

Abstract
This paper characterizes the complexes formed by a small protein, ubiquitin (UBI), and a negatively charged surfactant, sodium dodecyl sulfate (SDS), using capillary electrophoresis (CE), circular dichroism (CD), and amide hydrogendeuterium exchange (HDX; as monitored by mass spectroscopy, MS). Capillary electrophoresis of complexes of UBI and SDS, at apparent equilibrium, at concentrations of SDS ranging from sub-micellar and sub-denaturing to micellar and denaturing, revealed multiple complexes of UBI and SDS of the general composition UBI-SDSn. Examination of electrophoretic mobilities of complexes of UBI and SDS as a function of the concentration of SDS provided a new way to characterize the interaction of this protein with SDS and established key characteristics of this system: e.g., the reversibility of the formation of the complexes, their approximate chemical compositions, and the pathway of SDS binding to UBI. The work identified, in addition to SDS-saturated UBI, at least six groups of complexes of UBI with SDS, within which four groups were populated with complexes of distinct stoichiometries: UBI-SDS∼11, UBI-SDS∼25, UBI-SDS∼33, and UBI-SDS∼42. CD spectroscopy and amide HDX of the UBI-SDSn complexes suggested that many of the UBI-SDSn complexes (n > 11) have greater α-helical content than native UBI. Capillary electrophoresis provides a level of detail about interactions of proteins and SDS that has not previously been accessible, and CE is an analytical and biophysical method for studies of interactions of proteins and surfactants that is both convenient and practical. This study sheds light on the formation of the enigmatic protein-SDS complexes formed during SDS polyacrylamide gel electrophoresis and brings a new tool to the study of proteins and detergents.
Introduction
We have studied the association of a small protein, ubiquitin (UBI), with a negatively charged surfactant, sodium dodecyl sulfate (SDS), in aqueous buffers (e.g., tris-glycine, tris-borate, borate, phosphate). We chose SDS because it is a surfactant that is widely used in protein biochemistry [in SDS polyacrylamide gel electrophoresis (PAGE) and in studies of folding and unfolding]. Our analysis of mixtures of UBI and SDS by capillary electrophoresis (CE) revealed six groups of equilibrium complexes of composition UBI-SDSn. Analysis of these complexes by CE allowed us to (i) establish that they were formed reversibly, (ii) estimate (in some, exactly; in others, approximately) the number of equivalents of SDS bound in each complex, and (iii) propose a pathway for unfolding of UBI in presence of SDS. We also characterized elements of the structure of some of these complexes using circular dichroism (CD) and amide hydrogen-deuterium exchange (HDX). Structural analysis of these complexes of UBI and SDS - UBI-SDSn (where n increases approximately from 1 to 42 as the concentration of SDS increases from 0.05 to 10 mM) - by CD and HDX indicates that in some of these complexes the protein retained substantial native-like secondary structure (i.e., β-sheets and α-helices). Other complexes (e.g., those formed at denaturing levels of SDS as low as 3.0 mM) exhibited a surprisingly large amount of α-helical structure while retaining very little tertiary structure; this change in secondary structure suggests that the denaturation of ubiquitin by SDS involves substantial conversion of β-sheet to α-helical secondary structure.
Proteins and Lipids/Surfactants in Biochemistry
Complexes of proteins with lipids (and surfactants) are ubiquitous in biochemistry and are widely used in biotechnology. Examples range from structures of phospholipids bilayers with integral membrane proteins,1 to the surfactant-protein micelles present during the estimation of the molecular weight of proteins using anionic detergents (e.g., SDS-PAGE).2 Unfortunately, the technical difficulties in studying interactions of proteins and surfactants, the apparent low specificity of the binding of surfactant to proteins, and the stoichiometric and structural heterogeneity of protein-surfactant complexes have made this area of structural biology and biochemistry particularly trying to study.3 A better understanding of protein-surfactant interactions will certainly benefit from, and probably require, the development of new analytical tools and techniques (e.g., new methods of separation and analysis) in biochemistry and biotechnology.
Understanding protein-surfactant interactions might also contribute to understanding the hydrophobic effect,4,5 because protein denaturation is believed to expose hydrophobic amino acid residues that are generally buried in the hydrophobic core of the native protein. The interaction between a protein and a charged surfactant such as SDS is complex and involves both hydrophobic and electrostatic interactions.6 Hydrophobic (and other noncovalent) interactions are at the core of molecular recognition in biology. Surfactants, both individually and in aggregates, can modify the function, conformation, and activity of proteins.
Proteins and SDS
Understanding the interactions of SDS with proteins is also necessary to resolve the long-standing uncertainty surrounding the mechanism of SDS-PAGE and, perhaps, to suggest new analytical techniques based on protein-surfactant interactions. This legacy technique, which is based upon the binding of SDS to thermally and reductively denatured proteins, is arguably still one of the most widely used analytical tools in protein biochemistry.7 Its ubiquity not withstanding, the binding of SDS to proteins and the denaturation of proteins by SDS are not well understood.
During denaturing SDS-PAGE, proteins migrate as complexes with SDS (sometimes called, on the basis of little information, “SDS micelle-protein complexes”),8 roughly according to the molecular weight of the native protein; astonishingly, their mobility is largely insensitive both to amino acid sequence and to secondary, tertiary, or quaternary structure in the native conformation. The stoichiometry of SDS bound to proteins under denaturing conditions is relatively uniform: on average, 1.4 g of SDS associates with 1 gram of protein at saturation at concentrations of SDS greater than the critical micellar concentration ([SDS] > cmc).9,10 This consistent stoichiometry is the first rule of thumb for rationalizing behavior in SDS-PAGE; that is, on average, one molecule of SDS associates with two amino acid residues.11 The basis for this stoichiometry, and its apparent independence of structure of the proteins, remains a mystery.
Spectroscopic techniques (e.g., UV/visible, fluorescence, and CD spectroscopy) have been used to identify and study protein-SDS complexes that form under sub-denaturing SDS concentrations.12-17 Isothermal titration calorimetry (ITC) has also provided valuable information about the protein-SDS complexes that form under sub-denaturing concentrations of SDS.18 These techniques have approximated the stoichiometry of protein-SDS complexes and the thermodynamics of SDS binding;19-23 these methods do not directly observe distinguishable protein-SDS complexes.
We can obtain information about the stoichiometry of SDS complexes with proteins more directly by measuring the change in the electrophoretic mobility of a protein (using capillary electrophoresis) that occurs from the binding of a negatively charged SDS molecule to it. Capillary electrophoresis provides reproducible, quantitative information of a precision and resolution that cannot be achieved in gel electrophoresis.
