

Abstract
The expression of gene products in bacteria can be inhibited by the use of RNA external guide sequences (EGSs) that hybridize to a target mRNA. Endogenous RNase P cleaves the mRNA in the complex, making it inactive. EGSs participate in this biochemical reaction as the data presented here show. They promote mRNA cleavage at the expected site and sometimes at other secondary sites. Higher-order structure must affect these reactions if the cleavage does not occur at the defined site, which has been determined by techniques based on their ability to find sites that are accessible to the EGS oligonucleotides. Sites defined by a random EGS technique occur as expected. Oligonucleotides made up primarily of defined or random nucleotides are extremely useful in inhibiting expression of the gyrA and rnpA genes from several different bacteria or the cat gene that determines resistance to chloramphenicol in Escherichia coli. An EGS made up of a peptide-phosphorodiamidate morpholino oligonucleotide (PPMO) does not cleave at the same site as an unmodified RNA EGS for reasons that are only partly understood. However, PPMO-EGSs are useful in inhibiting the expression of targeted genes from Gram-negative and Gram-positive organisms during ordinary growth in broth and may provide a basis for broad-spectrum antibiotics.
Keywords: drug resistance, Gram-positive and Gram-negative bacteria, peptide-phosphorodiamidate morpholino oligonucleotide, RNase P
The utility of bacterial transformation for therapeutic purposes has been limited by the number of species that will undergo transformation and the frequency with which that event happens. To accommodate new therapies that involve small nucleic acids, a means has to be developed to enable bacterial species to take up these nucleic acids with relative ease. The covalent linkage of arginine-rich peptides to the ends of chemically-modified RNAs facilitates the uptake of the RNA analog (1, 2) and other similar molecules (4, 5). This methodology in combination with an effective means of inactivating gene expression has to be developed to make it useful for therapeutic agents. There are other processes that function in bacteria to inhibit gene expression (3, 6), but the external guide sequence (EGS) technology (7, 8) that is mediated by RNase P cleavage of the target RNA seems optimal in this regard.
RNAi and siRNA (ref. 9 and references therein) are not useful tools for the transformation of bacterial species because these RNAs rely on an intracellular complex, the Dicer complex (9) in particular, to release ssRNA that will base-pair with the target mRNA. EGS technology, which is just as effective as siRNA in mammalian cells in tissue culture (10), is very effective in Escherichia coli (11, 12) and Salmonella typhimurium (13). Bacterial cells can be altered from drug resistance to drug sensitivity with the methods generally described here (11), and a similar method has also been reported (14). Essential genes can also be inactivated in terms of their expression. The EGS method will allow 3-bp mismatches over 15 paired nucleotides (12) and still be effective providing the 3-bp mismatches are not contiguous in the target mRNA:EGS complex. Thus, 3 mutations in the target mRNA can still be overcome by the EGS technology.
We have studied the events in vitro subsequent to the use of small oligonucleotides to verify that the EGS technology is functional as envisaged, and we have also examined the use of modified RNA oligonucleotides with the same bases of EGSs to explore the effectiveness of the EGS technology. We determined that EGS oligoribonucleotides originally coded for by plasmids do enable cleavage by RNase P of the target mRNA at the same site as expected in vitro. The modified peptide-phosphorodiamidate morpholino oligonucleotides (PPMO-EGSs; ref. 1) also enable cleavage by RNase P but not always precisely at the same site as may have been predicted by the theoretical aspects of EGS technology. Nevertheless, these latter chemically-modified RNAs function as EGSs to inhibit expression of the target mRNA in prokaryotes. Other work with PPMOs that hybridize to sequences near the start codons of genes as antisense oligonucleotides has been somewhat successful in inhibiting gene expression (1, 2). The PPMO-EGSs enter several species of bacteria in broth, as do PPMOs (1), and broaden the possibility of EGSs and other methodologies as clinical tools.
Results
Design of EGSs.
