proof box until ready for scanning; however, slides should be scanned as soon as possible or at least on the same day.
(B) Automated array hybridization using 51 mm 20 × mm chambers
(i) Add the following solutions to separate 2 ml Eppendorf tubes, mix well and precipitate at −20 °C overnight or for 30 min at −70 °C.
Tube 1.
| Component | Amount (μl) |
|---|---|
| Cy3 and Cy5 labeled and cleaned test and reference DNAs | ~160 (entire cleaned labeling reaction) |
| Human Cot1 DNA (1 μg μl −1) | 135 |
| 3 M NaAc pH 5.2 | 35 |
| 100% EtOH (cold) | 1,000 |
Tube 2.
| Component | Amount (μl) |
|---|---|
| Herring sperm DNA (10 mg ml−1) | 80 |
| 3 M NaAc pH 5.2 | 10 |
| 100% EtOH (cold) | 500 |
(ii) For hybridization, spin the precipitated DNAs for 30 min at maximum speed at 4 °C in a refrigerated benchtop centrifuge (i.e., Eppendorf 5417R centrifuge).
(iii) Wash the pellets with 500 μl of 80% ethanol and spin at maximum speed for 5 min.
(iv) Remove the supernatant and re-spin the samples at maximum speed for 1 min.
(v) Take off the supernatant with a small tip to prevent accidentally discarding the DNA pellet. Repeat this process until the pellet is dry.
(vi) Individually resuspend the pellets in tubes 1 and 2 in 120 μl of hybridization buffer.
(vii) Denature both samples for 10 min at 70 °C.
(viii) Incubate tube 1 for 60 min in a 37 °C heat block to suppress repetitive sequences present in the labeled DNA (in the dark).
(ix) Perform hybridizations and washes on the slide processor as indicated in Table 1.
TABLE 1.
Setup for automated array hybridization (Step 33B(ix)).
| Step | Temperature (°C) | Options | |
|---|---|---|---|
| 1 | Wash | 37 | First: yes, channel: 1, runs: 1, wash time: 0:00:30, soak time: 0:00:00 |
| 2 | Injection | 37 | |
| 3 | Hybridization | 37 | Agitation frequency: medium, time: 0:45:00 |
| 4 | Injection | 37 | |
| 5 | Hybridization | 37 | Agitation frequency: medium, time: 21:00:00 |
| 6 | Wash | 37 | First: no, channel: 1, runs: 15, wash time: 0:00:30, soak time: 0:00:30 |
| 7 | Wash | 54 | First: no, channel: 2, runs: 5, wash time: 0:01:00, soak time: 0:02:00 |
| 8 | Wash | 23 | First: no, channel: 1, runs: 10, wash time: 0:00:30, soak time: 0:00:30 |
| 9 | Wash | 23 | First: no, channel: 3, runs: 1, wash time: 0:00:30, soak time: 0:00:00 |
| 10 | Slide Drying | 23 | Time: 0:02:30, final manifold cleaning: yes, channel: 3 |
Liquid identification: channel 1: PBS/0.05% Tween 20, 2 mM cysteamine; channel 2: 0.1× SSC; channel 3: pure water.
(x) Inject 100 μl of the denatured Herring sperm DNA (tube 2) into the station chamber containing the array slide, following the manufacturer's instructions displayed on the instrument.
CRITICAL STEP Prehybridization of the array is preferable in the automated procedure to ensure a homogenous distribution when injecting the labeled probe (Step xi).
(xi) After prehybridization, inject 100 μl of the hybridization solution (tube 1) into the chamber following the instructions displayed on the station.
(xii) When the run is finished, store slides in a light-proof box until scanning; however, slides should be scanned as soon as possible or at least on the same day.