Capillary Electrophoresis (CE)
The methods we applied here are, in principle, close to those used in affinity capillary electrophoresis24 and in surfactant capillary electrophoresis (SurfCE), a related technique that screens the conditions for the association of proteins with surfactants under non equilibrium conditions.25
CE separates molecular species according to their electrophoretic mobility (μ).26 To an (often quoted) approximation (eq 1), the electrophoretic mobility of a species is directly related to its net charge and inversely related to its hydrodynamic drag. In eq 1, Z equals the net charge of the protein or molecule, Cp and α are constants, and M is the molecular weight of the migrating species (here, protein or protein-SDS complexes).
| (1) |
We used CE to separate complexes (UBI-SDSn) having differences in their composition (e.g., different values of n). The migration and separation of different UBI-SDSn complexes depend on changes in electrophoretic mobility that result from the binding of SDS molecules (SDS is a negatively charged molecule, ZSDS ≈ -1) in a UBI-SDSn complex.
The sequential binding of SDS to a folded protein (which remains folded upon binding) is fundamentally similar to the removal of positive charges on a protein (e.g., charge ladders),27,28 at least in terms of charge and mass variations. We therefore use charge ladders as a tool with which to calibrate the number of molecules of SDS associated with UBI in UBI-SDSn.
Experimental Design
Selection of the Tris-Glycine Buffer, Its pH, and Working Temperature
We used a buffer that is typically used in SDS-PAGE (tris-glycine buffer; 25 mM tris, 192 mM glycine, pH 8.4). All experiments were carried at room temperature (22 < T < 24 °C). The results presented here, however, are not dependent upon the buffer; similar results were obtained using four different buffers (e.g., tris-borate, pH 8.1; 2-amino-2-methyl-1,3-propanediol-glycine, pH 8.7; phosphate, pH 8.4; borate, pH 8.3; see Supporting Information, Figure S1).
Selection of Ubiquitin (UBI)
Our primary motivation for using ubiquitin is that this protein exhibited a rich variety of complexes with SDS (as demonstrated in a survey using SurfCE).25 Our choice was also motivated by the extensive development of ubiquitin as a model system for studies of protein folding29 and NMR.30 Ubiquitin - as its name implies - is ubiquitous among all eukaryotic cells; its major role is to label other proteins for degradation by the ubiquitin/proteasome system (UPS) via an intracellular ATP-dependent polyubiquitination process.31
Ubiquitin (with indistinguishable structure, at least in bovine and human origins) is a polypeptide comprised of 76 amino acids; the native structure comprises one α-helix (three and one-half turns), one short 310 helix, a mixed β-sheet (comprising five β-strands), and seven reverse turns.32 Eighteen amino acid residues are involved in the α-helices (23%), 26 in the β-strands (∼34%), and 31 in coils and loops (41%) (PDB 1UBQ). Ubiquitin has no known metal binding sites, no cysteine residues, and a molecular weight of 8565 Da. Ubiquitin contains seven lysine (pKa ≈ 11.1), four arginine (pKa ≈ 12.5), six glutamic acid (pKa ≈ 4.5), five aspartic acid (pKa ≈ 4.5), one histidine (pKa ≈ 6.8), and one tyrosine residues (pKa ≈ 9.8) and is not acetylated at the methionine N-terminus (pKa ≈ 9.2). The number of positively charged residues (from the sequence) is thus 13 (α-NH3+ N-terminus included). The net charge (Z0) is estimated from the amino acid sequence to be -0.9 (Protein Calculator, http://www.scripps.edu). The measured net charge of UBI at pH 8.4 (determined by protein charge ladders and capillary electrophoresis) is Z0 = -0.2 (see Figure 4a, with ΔZ = -0.9).
Figure 4.

Superposed electropherograms of charge ladders of ubiquitin obtained respectively by acylation of ε-lysine residues with (a) acetic anhydride and (b) 4-sulfophenylisothiocyanate. Peaks marked with (#) disappear after extensive dialysis. The buffer is tris-glycine, pH 8.4. The inset in panel a corresponds to the plot of the electrophoretic mobilities of the rungs of the acetyl charge ladder of UBI as a function of acetylations n and allows the determination of the net charge of native UBI as a function of charge increment upon acylation, ΔZ. (c) Ruler 1 correlates experimental mobilities μn of the rungs of a UBI charge ladder to the number of acylations n. (d) Ruler 2 correlates the corrected mobilities (μUBI-SDSn, eq 4) of rungs of charge ladders to the number of acylations n. This second ruler permits the determination of the stoichiometry of UBI-SDSn complexes in panel e. Steps in gray in Ruler 2 were extrapolated from steps in black. (e) Superposition of CE electropherograms shown in Figure 1. Labels in circles indicate the stoichiometries of UBI-SDSn complexes within G1*, G2, and G3* according to Ruler 2.
Choice of SDS
SDS is the surfactant most used in studies of protein denaturation. The critical micelle concentration (cmc) of SDS in the tris-glycine buffer used here is 3.4 mM, as determined by CE (see the Supporting Information, Figures S2, S3).
The negative charge of SDS allows us to monitor its binding to UBI easily by CE: each bound SDS molecule will increase the negative charge of the resulting complex by ΔZ (which we assume to be indistinguishable from the charge increment in charge ladders). SDS is also transparent at the UV wavelength of the CE detector (214 nm); there is thus essentially no background in CE due to SDS, and the protein - in all of its conformations - can be studied without the requirement for an attached chromophore. This capability to observe the protein spectroscopically without interference from other species in the system is an enormous experimental convenience.
Quantitative Secondary Structure of UBI-SDSn Complexes by Circular Dichroism (CD)
We used CD to study the secondary structure of complexes of UBI with SDS. After their formation by equilibrium dialysis, we examined UBI-SDSn complexes in the range 205-260 nm (at a total protein concentration of ∼50 μM). The CD spectrum is a sensitive measure of the secondary structure of proteins (λ < 260 nm).33 We quantified the secondary structure of UBI-SDSn by using three independent programs for deconvolution (e.g., CDSSTR, SELCON3, and CONTINLL).33 These programs reconstruct the CD spectrum of a protein (here a UBI-SDS complex) with a linear combination of individual k’s (where k represents contributions from elements of secondary structure). Fractions of α-helices, β-sheets, turns, and random coils are the outputs from these three programs (fk, in %; see Figure 2b).
Figure 2.

(a) Circular dichroism spectra of an equilibrium mixture of UBI-SDSn obtained by unfolding native UBI by dialysis against tris-glycine buffers containing concentrations of SDS from 0 to 10 mM. (b) Fractions (fk) of individual secondary structure component k contributing to the CD spectra of complexes of UBI and SDS [where k represents respectively α-helices (●), β-strands (■), turns (○), and random coils (+) secondary structure]. Values were averaged from the output results from the secondary structure estimation of three programs (CDSSTR, SELCON3, and CONTINLL; see Supporting Information for details). (c) Kinetics of hydrogen-deuterium exchange for complexes of UBI and SDS with up to 3.0 mM SDS. Hydrogen exchange is expressed in terms of the total number of hydrogens that are protected from exchange, determined from the average of six molecular ions at [SDS] = 0.0 (□), 0.4 (●), 1.4 (○), and 3.0 mM (■); hydrogen exchange was not measured above 3.0 mM SDS due to the quenching of electrospray ionization by the nonvolatile SDS.