Two methods were used, one that relies on specific identification of accessible regions for the binding of oligonucleotides in target RNAs (7, 8), and another that relies on the use of a random mixture of EGS compounds (rEGS; ref. 15). Specific EGSs designed by the first method were used to inactivate expression of the cat gene in E. coli (11) and the gyrA and rnpA genes (12) in E. coli and S. typhimurium (13). The latter 2 are essential genes, and the details of these preparations are described in Materials and Methods. EGS sequences determined in the cat and gyrA cases (Tables 1 and 2) were used to design PPMO-EGSs that have identical sequences. A variation of the rEGS method that uses some defined oligonucleotides in certain genes (partially random EGS; prEGS) was also used to determine the cleavage site in the gyrA and the rnpA genes of E. coli in vitro and the inactivation of 2 other bacteria in vivo (Tables 3–6). Sequences of, e.g., gyrA were listed from a number of bacteria and a particular region was chosen (Table 2) that had a high percentage of homology. Some of the nucleotides were constant or were pyrimidines or purines, whereas others were distributed among 3 or 4 of the nucleotides. This latter case became N in the prEGS, indicating a prEGS, and the others were as constant, or Y or R.
Table 1.
gyrA sequences of mRNAs to be complemented with EGSs
| Organism | mRNAs |
|---|---|
| E. coli | 313GGUCAGGGUAACUUCGG329 |
| P. aeruginosa | GGGCAUGGUAACUUCGG |
| S. aureus | GGCCAAGGUAAcUUUgg |
| B. subtilis | GGUCACGGAAACUUCGG |
| E. faecalis | GGCCACGGAAACUUCGG |
| prEGS | CCNAANUUNCCNUGNCC |
mRNA targets and EGS identities of gyrA genes. mRNA sequences are listed as 5′ to 3′. EGS sequences are listed as 3′ to 5′. Nucleotide numbers are for the E. coli sequence. All other nucleotide numbers are close to the E. coli numbers but are not exact. N indicates any one of the 4 nt.
Table 2.
rnpA mRNAs (top line) and EGSs (bottom line) and Tm of hybrids
| Organism | mRNA-EGS hybrids | Tm (°C) of hybrids |
|---|---|---|
| E. coli |  |
59.0 |
| B. subtilis |  |
58.6 |
| E. faecalis | 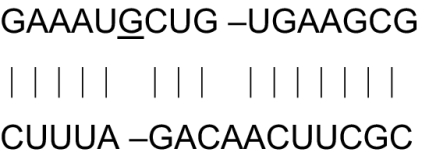 |
39.1 |
| Partially random EGS | CGYUUNAUCNGAUUNC |
mRNA targets and EGS identities of rnpA genes. mRNA sequences are listed as 5′ to 3′. EGS sequences are listed as 5′ to 3′. Nucleotide numbers are for the E. coli sequence. All other nucleotide numbers are close to the E. coli numbers but are not exact. N indicates any one of the 4 nt.
Table 3.
Inactivation of chloramphenicol resistance gene in E. coli in vivo [50 μM PPMO-EGS each; 4 hr after administration (LB broth-chloramphenicol (50 μM)]
| PPMO-EGS | Colony counts | Fraction of survival |
|---|---|---|
| Control | 4.1 × 105 | 1.0 |
| Scrambled | 4.0 × 105 | 0.98 |
| EGS 1 | 3.9 × 104 | 0.098 |
| EGS 2 | 6.5 × 103 | 0.016 |
| EGS 1 + 2 | 6.0 × 102 | 0.0015 |
Scrambled PPMO-EGS contains a sequence that has no relation to the targeted mRNA. At least 2 experiments were done, and averages are presented. PPMO-EGS concentrations are accurate within 10%. Naked RNA EGSs were not tested because they have little probability of entering the cells under test.
Table 4.
Inactivation of gyrA gene in E. coli [50 μM PPMO-EGS each: 4 hr after administration (LB broth)]
| PPMO-EGS | Colony counts | Fraction of survival |
|---|---|---|
| Control | 2.3 × 105 | 1.0 |
| Scrambled | 2.2 × 105 | 0.96 |
| EGS241 | 1.7 × 105 | 0.74 |
| PrEGS313 | 6.8 × 104 | 0.29 |
| 241 + 313 | 8.3 × 103 | 0.036 |
See legend for Table 3.
Table 5.
Inactivation of gyrA gene in B. subtilis in vivo [15 μM PPMO-EGS313; 4 hr after administration (LB broth)
| PPMO-EGS | Colony count | Fraction of survival |
|---|---|---|
| Control | 7 × 106 | 1.0 |
| Scrambled | 7.3 × 105 | 0.1 |
| PrEGS313 | 1 × 105 | 0.014 |
See legend for Table 3.