TROUBLESHOOTING
TIMING
Steps 1–10, DNA preparation of large-insert clones: approximately 36 h
Steps 11–27, array production: approximately 3 d for 24 96-well plates (steps 11–19); approximately 3 d (steps 20–27)
Steps 28–39, DNA probe labeling: approximately 16 h
Step 40A, manual hybridization: approximately 24 h
Step 40B, automated hybridization: approximately 24 h
TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Solution |
|---|---|---|
| Step 13: DOP-PCR amplification | No amplification products | Ensure that BSA and β-mercaptoethanol have been added to the reaction and the pH of the TAPS solution has been adjusted to 9.3 |
| Negative control contaminated | It is crucial to UV-sterilize reagents as well as plastic ware before use. To rule out possible contamination of primers, we order our primers freeze-dried and dilute them before use in UV-sterilized water | |
| Step 17: amino-linking PCR | Amino-linking PCR: no amplification product | Ensure that the amino-linking buffer has been prepared before use. Storage below room temperature results in inefficient amplification |
| Step 17: amino-linking PCR | Amino-linking PCR: negative control contaminated | Order primer freeze-dried and dilute before use in UV-sterilized water. Ensure you are working with sterile reagents and plastic ware |
| Step 39: random primed labeling | Fragments too small after random primer labeling | Ensure that DNA is not degraded, if necessary repeat DNA isolation |
| Step 39: random primed labeling | Insufficient incorporation of Cy-dyes | This can be due to degraded DNA used as a template or low DNA quality. If necessary, clean up DNA by, for example, phenol–chloroform extraction or repeat DNA isolation |
| Step 40: hybridization | Compressed hybridization ratios/increased ratio variability | A critical factor for the reliable detection of copy number changes is the sufficient suppression of repeat sequences during hybridization. This is dependent on the quality of the Cot1 DNA, which mainly consists of LINE and SINE repeats. When using high-quality Cot1 DNA, single copy changes are reported by log 2 ratio values close to 0.58 (single copy gain) and −1 (single copy loss). Low-quality Cot1 DNA will fail to suppress repeats completely, thus resulting in compressed abnormal ratios and increased background ratio variability The conditions of the second wash step (42 °C in 50% formamide/2× SSC for the manual procedure, 54 °C in 0.1× SSC for the automated one) are critical for efficient removal of nonspecific probe hybridization. Carefully adjust these conditions in case of compressed abnormal ratios, increased background ratio variability or low spot intensities |
| Step 40: hybridization | Noisy hybridization profiles | There are many factors contributing to noisy hybridization profiles. However, DNA quality, array quality and hybridization conditions (e.g., high humidity levels throughout hybridization, inappropriate washing conditions) as well as environmental factors such as temperature, humidity and high ozone levels are the main causes. Although environmental factors seem to influence the signal intensities especially with regard to Cy5, problems with hybridization conditions often result in speckly images. The addition of antioxidant in hybridization and wash solutions (such as cysteamine; see automated procedure) can help protect Cy5 from degradation and limit experimental variability It should also be noted that commercially available scanners without dynamic focusing if poorly adjusted can introduce spatial artifacts into the data especially when slide surfaces are uneven. This effect however can be reduced by performing block normalization analyses |
ANTICIPATED RESULTS
DOP-PCR
Depending on the size of the template DNA, electrophoresis (2.5% agarose gel) will generate either a smear (genomic DNA as a template) or distinct banding patterns (cosmids, fosmids, BACs and PACs as templates (Fig. 3a).
Figure 3.

Distinct banding patterns of DOP-PCR and amino-linking PCR products using long-insert genomic clones as templates. Note the much brighter smears—due to the increased DNA concentration—obtained after the secondary amino-linking PCR (b) compared with primary DOP-PCR (a). Negative controls for both PCRs are running in lanes 16 (a) and 16 (b).
Amino-linking PCR
After amino-linking PCR, the previously observed smear/banding pattern after electrophoresis should now appear much brighter due to the increased DNA concentration after the amino-linking reaction (Fig. 3b).
DNA probe labeling by random priming
Electrophoresis (2.5% agarose gel) should generate a smear with the majority of fragments around 300–600 bp.
Hybridization results
With good-quality DNA and optimal hybridization conditions, single-copy changes should be reported by log 2 ratio values being close to 0.58 (single copy gain) and −1 (single copy loss), as shown in Figure 4a. Standard deviations of autosomal log 2 ratio values typically range from 0.03 to 0.08 for normal individuals or patients with small constitutional rearrangements. Hybridization results with s.d. above 0.08 in our hands are discarded and have to be repeated. For analysis of tumor samples, s.d. can be higher depending on the amount of copy number aberrations present in the test genome. Factors that contribute to hybridization failures—a typical example is shown in Figure 4b—include poor DNA quality, poor suppression of repetitive sequences by Cot1 DNA, low signal intensities due to, for example, labeling failures, laser misalignments in scanners and environmental factors such as high humidity or ozone level (see TROUBLESHOOTING for more detailed information).
Figure 4.

Chromosome 10 array-CGH profile of a colon cancer cell line on a whole-genome tiling path microarray. (a) The profile indicates a single copy gain (highlighted in green) and a single copy deletion (highlighted in red) on the q-arm of chromosome 10. (b) Genomic profile with the same DNA after an unsuccessful array hybridization.