Structure of UBI-SDSn Complexes by Amide Hydrogen-Deuterium Exchange (HDX) in SDS, Monitored by Electrospray Ionization Mass Spectroscopy (ESI-MS)
We also examined the structure of ubiquitin at various concentrations of SDS by measuring the rate of amide HDX using mass spectrometry.34 This rate is a sensitive method for detecting minor changes in the secondary and tertiary structured of proteins that arise from changes in hydrogen bonding or hydrophobic interactions (i.e., during thermal or chemical unfolding, ligand binding, or as a result of amino acid substitution).35
Charge Ladders: A Method for Evaluating the Stoichiometry of UBI-SDSnComplexes
A protein charge ladder is a set of protein derivatives obtained by the conversion of charged residues (typically lysine ε-NH3+ and N-terminus α-NH3+) into electrically neutral (α- and ε-NHCOCH3) or negatively charged (α- and ε-NHCO-R-X-) residues (R is an organic spacer). Charge ladders make it possible to measure experimentally the change in mobility Δμ associated with the chemical removal of positive charge on UBI (either by acetylation or by acylation with reagents with negatively charged sulfonate groups) and to compare them to the values of Δμ obtained on the addition of a negative charge (by association with SDS). The mobility μn of the rung n of a protein charge ladder (here a UBI charge ladder) can be expressed as a function of the net charge of the unacylated protein (Z0), its molecular weight (MUBI), the number of acylated groups (n), the change in charge (ΔZ) resulting from acylation, the added mass (MX), and Cψ,n a correction factor accounting for nonlinearities in surface potentials associated with each acylation (eq 2; more details in the Supporting Information).
| (2) |
The value of ΔZ observed on adding one unit of charge is less than one unit, due to so-called charge regulation (which is an adjustment of the extent of protonation of ionizable residues on the protein as a response to a change in electrostatic potential).36,37 One plausible origin of the difference between the theoretical value of ΔZ = -1.0, expected on changing the charge of a protein by one unit of charge (e.g., by converting Lys-ε-NH3+ into Lys-ε-NHCOCH3), and the inferred value of ΔZ = -0.9 (as established using charge ladders of BCAII)37 is a small change in local pH, or a shift in the pKa, of some ionizable residues (the two are equivalent analytically). The effect of charge regulation would be suppressed (e.g., ΔZ ≈ -1) if pKa values were significantly different from the working pH (at least, different by more than three pH units), according to the “charge-regulation model” proposed by Menon and Zydney36 and applied to BCAII.37 The distribution of values of pKa in a protein is such that this condition cannot, in practice, be met. Rather than working with an absolute relationship between ΔZ and Δμ, in this work we use relationships calibrated empirically using charge ladders of UBI.
The values of mobility (μn) of each rung of the charge ladder cannot be used directly (e.g., superposed) to determine the stoichiometry n for a species with composition UBI-SDSn which has an equivalent electrophoretic mobility: while we expect the association of SDS with UBI, and the neutralization of Lys-ε-NH3+ and N-terminal-NH3+ via acetylation, to increase the net negative charge by an indistinguishable amount, the molecular weight (and hydrodynamic drag) of two equally charged species (here UBI-SDSn and the nth-rung of a UBI charge ladder) are significantly different (dodecyl sulfate, DS-, has a molecular weight of MDS- = 265.4 Da; acetylation of lysine by acetic anhydride increases the mass by MX = 42.0 Da; chemical modification of lysine by 4-sulfophenylisothiocyanate increases the mass by MX = 214.2 Da).38 For expressing μUBI-SDSn (e.g., the mobility of a UBI-SDSn complex; eq 3) as a function of μn, we replaced Z0 + nΔZ in eq 3 by its expression in eq 2, assuming that the binding of a SDS molecule is undistinguishable electrostatically from acylation of Lys-ε-NH3+). These manipulations resulted in eq 4.
| (3) |
| (4) |
Equation 4 produces predicted values of μUBI-SDSn and describes complexes in which ubiquitin bears both additional negative charges and additional mass. We assume that the nonlinear dependence of the surface potential, Cψ,n, is indistinguishable for equally charged species in UBI-SDSn and UBI charge ladders. We also assume, in the absence of other information, that α = 2/3.39 Charge regulation is empirically included in this analysis, as we use experimental values of the mobilities of the rungs of UBI charge ladder where ΔZ ≈ 0.9 is assumed to be the same for both UBI charge ladders and UBI-SDSn.
For increasing numbers of associated SDS molecules (or for higher rungs of the charge ladder), however, ubiquitin becomes highly negatively charged, which might, in principle, affect the folding of UBI. We neglect this effect in light of the work by Makhatadze demonstrating that the neutralization of all arginines and lysines on the surface of UBI left the protein folded.40
Analysis of the Thermodynamics of Formation of UBI-SDSn by CE
We quantified the stoichiometry n in UBI-SDSn complexes by assuming the association of UBI and SDS to be at equilibrium, and by considering eqs 5 and 6.
| (5) |
| (6) |
The linear least-squares fit regression of log([UBI-SDSn]/[UBI]) as a function of log([SDS]) yields both the stoichiometry n and the binding constant Kn for the transition; [SDS] is exactly known if the protein is equilibrated with SDS by dialysis. The ratio [UBI-SDSn]/[UBI] can be determined by measuring the relative concentrations of UBI and of UBI-SDSn at a given value of [SDS] using the UV/visible detector in CE.
In order to treat the formation of UBI-SDSn complexes as reactions that have reached equilibrium, we first proved the reversibility of their formation. For this proof, we dialyzed native UBI (N) and fully denatured UBI (D) for 170 h against buffers containing SDS in the range between 0 and 10 mM (the time required for UBI denatured in 10 mM SDS - that is, D - to refold into a species having a mobility indistinguishable from that of the native protein under these dialysis conditions is 170 h). By generating a particular UBI-SDSn complex, at a specific concentration of SDS, in two different ways (i.e., starting from N or starting from D) and then characterizing these proteins (with CE, CD, and HDX), we established that the formation of UBI-SDSn was reversible (see Figure 3 for CE data, and Supporting Information for CD and HDX data, Figure S4).
Figure 3.

CE traces demonstrating reversibility and equilibrium. Electro-pherograms of UBI-SDSn complexes obtained after dialyzing native (N) and SDS-denatured UBI (D, incubated in 10 mM of SDS in tris-glycine buffer for 170 h prior refolding) against tris-glycine buffer containing different concentrations of SDS over 170 h. “Unfolding” experiments correspond to the dialysis of native UBI against higher SDS concentration, and “Refolding” experiments correspond to dialysis of unfolded/denatured UBI (initially in 10 mM SDS) against lower concentrations of SDS. The peak marked (#) was an artifact. Electropherograms were mirrored across the x-axis to facilitate comparison.