Table 6.
Inactivation of gyrA gene in E. faecalis in vivo [100 μM PPMO-prEGS313: 4 hr after administration (BHI medium)]
| PPMO-EGS | Colony count | Fraction of survival |
|---|---|---|
| Control | 8.6 × 105 | 1.0 |
| Scrambled | 6.15 × 105 | 0.72 |
| PrEGS313 | 2.15 × 105 | 0.25 |
See legend for Table 3.
The rEGS method was also used to design EGSs to attack the mRNA of the gyrA and rnpA genes in several bacteria, both Gram-positive and Gram-negative, as will be described below.
Cleavage of Target mRNAs by EGSs and PPMO-EGSs in Vitro.
The identification of the cleavage sites in CAT mRNA and gyrA mRNA by EGSs and PPMO-EGSs are illustrated in Figs. 1 and 2. The target RNAs were internally or end-labeled with 32P, and the RNase P assay was standard (see Materials and Methods), and all of the components of these reactions were from E. coli. Precise identification of the cleavage sites was aided by running gels for different lengths of time and the utilization of marker RNAs (Fig. S1 and S2). Subsequently, the cleavage products isolated from the gels can be further identified by running complete RNase T1 digestion or primer extensions, as shown in Fig. S1 and Fig. S2. Although there may be an uncertainty in cleavage site identification by 1 nt in some cases, the results of cleavage site identity are clear enough, with RNA marker identity (Fig. S1) to allow further observations. We note, for example, that the CAT mRNA was cleaved at position 67 (1 is the identity of the first nucleotide in the coding mRNA sequence) by EGSCAT1, which is the expected site from the design of the EGSs. With PPMO-EGSs there was miscleavage 20 nt upstream of the “expected” site as indicated (Fig. 1B). With respect to the gyrA mRNA, specific EGS cleavage at the expected site is found (site 241) and a weak secondary cleavage site (site 341). The PPMO-EGS also has 2 cleavage sites for gyrA mRNA, 171 and a weak one at 276 and a prEGS cleaves at the expected site, 313 (Fig. 2). In preliminary experiments, the rnpA mRNA was cleaved only at the specific, expected site by EGSs designed to do that. The PPMO-EGS for rnpA mRNA directed at −2 and 32 showed no strong cleavage at all under the conditions we used in the initial experiments with a shortened version of the rnpA mRNA but an mRNA with a longer 5′ sequence upstream of the coding region was cleaved at A −18 by a PPMO-EGS designed for C −2. A prEGS against the rnpA mRNA cleaved at G 185 and the PPMO-EGS cleaved at G173, consistent with other results of PPMO-EGSs in preliminary experiments.
Fig. 1.

Cleavage by RNase P of complexes of mRNAs and EGSs or PPMO-EGSs. (A) EGS and PPMO-EGS directed cleavage of CAT mRNA in vitro. An internally-labeled fragment of CAT mRNA (20 nM) was incubated with E. coli RNase P holoenzyme (20 nM M1 RNA, 200 nM C5 protein). Lane 1: CAT mRNA alone; lane 2: CAT mRNA with E. coli RNase P holoenzyme; lane 3: the same as in lane 2 in the presence of EGSCAT1 (200 nM); lane 4: with EGSCAT1 (2 μM); and lane 5, PPMO-EGSCAT1 (500 nM), all for 30 min at 37 °C. All products were run on a 5% denaturing PAGE. Dots indicate the positions of cleavage products. (B) Location of RNase P cleavage sites in different mRNAs. Shown are 3 nucleotide sequences of the 3 target mRNAs. The arrows indicate cleavage sites induced either by EGSs or PPMO-EGSs. Small arrows indicate weak second site cleavages that were not analyzed further.
Fig. 2.

RNase P cleavage in vitro of complexes formed by target mRNAs from the gyrA gene. EGS and PPMO-EGS directed cleavage of gyrA mRNA in vitro. Control lanes are: 1, gyrA mRNA alone; 2, gyrA mRNA with E. coli RNase P holoenzyme. An internally-labeled fragment of gyrA mRNA (10 nM) was incubated with E. coli RNase P holoenzyme (10 nM M1 RNA, 100 nM C5 protein) in the presence of increasing amounts (100 nM, 500 nM, 1 μM) of EGS241 (lanes 3–5), PPMO-EGS241 (lanes 6–8) or rEGS313 (lanes 9–11); lanes 12–14: gyrA mRNA with 1 μM EGS241, PPMO-EGS241, or prEGS313, respectively without RNase P. Reactions were incubated for 30 min at 37 °C and analyzed on 5% denaturing PAGE. Dots indicate the positions of cleavage products.