ACKNOWLEDGMENTS
The protocols described in Steps 1–9 were originally developed by Sean Humphray and members of the Wellcome Trust Sanger Institute Core Mapping Group; Steps 15–27 were originally developed or are modified from protocols developed by David Vetrie, Cordelia Langford and other members of the Wellcome Trust Sanger Institute Microarray Facility. The original protocols are available at http://www.sanger.ac.uk/Projects/Microarrays/arraylab/protocol3b.pdf and http://www.sanger.ac.uk/Projects/Microarrays/arraylab/protocol4.pdf. The authors are supported by the Wellcome Trust.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Kallioniemi A, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258:818–21. doi: 10.1126/science.1359641. [DOI] [PubMed] [Google Scholar]
- 2.Lichter P, Joos S, Bentz M, Lampel S. Comparative genomic hybridization: uses and limitations. Semin. Hematol. 2000;37:348–57. doi: 10.1016/s0037-1963(00)90015-5. [DOI] [PubMed] [Google Scholar]
- 3.Pinkel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- 4.Solinas-Toldo S, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- 5.Geigl JB, et al. Multiplex-fluorescence in situ hybridization for chromosome karyotyping. Nat. Protoc. 2006;1:1172–1184. doi: 10.1038/nprot.2006.160. [DOI] [PubMed] [Google Scholar]
- 6.Douglas EJ, et al. Array comparative genomic hybridization analysis of colorectal cancer cell lines and primary carcinomas. Cancer Res. 2004;64:4817–4825. doi: 10.1158/0008-5472.CAN-04-0328. [DOI] [PubMed] [Google Scholar]
- 7.Hurst CD, et al. High-resolution analysis of genomic copy number alterations in bladder cancer by microarray-based comparative genomic hybridization. Oncogene. 2004;23:2250–63. doi: 10.1038/sj.onc.1207260. [DOI] [PubMed] [Google Scholar]
- 8.Mulholland PJ, et al. Genomic profiling identifies discrete deletions associated with translocations in glioblastoma multiforme. Cell Cycle. 2006;5:783–791. doi: 10.4161/cc.5.7.2631. [DOI] [PubMed] [Google Scholar]
- 9.Koolen DA, et al. A novel microdeletion, del(2)(q22.3q23.3) in a mentally retarded patient, detected by array-based comparative genomic hybridization. Clin. Genet. 2004;65:429–432. doi: 10.1111/j.0009-9163.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 10.Koolen DA, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006;38:999–1001. doi: 10.1038/ng1853. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg C, et al. Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J. Med. Genet. 2006;43:180–186. doi: 10.1136/jmg.2005.032268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 13.Shaw-Smith C, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 14.Shaw-Smith C, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vissers LE, et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 17.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 18.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 20.Locke DP, et al. Large-scale variation among human and great ape genomes determined by array comparative genomic hybridization. Genome Res. 2003;13:347–357. doi: 10.1101/gr.1003303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry GH, et al. Hotspots for copy number variation in chimpanzees and humans. Proc. Natl. Acad. Sci. USA. 2006;103:8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson GM, et al. Identification by full-coverage array CGH of human DNA copy number increases relative to chimpanzee and gorilla. Genome Res. 2006;16:173–181. doi: 10.1101/gr.4456006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fiegler H, et al. Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res. 2006;16:1566–1574. doi: 10.1101/gr.5630906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichimura K, et al. Small regions of overlapping deletions on 6q26 in human astrocytic tumours identified using chromosome 6 tile path array-CGH. Oncogene. 2006;25:1261–1271. doi: 10.1038/sj.onc.1209156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishkanian AS, et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nat. Genet. 2004;36:299–303. doi: 10.1038/ng1307. [DOI] [PubMed] [Google Scholar]
- 26.Redon R, et al. Tiling path resolution mapping of constitutional 1p36 deletions by array-CGH: contiguous gene deletion or “deletion with positional effect” syndrome? J. Med. Genet. 2005;42:166–171. doi: 10.1136/jmg.2004.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snijders AM, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat. Genet. 2001;29:263–264. doi: 10.1038/ng754. [DOI] [PubMed] [Google Scholar]
- 28.Buckley PG, et al. A full-coverage, high-resolution human chromosome 22 genomic microarray for clinical and research applications. Hum. Mol. Genet. 2002;11:3221–3229. doi: 10.1093/hmg/11.25.3221. [DOI] [PubMed] [Google Scholar]
- 29.Hodgson G, et al. Genome scanning with array CGH delineates regional alterations in mouse islet carcinomas. Nat. Genet. 2001;29:459–464. doi: 10.1038/ng771. [DOI] [PubMed] [Google Scholar]
- 30.Fiegler H, et al. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer. 2003;36:361–374. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- 31.Telenius H, et al. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–725. doi: 10.1016/0888-7543(92)90147-k. [DOI] [PubMed] [Google Scholar]
- 32.Foreman PK, Davis RW. Real-time PCR-based method for assaying the purity of bacterial artificial chromosome preparations. Biotechniques. 2000;29:410–412. doi: 10.2144/00293bm01. [DOI] [PubMed] [Google Scholar]
- 33.Cai WW, et al. Genome-wide detection of chromosomal imbalances in tumors using BAC microarrays. Nat. Biotechnol. 2002;20:393–396. doi: 10.1038/nbt0402-393. [DOI] [PubMed] [Google Scholar]
- 34.Fare TL, et al. Effects of atmospheric ozone on microarray data quality. Anal. Chem. 2003;75:4672–4675. doi: 10.1021/ac034241b. [DOI] [PubMed] [Google Scholar]