Results and Discussion
Capillary Electrophoresis Detects Multiple Complexes of UBI-SDSn with Distinct Electrophoretic Mobilities after Equilibrium Dialysis against Concentrations of SDS Ranging from 0 mM to Greater than the Critical Micelle Concentration (cmc)
Analysis of UBI dialyzed against buffered SDS by CE allowed us to observe distinct complexes of UBI and SDS, with electrophoretic mobilities (μ) that are intermediate between the value of mobility for native UBI in the absence of SDS (UBI in 0.0 mM SDS, denoted N; μ = 0.3 cm2 kV-1 min-1) and fully denatured41 UBI (UBI in 10 mM SDS, denoted D; μ = 22.6 cm2 kV-1 min-1) (Figure 1).42 We classified these intermediate complexes subjectively (based on the appearance and shape of peaks) into six groups of complexes which we denote G1*, G2, G3*, G4, G5, and G6* in addition to N (native UBI) and D (SDS-saturated UBI).
Figure 1.

(a) Electropherograms of UBI-SDSn complexes obtained after dialyzing native ubiquitin (UBI) against tris-glycine (TG) containing concentrations of SDS (from 0 to 10 mM) for 170 h. The total concentration of UBI and UBI-SDSn complexes in the injected plug is 50 μM. Capillary electrophoresis (CE) was performed with a “short” capillary (total length of 60.2 cm; length from injection to the detector of 50.0 cm), with an applied voltage of 30 kV. Each of the electropherograms is labeled with the concentration of SDS. The critical micelle concentration (cmc) of SDS in this medium is 3.4 mM, as determined by CE using 2-hydroxymethylenenaphthalene (plain bracket; inferred from the results in Figure S3). The cmc is also inferred by observing the formation of a shoulder on the neutral marker peak due to an impurity, either in the presence or in the absence of proteins (dotted bracket; inferred from the results in Figure 1e (■) and Figure S3). Native UBI (e.g., N) has a net charge close to zero; the peak for N is therefore superimposed on the neutral marker DMF. The peak marked (#) was not reproducible. Dotted vertical lines represent the electrophoretic boundaries we used to define each group (G) of UBI-SDS complexes (including N and D). We used these boundaries in the thermodynamic analysis. (b,c) Electropherograms in specific regions of interest (with the “short” capillary). (d) High-resolution electropherograms of UBI-SDS complexes obtained at concentrations of SDS ranging from 2.6 to 10 mM and analyzed with a “long” capillary (total length of 110.0 cm, length to the detector of 100.0 cm).
The observation of a number of stable (over the time required for a CE experiment) complexes of UBI with SDS establishes a multistep pathway for the unfolding of UBI in SDS. The appearance and disappearance of discrete peaks (e.g., N, G2, G4,G5, and D) as the concentration of SDS increases demonstrates the formation of UBI-SDSn species with distinct composition.
Increase in Mobility of UBI-SDSn in G1* Proceeds through Multiple Overlapping Peaks until the First Discrete Complex (G2) Forms at [SDS] = 0.8-1.4 mM
The association of a small number of SDS molecules with UBI takes place at concentrations of SDS as low as 0.4 mM. We interpret the conversion of the peak at μ ≈ 0cm2 kV-1 cm-1, N, to broad peaks spanning 1-14 cm2 kV-1 cm-1 to represent the formation of a range of different species of UBI-SDSn. These various species, G1*, all convert to a well-defined, discrete intermediate, G2, at [SDS] = 1.0 mM. G2 is a stable species at concentrations of SDS from 0.8 to 1.4 mM, beyond which the association of a discrete number of SDS molecules converts it into G3* and G4.
A Second Discrete Group of Intermediates (G4) Dominates at [SDS] = 2.0-3.0 mM
In this region, a second discrete peak appears at μ ≈ 20 cm2 kV-1 cm-1 (e.g., G4). The transition from G2 to G4 proceeds via G3* (Figure 1c). This transition also involves an additional complex observed as a shoulder on the G2 peak at 1.7 mM SDS (μ ≈ 16 cm2 kV-1 min-1; this shoulder is not observed at any other SDS concentration) (Figure 1c). The presence of this shoulder indicates that there is at least one additional species on the path from G2 to G4 (differing, we estimate later using charge ladders, by Δn ≈ 1). The mobilities of both major peaks in G2 and G4 become higher with increasing concentrations of SDS (Figure 1b,d); this behavior suggests the presence of species in which the number of associated SDS increases with the concentration of SDS but does not result in a transition to another distinct structure (e.g., another major unfolding or restructuring event).
A Third Discrete Group of Complexes (G5) Dominates at [SDS] = 3.8 mM
The electropherograms in Figure 1d show a discrete transition from G4 to G5:G (μ ≈ 20.6 cm2 kV-1 min-1) converts to G5, a group of complexes with higher mobility: μ ≈ 21.5 cm2 kV-1 min-1. The transitions generating D from G5 (occurring in the group G6*) are less distinct (although still analyzable) than the transitions from G2 to G4 or G4 to G5. These peaks also undergo a distinct transition in shape: from triangular and sloping to the left (G4 and G5 are “right-handed” at 2.0 < [SDS] < 3.8 mM) to a peak sloping to the right (D is “left-handed” at [SDS] > 7.0 mM; we discuss these asymmetries in peak shape in terms of electrodispersion in the Supporting Information, Figure S7). Since the micellar phase begins to appear significantly at [SDS] > 3.4mM, those transitions - observed here above the cmc - suggest that SDS continues to associate with UBI-SDSn, while the literature generally states that, beyond the cmc, no further unfolding occurs and excess surfactant simply leads to further formation of micelles.11
Structure of UBI-SDSn Complexes on the Path from N to D by Circular Dichroism
As the concentration of SDS increases, the average ellipticity generally increases for UBI-SDS complexes (Figure 2a). As estimated from the deconvolution of CD spectra, the UBI-SDSn complexes in groups G1* and G2 ([SDS] < 1.4 mM) have secondary structure that is indistinguishable from native UBI (Figure 2b). For concentrations of SDS higher than 1.5 mM, we observe an approximately 2-fold increase in α-helical structure and a 2-fold decrease in β-strand structure (we infer a maximal content of α-helical secondary structure at [SDS] = 2.6 mM).
The overall structural picture that emerges from CD spectroscopy is that, as the concentration of SDS increases, UBI loses β-strands (and possibly loop structures) in favor of α-helices, with the maximum amount of α-helical structure observed at 2.6 mM SDS (G4). This increase is consistent with previous investigations into the structural effects of SDS on binding to proteins.8
While a small (∼1.2-fold) decrease in ellipticity is observed in the conversion of G4 to G5 (e.g., 2.6 < [SDS] < 3.5 mM), no significant variation in secondary structure was detected for the transition from G5 to D; this observation is in agreement with previous literature reporting that no further unfolding is observed above the cmc.11 This absence of variation in structure in the micellar regime could be due to a lack of sensitivity of CD to this transition, or perhaps because the secondary structure does not change once SDS micelles form.