There is no required binding to the same site between a prEGSs and specifically-designed EGSs and PPMO-EGSs designed for the same accessible site. Partial homologies in parts of the mRNA targeted sequence may play a role in determining a cleavage sites in both of these cases as is discussed below, but the PPMO-EGS may choose another locale in the mRNA with similar homologies. The prEGS cleavage site was determined in 1 rapid experiment, and there is no expectation of miscleavage in that case nor is there any expectation of exact determination of the locale of accessibility as with the other EGSs with a prEGS.
The PPMO-EGS cleavage sites are as yet unclear in terms of their selection of cleavage sites. The basis of miscleavage by PPMO-EGSs is not understood, although we note that the sequence GGCAU occurs near the EGS CAT1 cleavage site in the CAT mRNA and in the PPMO-EGS cleavage site. Quite remarkably, there is an apparent similarity in the cleavage of the gyrA sequence. In this case, the sequence GGUGACU (UG is looped out of a base-paired region in the secondary structure of M1 RNA) occurs near both sites. The EGS sites are where we would expect them to be and they are close to the terminus of the 5′ side of a helical segment of the mRNAs. The PPMO-EGSs are near the beginning of a loop region and are not at a canonical RNase P cleavage site. The presence of the same sequence near each cleavage site may be an indication of binding stability of the EGSs or PPMO-EGSs to the target mRNA or to part of the RNase P RNA but this is not known. If PPMO-EGSs that target the CAT mRNA are used that differ in the position of the peptide, either the 5′ or the 3′ end of the PMO-EGS, then a very small difference in gel mobility is seen (Fig. S3). This difference, on further analysis in PAGE of the cleavage fragments is caused by a 1-nt difference in the molecular weight of the compounds.
The positively-charged peptide of a PPMO-EGS could interact with the target mRNA or the RNase P RNA (by competing with the conserved sequence of positively-charged amino acids in the RNase P protein) and change their structures to lead to a nonobvious cleavage site becoming accessible. However, the PPMO-EGS structure involved in the complex is not apparent. It is unknown whether it is a helical structure or an alternative that is more extended in space. The details of the energy of interaction cannot solely be attributed to hydrogen-bonding contacts between the bases in the PPMO-EGS and the target mRNA. Aspects of tertiary structure or details of the secondary structure that cannot be included in the mfold program (16) must also be significant in these cases in determining cleavage sites.
We note also that RNase P RNA from E. coli (M1 RNA) is active in cleaving the target mRNA in the company of an EGS. However, the RNase P RNA is not capable of cleaving a target mRNA in the presence of a PPMO-EGS (Fig. S3) nor is it capable of cleavage alone on a target mRNA.
Theoretical Prediction of Cleavage Sites Using Partially Random Primers for Target mRNAs and rEGSs.
The sequences for target mRNAs of gyraseA genes (www.ncbi.nim.nih.gov, National Center for Biotechnology Information Genbank gyrA gene ID 946614), 5 bacteria, and rnpA genes (www.ncbi.nim.nih.gov National Center for Biotechnology Information Genbank rnpA geneID 948215), 3 bacteria, are listed for both Gram-negative and Gram-positive ones, as shown in Tables 1 and 2. In the case of the gyrA gene there is sufficient homology among the bacteria to predict a single partially-random set of primers for use in cloning fragments that code for all mRNAs from genomic DNA. In the case of rnpA, the mRNA in each case must be cloned separately because of differences in sequences adjacent to the region that is to be cloned. In both cases, there is sufficient homology to use a partially-random sequence to design prEGS primers for each gene (see Tables 1 and 2). Once an mRNA fragment has been identified, an rEGS or a prEGS can be found immediately.