Structure of UBI-SDSn Complexes on the Path from N to G4, Examined by Hydrogen-Deuterium Exchange (HDX)
Mass spectrometric results revealed that the binding of SDS to UBI decreases the rate of amide HDX as the concentration of SDS increased up to 1.4 mM (Figure 2c). The number of unexchanged or “protected” hydrogens therefore increased along the pathway from N to G2 (the increase in protected hydrogens is approximately linear with respect to [SDS], see Figure S4d). For example, under completely native conditions (e.g., [SDS] = 0 mM), the ubiquitin polypeptide exchanges ∼50 amide hydrogen very quickly (i.e., t < 10 min) and retains ∼10 protected hydrogens at t = 30 min. These protected hydrogens remained unexchanged with solvent throughout the course of the experiment (60 min). At higher concentrations of SDS (1.4 mM), the UBI polypeptide undergoes slower amide hydrogen exchange than at [SDS] = 0 mM: ∼21 hydrogens are protected for [SDS] = 1.4 mM. We speculate that the increase in protection on going from [SDS] = 0 to 1.4 mM is due to electrostatic interactions between the negative charge on SDS and OH-.43
HDX has been previously used in combination with CD spectroscopy to demonstrate that proteins (in the form of molten globules or partially folded states)44 can possess substantial secondary structure (as measured by CD spectroscopy) while also lacking tertiary structure (as indicated by a lack of protection from H/D exchange). Molten globule states are generally observed at partially denaturing conditions (e.g., mild denaturants or low pH) and are characterized by having minimal tertiary structure (e.g., fast HDX rates) and substantial, native-like secondary structure (e.g., a compact, partially folded state).45 The presence of secondary structure at 3.0 mM in conjunction with the rapid HDX kinetics makes it is tempting to hypothesize that UBI is populating a molten globule state at 3.0 mM SDS. The CD spectrum of UBI at 3.0 mM SDS, however, is different than the spectrum of native, SDS-free UBI; UBI could thus be considered a molten globule, but not a molten globule with native-like secondary structure.45
Formation of UBI-SDSn Complexes Is Reversible According to Capillary Electrophoresis, Circular Dichroism, and Amide Hydrogen-Deuterium Exchange
We investigated the reversibility of formation of UBI-SDSn with CE (Figure 3), CD spectroscopy (Figure S4a), and HDX measured by electrospray ionization MS (Figure S4b-d). In each experiment, the native (N) or denatured (D) UBI was dialyzed against sub-denaturing concentrations of SDS, so that the UBI-SDS complex was formed from native UBI (i.e., formed by unfolding UBI) or from denatured UBI (i.e., formed by refolding UBI).46 Figure 3 shows the similarity of the electropherograms for UBI-SDSn mixtures at various concentrations of SDS. To facilitate a comparison of the mixtures of UBI-SDSn formed from native and denatured UBI, the electropherograms from both the unfolding and refolding experiments are shown at each concentration of SDS, mirrored across the x-axis. The results demonstrate that the UBI-SDSn complexes produced from either refolding or unfolding have similar electrophoretic mobilities at each concentration of SDS. While they are not identical, the similarities in the mirrored electropherograms indicate that the association and dissociation of SDS and UBI are reversible under these conditions (0-10 mM SDS; 170 h of dialysis).
Determination of the Stoichiometry of UBI-SDSn Complexes in G1* and G2 by Comparing Electropherograms of UBI-SDSn with UBI Charge Ladders
A lysine-ε-acetyl and N-terminal-α-acetyl protein charge ladder of UBI, prepared by allowing UBI to react with acetic anhydride, provides a useful “charge ruler” for correlating the electrophoretic mobility to the net negative charge on UBI (Figure 4a). We used the acetyl charge ladder to estimate the charge (and thus the composition) of UBI-SDSn complexes having less than eight added charges. To extend the range of negative charges beyond eight, we also prepared a charge ladder derivatized with 4-sulfophenylisothiocyanate (Figure 4b). Modification of reactive amino groups with this compound results in a change in charge of ∼ -2 (ignoring the effect of charge regulation) per acylation, making it possible to determine the stoichiometry of UBI-SDSn complexes associated with up to 14 SDS.
The first ruler (Figure 4c, Ruler 1) correlates the experimental values of mobility, μn, of rungs of UBI charge ladders with the number of acylations, n, for each rung of the various charge ladders. This ruler includes a total of 12 points (i.e., from 0-8, 10, 12, 14).47 The second ruler (Figure 4d, Ruler 2) represents the values of mobility μn that we estimated after using eq 4 to take into account the change in mass associated with the binding of n SDS molecules. This ruler can be used to determine the number of SDS molecules associated with UBI-SDSn complexes within G1* and G2 (assuming Cp and α are the same for all the protein-derived species to which we apply this equation; Figure 4e).
The second ruler shows that the conversion of N to G2 (via G1*) involves the stepwise binding of ∼11 SDS molecules to N with no significant changes of secondary structure (according to CD). The estimation of 11 SDS molecules associated with UBI in G2 is also in good agreement with previous work, stating that several proteins (i.e., lysozyme, ovalbumin)10 associate with SDS with the stoichimetry of 0.4 g of SDS per gram of protein at sub-denaturing concentrations of SDS (e.g., [SDS] < cmc).48 One hypothesis to rationalize the results is that the cationic residues nucleate the condensation of SDS in the transition from N to G2, yielding UBI-SDS∼11 (e.g., UBI contains 13 cationic sites, e.g. 7 lysines, 4 arginines, 1 histidine, and 1 unacetylated N-terminus; the extent of protonation of the single histidine and of the N-terminus at pH 8.4 is lower than that of arginine and lysine, and Lys27 is involved in a salt bridge with Asp52;49 these details might explain why fewer than 13 SDS molecules bind in this first transition).
If the electrostatic binding of SDS to cationic residues is the primary interaction between proteins and SDS at very low concentrations (i.e., the G2 state of UBI), then such electrostatic interactions might help to rationalize the retention of substantial native structure in UBI upon the binding of these first 11 SDS. This interaction would, in effect, neutralize the charge contributed by each lysine group; we know from the work of Makhatadze that the neutralization of lysine and arginine residues in UBI does not change its native structure.40
Determination of the Number of SDS Molecules Involved in the Transitions from G2 to G4 and in the Transition from G4 to G5 by Equilibrium Analysis
The equilibrium analysis aims at determining the stoichiometry of Gi (e.g., i = 4 for G4 and i = 5 for G5) by determining the number of SDS molecules involved in the transitions from Gi to Gj, e.g., Δni→j; the stoichiometry of G2 (UBI-SDS∼11) is already known from the charge ladder analysis. For this purpose, we first determined the relative concentrations of UBI-SDS complexes within each group (e.g., [G2], [G4], and [G5]) by integrating the peaks for each group in Figure 1.50 Second, we calculated log([Gj]/[Gi]) (that is, log([G4]/[G2]) for G2→G4, and log([G5]/[G4]) for G4→G5) for each concentration of SDS and plotted these values as a function of log([SDS]). Third, we generated a linear least-squares fit of log([Gj]/[Gi])= Δni→j log([SDS]) + log(Ki→j); this fit made it possible to estimate the slope, Δni→j, and the y-intercept, log(Ki→j). These numbers are, respectively, the number of SDS molecules that bind Gi to form Gj and the logarithm of the equilibrium constant for the transition from Gi to Gj (Figure 5).