Particular individual gels for assay of the rnpA prEGSs, in combination with labeled mRNA and RNase P from E. coli, are shown in Fig. 3. Successful cleavage is indicated by an increase in the intensity of the band indicated in the prEGS lanes. The theoretical and experimental targets of several sequences are shown in Table 2. For the case of rnpA, additional experimental sequences that arise from the rEGS initial assays are shown and the amount of mismatches from the theoretical sequence are also indicated. Note the mismatches shown in Fig. 3, in which cases we never see >3 mismatches as expected. G–U pairs and A–C pairs do occur in several cases. Although our previous estimates of mistargeting mRNAs with EGSs have not revealed any such interactions, in the cases discussed here, mismatches that affect genes other than the target identified can only be of some aid in inactivating bacteria under study.
Fig. 3.

Cleavage in vitro of the target rnpA mRNAs by rnpA-specific prEGS libraries. (A) E. faecalis rnpA mRNA. (B) B. subtilis rnpA mRNA. PAGE of cleavage products of 5′end-labeled rnpA mRNAs generated by RNase P holoenzyme and prEGS libraries. The RNase P holoenzyme was reconstituted by 10 nM M1 RNA and 100 nM C5 protein. rnpA mRNA cleavage products were separated on 8% polyacrylamide/7 M urea gels together with an alkali ladder, represented by OH, and a partial RNase T1 digest, represented by T1, of the same mRNA. The black triangles with prEGS labeled above represent increasing concentrations of prEGSs (10-, 100-, and 1,000-fold molar excess to the rnpA mRNAs) incubated with RNase P holoenzyme. Lane P represents reaction with RNase P holoenzyme only. Lane control means that the mRNA only was present. The numbers indicate the positions of cleavage relative to the rnpA translational start codon. The target mRNA sequences starting from the numbers (5′ to 3′) are best base-paired by the bottom EGS candidates (3′ to 5′).
The assay for prEGS for gyrA mRNA showed the correct, expected cleavages in vitro and some secondary cleavages for Staphylococcus aureus and Pseudomonas aeruginosa (Fig. S4), in which case 2 or 3 mismatches in the hybrids exist.
Using the mfold program (16) for energy of interactions in hybrids for rnpA prEGSs, we find that most additional mismatched sequences are “acceptable” target RNAs as judged by the parameters of the oligonucleotide complexes (reaction at 37 °C; CT = 0.01 μM; Na+ = 1 M; Mg2+ = 0 M; CT = target RNAs:EGS concentration). Some sequences that were not perfect matches had melting temperatures closer to 37 °C but still were cleaved in vitro (Fig. 3 A and B). In the case of rnpA in Enterococcus faecalis, mRNAs:EGS complexes that contain 2 hybrids that are mismatched are shown in the gel, indicating cleavage of the complexes in vitro (Fig. 3A). However, the theoretical comments apply only to naked RNAs that would be delivered by plasmids that contained these sequences in vivo. The theoretical calculations do not precisely reflect what happens in solution with the fragment of the mRNA used as a target: the experimental results indicate more stable pairings between EGSs and the whole fragment of the mRNA targets. Sequences in the target mRNAs other than in the hybrid regions obviously provide more stability to the EGS–mRNA complexes than anticipated from the limited theory of the association of the hybrid complexes. We have not determined the binding sites as a consequence of changes in the salt composition of the buffers.
Utilization of PPMO-EGSs.
PPMO-EGSs were mixed directly with cells for uptake in rich medium. This is not a conventional transformation procedure. An electrophoretic transformation step was carried out briefly in 1 case with E. coli but it proved not to have properties that were effective over the simple mixing procedure. The PPMO-EGSs do not have the negative charge of naked DNA or RNA. No other polymers, like PNA polymers, have been tried in these experiments. However, other researchers have also tried PPMO antisense molecules, targeted to the initiation site for translation, with some success (1, 6).
We had shown that 2 EGSs used simultaneously to inhibit gene expression were very effective in converting drug resistance to drug sensitivity by transformation in E. coli (11). Two cat PPMO-EGSs targeting 2 different sites in the cat mRNA were also effective in converting drug resistance to drug sensitivity (Table 3). EGSs against gyrA mRNA were also effective (12, 13; all experiments with EGSs per se were done with plasmids that contained genes coding for the EGSs) as were their respective PPMO-EGSs (Tables 4 and 5). The PPMO-EGSs worked very efficiently in this case but the temporal stability of this effect does not seem promising at this time. The time of effectiveness of the PPMO-EGS solution procedure was optimal ≈4 h after mixing. After 4 h the PPMO-EGSs were less effective in our experiments, either because the peptide fragments were degraded or the fraction of cells that ingested them was already saturated and their roles were superfluous. Another possibility is that the PPMO-EGSs encourage uptake and discharge of these molecules and perhaps, after 4 h, there is less PPMO-EGS inside the cells.
Plasmids that contained 2 gyrA EGS components and 2 rnpA EGS components were quite effective in transformation experiments (12). However, a mixture of 2 different gyrA PPMO-EGSs was also effective, but somewhat less so than the cat PPMO-EGSs (Table 4). PPMOs against the AUG initiator region were successful in many different bacteria (1), both Gram-positive and Gram-negative, but quantitative results are not available. Two Gram-positive bacteria were also assayed in our system with the gyrA PPMO-EGSs.
PPMO-EGSs successfully inhibited the growth of Bacillus subtilis and E. faecalis to ≈10–30% of the control culture when the gyrA gene was targeted (Tables 3–6). In the case of B. subtilis, the effective concentration of PPMO-EGSs was ≈10-fold lower than that used for E. coli. Although it is clear that the cell walls of the 2 bacteria are different, it is not known whether this specifically affects our results.
The timing of future PPMO-EGS doses to completely inhibit the expression of the genes listed here remains to be explored.
Discussion
We have provided data regarding the nature of the biochemical interaction between an EGS, a target RNA, and RNase P in prokaryotes in vitro. Although the results of the phenotypes of such interactions have been published (11–13), the specific identification of cleavage sites in the mRNAs and the probability of their functioning has not been measured as we show here. The presumed interaction of these elements occurs as previously imagined and seems to occur in the same manner in all of the bacteria tested. Only E. coli RNase P was used in these experiments but previous experience indicates that other species of RNase P from different bacteria will work in the same fashion.
The correlation between the precise numbering of the nucleotides in the mRNAs for cleavage at expected sites and nonexpected cleavages by PPMO-EGSs in vitro indicates that there are significant cleavages aside from the expected ones. We did verify that 3 mismatches between an EGS and the target RNA still function to inactivate the target mRNA as has been shown before with genetic experiments (see Tables 1 and 2 and ref. 12). We actually analyzed the mRNA sequences of ≈10 bacteria in each case although fewer are shown in Tables 1 and 2. The rules of hydrogen bonding, including wobble, are apparent in examining the data in Fig. 3. Weak interactions between A–C base pairs apparently contribute to the stability of the hybrids. Are results in vivo different? In keeping with our methodology, it is actually very challenging, if not impossible, to determine the cleavage sites in vivo because the remaining living cells would not contain the cleaved mRNA or would compose a very small fraction of the living cells.
The 2 methods used to design EGSs (7, 8, 15) do not always correlate with accessible sites for oligonucleotide hybrids in the target mRNA as it is calculated from the mfold method (16). The same is true for PPMO-EGSs, where an even greater difference is found between the expected site and the site of cleavage. Previous data regarding the minimal substrate features that guide cleavage of a substrate by RNase P (7), i.e., a short portion of a double helical RNA with a 5′ single-stranded segment, suggest that the structure cleaved in a PPMO-EGS complex with an RNA is unusual because the PPMO segment is an unknown quantity in solution in a hybrid with the target RNA. It is always possible to assign some factor of higher-order (tertiary) structure in solution to explain similar phenomenon but no available data are evident yet in these cases. With respect to PPMO-EGSs, there must be other factors involved including ionic interaction between the arginine-rich peptides and the negative charge of the phosphodiester backbone of the target mRNA. Of course, if the positively-charged peptide substitutes in some manner for the typical conserved positively-charged region of amino acids in the protein component of the RNase P holoenzyme, then one could imagine a binding to a part of RNase P RNA by the cationic segment of the PPMO-EGS and an anomalous shift in the cleavage site that the enzyme should make. A PMO-EGS, the morpholino oligonucleotide with a sequence complementary to the main site cleaved in the CAT mRNA, however, also cleaved at an unusual site, T38, but at a rate 10-fold slower than the PPMO-EGS in preliminary experiments. A subsequent problem is the design of the newly-recognized cleavage site. If the PPMO-EGSs are degraded in vivo, the mechanism of action of cleavage site selection by the PPMO-EGS may be affected by a degraded PPMO-EGS in which the peptide fragments have been removed (1) and the cleavage sites selected in vivo may be different from the ones identified in vitro.