Figure 5.
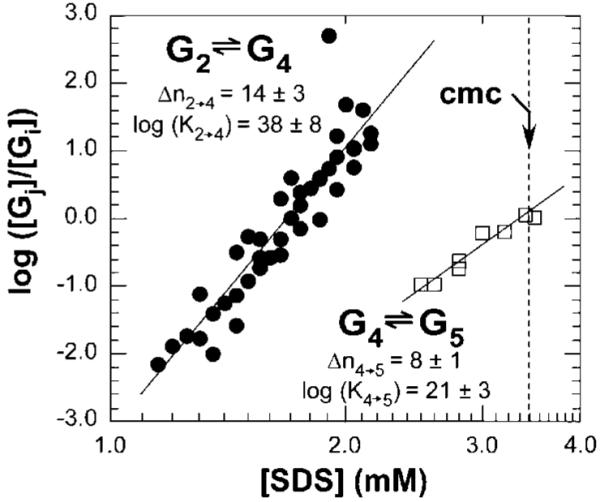
Plot of log([G4]/[G2]) and log([G5]/[G4]) vs log [SDS] for four sets of experiments for the transition G2→G4 (●) and two sets for the transition G4→G5 (□), where [G2], [G4], and [G5] are the relative concentrations of all UBI-SDSn complexes within their respective groups. Values Δni→j correspond to the number of SDS molecules involved for the transition from Gi to Gj (i and j represent the label of the group). For example, in the transition from G2 to G4, the value Δni→j corresponds to the number of SDS that bind G2 to yield G4, and Ki→j refers to the thermodynamic equilibrium constant for this transition (Ki→j has units of (mol/L)-Δni→j). Relative concentrations were determined by integrating electropherograms in the mobility range attributed to each group (integration was performed as a function of time, t). We fitted the data to the linearized expressions of the law of mass action described in eqs 5 and 6. The errors ((±) correspond to half of the amplitude between the highest and lowest values of the slope and y-intercept fitting the data points.
Determination of the Stoichiometry of G4 and G5 by Extrapolation of Mobilities of 4-Sulfophenylisothiocyanate Charge Ladders in the Range of Mobility Defining G4, G5, and D
Data for μUBI-SDSn(○, obtained using eq 4) and D (□, n = 42)51 were plotted versus n and fitted with a log-normal algorithm. The fitted curve was used to correlate the mobilities of complexes of UBI and SDS within each group to their stoichiometry n (Figure 6a). Interestingly, the stoichiometry of G4 (respectively G5) obtained from this independent analysis based on charge ladders agrees very well with the values from equilibrium analysis (Figure 6b).
Figure 6.

Extrapolation of mobilities of charge ladders for the determination of the stoichiometry n of UBI-SDSn complexes within each group of complexes. (a) The electrophoretic mobilities of the rungs of the 4-sulfophenylisothiocyanate charge ladder (μn) corrected by the factor defined in eq 4 are represented by (○) and are plotted versus n (e.g., the number of SDS molecules bound to UBI). The SDS-saturated UBI (□, 0) is represented in this plot at n = 42.51 Data points (○) and (□) were fitted from n = 2 to n = 42 with a log-normal algorithm (e.g., μ(n) = μ0 + A exp(-(ln(n/n0)/k)2), with μ0 = 25.0, A = -21.4, n0 = 1.25, and k = 2.39; β2 = 0.16). The result of the fit is represented by the plain line. Each group of UBI-SDSn complexes, within its mobility range, was highlighted in gray and the corresponding number of SDS was included on the plot (indicated by arrows). (b) Summary of the stoichiometries of UBI-SDSn complexes within each group according to the extrapolated charge ladder (in parentheses: number of SDS molecules obtained by the equilibrium analysis described in Figure 5).
The conversion of G2 to G4 (via G3*) involves the binding of ∼14 additional SDS to G2 (G4 is UBI-SDS∼25). The binding of these 14 SDS molecules to UBI-SDS∼11 causes a major change in the secondary structure (e.g., a 2-fold increase in ellipticity at 220 nm) and in the tertiary structure (e.g., faster rates of HDX) of UBI. We therefore conclude that the binding of a total of ∼25 SDS molecules to UBI is sufficient to “denature” the UBI polypeptide per se (at 25 °C, pH 8.4) into G4, perhaps a non-native molten globule state. The conversion from G4 to G5 occurs at concentration of SDS close to the cmc and was shown to involve the binding of ∼8 SDS to G4 according to the equilibrium analysis (respectively ∼9 SDS according to the charge ladder analysis).
Above the cmc (3.4 mM), the thermodynamic activity of SDS present in monomeric form is constant. Nonetheless, as [SDS] increases beyond the cmc, more molecules of SDS bind to UBI as we see a subsequent transition from G5 to D (via G6*) involving ∼9 molecules of SDS, based on the use of charge ladders to calibrate the stoichiometry of complexes within G5 and G6*. These last SDS molecules appear to bind to an unfolded (but still structured), non-native polypeptide, and the binding is accompanied by no significant conformational changes (as measured by CD).
Conclusions
Capillary electrophoresis, coupled with equilibrium dialysis and charge ladders, was used to characterize a multistep process for the denaturation of ubiquitin by SDS (Figure 7). The data show the formation of six distinguishable groups of complexes in the presence of sub-denaturing to denaturing concentrations of SDS, four of which have approximately defined compositions (e.g., G2, G4, G5, and D). Three of these UBI-SDSn complexes (G2, G4, and G5) have stoichiometries that involve smaller numbers of SDS molecules than required to saturate the protein (e.g., ∼42 SDS, as estimated from the “1.4:1 rule of thumb” applied to UBI).
Figure 7.

Pictures describing the hypothetical structure of UBI-SDSn complexes within groups N, G2,G4,G5, and D. The 11 cationic sites (e.g., 7 lysines and 4 arginines; we consider the unique histidine and the amino N-terminus to be uncharged at pH 8.4) are represented by positive charges along the protein backbone. N represents the native UBI (PDB 1UBQ; one turn of the α-helix contains four amino acids). G2 is a native-like UBI-SDS complex (as revealed by CD, Figures 2a,b and S4a) associated with 11 bound SDS molecules (as determined from the charge ladder analysis, Figure 4e). G4 is less organized (perhaps a molten globule, Figure 2c), obtained after the binding of ∼14 SDS to G2 (cf. Figure 6b) with the conversion of about half of the β-stands into α-helices (cf. Figure 2b). Transitions from G4 to G5 and from G5 to D respectively involve ∼8 and ∼9 molecules of SDS (cf. Figures 5 and 6b) with no major change in secondary structure as revealed by CD; however, D might be best represented by an unfolded, SDS-saturated polypeptide with extensive α-helical structure, represented in this cartoon by the disruption of the remaining β-strand in G5 (*).
Some of the complexes (UBI-SDS1, UBI-SDS2, ..., UBI-SDS10, UBI-SDS11) have native-like structure; others (UBI-SDS∼25) appear to have a more loosely organized structure (perhaps a molten-globule); still others (UBI-SDSn>25) appear to be completely non-native but still have more α-helical structure than the SDS-free, native protein. None of the complexes appeared to be “unstructured” (e.g., none lacked secondary structure). More importantly, the pathway described in Figure 7 is an oversimplication of what CE is able to detect (“unresolved” groups-e.g., G1*, G3*, G6* - and the shoulder on the G2 peak at 1.7 mM of SDS are not included in this unfolding pathway; we do not know with precision what those complexes are).
While we initially set out to answer the question of why most proteins show the same ratio of binding (about one SDS bound for every two amino acids at [SDS] > 10 mM), we found that the equilibrium pathway of SDS binding can be complicated (for example here with UBI), and we know from SurfCE25 that it can be significantly different among proteins at sub-denaturing levels of SDS. There must be, therefore, at least two regimes of binding: a sub-micellar regime where proteins show protein-specific pathways of interaction with SDS, and a micellar regime where, to the precision revealed in SDS-PAGE, proteins bind SDS independently of their primary, secondary, and tertiary structures.
The total number of solvent-accessible positive charges may determine (at least in part) the pathway of binding of SDS to proteins and the composition of intermediates. This type of electrostatic correlation could explain the observation that proteins with a higher content of β-sheets are more resistant to denaturation by SDS than proteins with a higher content of α-helices.52 Several studies that report the thermodynamic β-sheet and α-helical propensities for all 20 amino acids have shown that (i) positively charged residues stabilize α-helical structures (to unfolding by urea) more so than negatively charged and most uncharged residues53,54 and (ii) uncharged and bulky hydrophobic residues (W, Y, F, and I) stabilize β-sheet structures more than positively charged residues.55
Capillary electrophoresis has the potential to help to answer questions concerning the binding of proteins and SDS (and other charged surfactants, lipids, or other molecules) while providing a level of detail not accessible from other techniques such as 1D or 2D SDS-PAGE, circular dichroism, ITC, or fluorescence binding assays.
The study reported here suggests answers (or perhaps, more accurately, hypotheses) describing the interaction of UBI, a representative small protein, with SDS. It also demonstrates that CE is a powerful method to monitor the interaction of proteins with small charged molecules. We believe, but cannot yet prove, that the accessibility of charged residues on proteins, combined with secondary structure (α-helical vs β-sheet content) and conformational flexibility, strongly influences the sequence of intermediates formed on binding of SDS to proteins.
Acknowledgment
This research was funded by NIH award GM 051559. G.F.S. is supported by a “Lavoisier Générale” postdoctoral fellowship (Ministère des Affaires Etrangères Français). B.F.S. is supported by a NIH Ruth L. Kirchstein NRSA postdoctoral fellowship. E.C. is supported by a visiting scholar fellowship (Fundação de Amparo à Pesquisa doEstado de São Paulo-FAPESP). The authors thank Steve P. Gygi for use of an electrospray ionization mass spectrometer. The authors also thank Phil Snyder and Demetri Moustakas for helpful discussions.
Footnotes
Supporting Information Available: Chemicals used; experimental protocols; electropherograms in buffers other than tris-glycine; isothermal titration calorimetry; conductivity and CE data for the determination of the cmc of SDS in tris-glycine; reversibility experiments (CD, HDX); SDS-PAGE analysis of UBI; effect of electrodispersion on peak shapes; CE, CD, and HDX procedures; protocols for the acylation of UBI; analysis of the purity of UBI by reverse-phase high-pressure liquid chromatography; and baseline electropherograms. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Weber K, Osborn M. J. Biol. Chem. 1969;244:4406–4412. [PubMed] [Google Scholar]
- (3).La Mesa C. J. Colloid Interface Sci. 2005;286:148–157. doi: 10.1016/j.jcis.2004.12.038. [DOI] [PubMed] [Google Scholar]
- (4).Dill KA, Truskett TM, Vlachy V, Hribar-Lee B. Annu. Rev. Biophys. Biomol. Struct. 2005;34:173–199. doi: 10.1146/annurev.biophys.34.040204.144517. [DOI] [PubMed] [Google Scholar]
- (5).Southall NT, Dill KA, Haymet ADJ. J. Phys. Chem. B. 2002;106:521–533. [Google Scholar]
- (6).Gudiksen KL, Gitlin I, Moustakas DT, Whitesides GM. Biophys. J. 2006;91:298–310. doi: 10.1529/biophysj.106.081547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Righetti PG. J. Chromatogr. A. 2005;1079:24–40. doi: 10.1016/j.chroma.2005.01.018. [DOI] [PubMed] [Google Scholar]
- (8).Parker W, Song PS. Biophys. J. 1992;61:1435–1439. doi: 10.1016/S0006-3495(92)81949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pitt-Rivers R, Impiombato FSA. Biochem. J. 1968;109:825–830. doi: 10.1042/bj1090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Reynolds JA, Tanford C. Proc. Natl. Acad. Sci. U.S.A. 1970;66:1002–1007. doi: 10.1073/pnas.66.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jones MN. Chem. Soc. Rev. 1992;21:127–136. [Google Scholar]
- (12).Ferreon ACM, Deniz AA. Biochemistry. 2007;46:4499–4509. doi: 10.1021/bi602461y. [DOI] [PubMed] [Google Scholar]
- (13).Maestro B, Sanz JM. FEBS Lett. 2007;581:375–381. doi: 10.1016/j.febslet.2006.12.042. [DOI] [PubMed] [Google Scholar]
- (14).Miksovska J, Yom J, Diamond B, Larsen RW. Biomacromolecules. 2006;7:476–482. doi: 10.1021/bm0506703. [DOI] [PubMed] [Google Scholar]
- (15).Moosavi-Movahedi AA, Gharanfoli M, Nazari K, Shamsipur M, Chamani J, Hemmateenejad B, Alavi M, Shokrollahi A, Habibi-Rezaei M, Sorenson C, Sheibani N. Colloids Surf., B. 2005;43:150–157. doi: 10.1016/j.colsurfb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- (16).Naeem A, Khan RH. Int. J. Biochem. Cell Biol. 2004;36:2281–2292. doi: 10.1016/j.biocel.2004.04.023. [DOI] [PubMed] [Google Scholar]
- (17).Otzen DE, Oliveberg M. J. Mol. Biol. 2002;315:1231–1240. doi: 10.1006/jmbi.2001.5300. [DOI] [PubMed] [Google Scholar]
- (18).Lad MD, Ledger VM, Briggs B, Green RJ, Frazier RA. Langmuir. 2003;19:5098–5103. [Google Scholar]
- (19).Andersen KK, Westh P, Otzen DE. Langmuir. 2008;24:399–407. doi: 10.1021/la702890y. [DOI] [PubMed] [Google Scholar]
- (20).Nielsen AD, Arleth L, Westh P. Biochim. Biophys. Acta. 2005;1752:124–132. doi: 10.1016/j.bbapap.2005.08.001. [DOI] [PubMed] [Google Scholar]
- (21).Nielsen AD, Arleth L, Westh P. Langmuir. 2005;21:4299–4307. doi: 10.1021/la047299+. [DOI] [PubMed] [Google Scholar]
- (22).Nielsen MM, Andersen KK, Westh P, Otzen DE. Biophys. J. 2007;92:3674–3685. doi: 10.1529/biophysj.106.101238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Nielsen AD, Borch K, Westh P. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2000;1479:321–331. doi: 10.1016/s0167-4838(00)00012-1. [DOI] [PubMed] [Google Scholar]
- (24).Chu YH, Avila LZ, Gao JM, Whitesides GM. Acc. Chem. Res. 1995;28:461–468. [Google Scholar]
- (25).Gudiksen KL, Gitlin I, Whitesides GM. Proc. Natl. Acad. Sci. U.S.A. 2006;103:7968–7972. doi: 10.1073/pnas.0602816103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Grossman PD. Capillary Electrophoresis: Theory and Practice. Academic Press; San Diego, CA: 1992. [Google Scholar]
- (27).Colton IJ, Anderson JR, Gao JM, Chapman RG, Isaacs L, Whitesides GM. J. Am. Chem. Soc. 1997;119:12701–12709. [Google Scholar]
- (28).Gitlin I, Carbeck JD, Whitesides GM. Angew. Chem., Int. Ed. 2006;45:3022–3060. doi: 10.1002/anie.200502530. [DOI] [PubMed] [Google Scholar]
- (29).Jackson SE. Org. Biomol. Chem. 2006;4:1845–1853. doi: 10.1039/b600829c. [DOI] [PubMed] [Google Scholar]
- (30).Jonas J. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2002;1595:145–159. doi: 10.1016/s0167-4838(01)00341-7. [DOI] [PubMed] [Google Scholar]
- (31).Glickman MH, Ciechanover A. Physiol. Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- (32).Vijay-Kumar S, Bugg CE, Cook WJ. J. Mol. Biol. 1987;194:531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- (33).Bulheller BM, Rodger A, Hirst JD. Phys. Chem. Chem. Phys. 2007;9:2020–2035. doi: 10.1039/b615870f. [DOI] [PubMed] [Google Scholar]
- (34).Shaw BF, Durazo A, Nersissian AM, Whitelegge JP, Faull KF, Valentine JS. J. Biol. Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- (35).Busenlehner LS, Armstrong RN. Arch. Biochem. Biophys. 2005;433:34–46. doi: 10.1016/j.abb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- (36).Menon MK, Zydney AL. Anal. Chem. 2000;72:5714–5717. doi: 10.1021/ac000752b. [DOI] [PubMed] [Google Scholar]
- (37).Gitlin I, Mayer M, Whitesides GM. J. Phys. Chem. B. 2003;107:1466–1472. [Google Scholar]
- (38).We did not account for the sodium ion in SDS for these corrections, as only the dodecyl sulfate moieties (DS-) associate with UBI; Na+ is the counterion.
- (39).Basak SK, Ladisch MR. Anal. Biochem. 1995;226:51–58. doi: 10.1006/abio.1995.1190. [DOI] [PubMed] [Google Scholar]
- (40).Loladze VV, Makhatadze GI. Protein Sci. 2002;11:174–177. doi: 10.1110/ps.29902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Figure S5 in the Supporting Information provides the SDS-PAGE analysis of ten proteins with molecular weights from 2.5 to 20 kDa, including UBI (equilibrated against both 10 and 30 mM SDS and denatured at both 22 and 100 °C). On the basis of the SDS-PAGE results, it is reasonable to conclude that D is fully denatured and saturated with SDS at the ratio of 1.4 g of SDS per gram of protein.
- (42).At [SDS] > 2.8 mM, a peak appears at μ < 1.2 cm2 kV-1 min-1, directly after the neutral marker (DMF) peak (Figure 1a,d, ■). We observed the same peak under similar conditions when only DMF was injected (see Supporting Information, Figures S3a and S8). We hypothesize that this signal arises from the partitioning of an impurity within SDS micelles (Figure 1, cmc labeled with a dotted bracket). To verify this hypothesis, we analyzed injections of a UV-absorbent hydrophobic marker into the buffer (20 μM of 2-hydroxymethylenenaphthalene, Figure S3) and determined a cmc for SDS at 3.4 mM (Figure 1, cmc labeled with a plain bracket).
- (43).Perrin CL, Chen JH, Ohta BK. J. Am. Chem. Soc. 1999;121:2448–2455. [Google Scholar]
- (44).Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, Fraser PE, Hawkins PN, Dobson CM, Radford SE, Blake CCF, Pepys MB. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- (45).Arai M, Kuwajima K. Adv. Protein Chem. 2000;53:209–282. doi: 10.1016/s0065-3233(00)53005-8. [DOI] [PubMed] [Google Scholar]
- (46).Stutz H, Wallner M, Jr., Bordin G, Rodriguez AR. Electrophoresis. 2005;26:1089–1105. doi: 10.1002/elps.200406195. [DOI] [PubMed] [Google Scholar]
- (47).We were not able, under the experimental conditions presented here, to obtain peracylated UBI when using 4-sulphophenylisothiocyanate (e.g. the 16th rung).
- (48).A stoichiometry of 0.4 g of SDS per gram of protein corresponds to a composition of UBI-SDS∼12. This composition is in good agreement with that obtained from our analysis with charge ladders (complexes in G2 are UBI-SDS∼11).
- (49).Macdonald JM, LeBlanc DA, Haas AL, London RE. Biochem. Pharmacol. 1999;57:1233–1244. doi: 10.1016/s0006-2952(99)00039-8. [DOI] [PubMed] [Google Scholar]
- (50).The integration was performed with respect to the migration time; details available in the Supporting Information.
- (51).The number of SDS molecules expected to bind UBI in D is 42, based on the approximation that 1 g of protein associates with 1.4 g of SDS.
- (52).Manning M, Colon W. Biochemistry. 2004;43:11248–11254. doi: 10.1021/bi0491898. [DOI] [PubMed] [Google Scholar]
- (53).O’Neil KT, DeGrado WF. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- (54).Lyu PC, Liff MI, Marky LA, Kallenbach NR. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- (55).Kim CA, Berg JM. Nature. 1993;362:267–270. doi: 10.1038/362267a0. [DOI] [PubMed] [Google Scholar]