Our experiments with PPMO-EGSs on the viability of wild-type bacteria grown in broth are simple: mix the gene-targeted PPMO-EGSs with the broth and assay cell counts after a period. The most efficient experiments are on the inactivation of drug resistance although there is considerable loss of viability with essential genes. In fact, the loss of drug resistance is remarkable (10−3 inactivation with 1 dose) with the PPMO-EGSs and supports earlier experiments in which plasmids that coded for the attack on cat resistance genes by transformation were also promising (11). Similarly, 2 copies of PPMO-EGSs attacking different sites in the gyrA gene are much more effective than 1 PPMO-EGS attacking a single site.
Several researchers have investigated the effects of peptide-linked oligonucleotides on gene inactivation in bacteria (1, 2). In some cases peptide nucleic acids (PNAs) have been used and PPMOs (4–6). Both of these polymers are antisense molecules that hybridize to regions near the initiator codon of genes in bacteria. These data are comparable to ours and strengthen the notion that the facilitating agents are useful clinical tools if their efficiency in terms of the effective molarity can be improved. It does seem apparent that the PPMO-EGS method is more efficient in curing cells of drug resistance than it is to stop cell growth by inhibiting the production of essential proteins (Tables 3–6). We note again that 2 EGS attacking different sites in the same target mRNA are dramatically better than 1 EGS.
PPMOs can be used to cure mice of bacterial infections (2). Our data lend support to the further exploration of EGSs and PPMO-EGSs to cure not only infections of mice but also of humans with respect to both internal and external (skin) infections.
Materials and Methods
Bacterial Strains and Plasmids.
Strains used were: E. coli SM105, thr-1, araC14, tsx-78, Δ(galK-attLAM)99, hisG4(Oc), rfbC1, rpsL136(strR), xylA5, mtl-1, thi-1; and E. coli DH5α, F− ϕ80 dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rk− mk+) phoA supE44 λ− thi-1 gyrA96 relA1. Ampicillin or carbenicillin (100 μg/mL) was used for plasmid selection and maintenance in E. coli. Plasmids: pUCT7/AEFRNAHHT7T (17, 18), pGEM-T (Promega), and pBluscript II SK(+) (Stratagene).
Oligonucleotides.
Oligonucleotides were made by the Yale University Keck synthesis facility.
Enzymes, Kits, and Labeled Nucleotides.
Restriction endonucleases, DNA polymerase I Klenow fragment, T4 polynucleotide kinase, and T4 DNA ligase were purchased from New England Biolabs. SP6 and T7 RNA polymerases were purchased from Promega. Qiaquick PCR purification kit, Qiaquick gel extraction kit, and Qiaprep spin miniprep kit were purchased from Qiagen. AmpliTaq DNA polymerase was purchased from Roche. [α-32P]GTP and [γ-32P]ATP were purchased from PerkinElmer.
Cloning of gyrA Fragments and Preparation of mRNAs and EGSs.
We amplified the gyrA gene 5′ fragment by PCR using universal primers GyrA F (5′-GTN NTN GGN AAN TAN CAN CCN CA-3′) and GyrA R (5′- GGN GGN ATN TTN GTN GC-3′). The resulting PCR product was directly ligated into pGEM-T vector, and the sequence was confirmed by DNA sequencing. The vector was used as the template for transcription in vitro. The transcribed mRNA fragments are of ≈340 nt, depending on the source of the bacteria species, and started at approximately +220 nt in the gene sequences. The prEGS1 (5′-CCNAANUUNCCNUGNCCACCA-3′) was generated as described (15). The prEGS1 sequence contains 5 random nt of 21 nt. It was designed to target the gyrA gene from different bacterial strains. Oligonucleotide G241U 5′-gga ccg ccg agt cac cac cag-3′ and G241D 5′-gtg acc tgg tgg tga ctc ggc ggt cct gca-3′ were used for construction of the E. coli plasmid that contained the EGS241 sequence. For EGS313, the oligos were UP 5′-GCCGAAGTTACCCTGACCACCAG-3′ and DOWN 5′-GTGACCTGGTCAGGGTAACTTCGGCTGCA-3′. The constructions of the plasmids were performed with the annealed products of the oligos and pUC/T7AEFRNAHHT7T as described (17).
Mapping of Accessible Sites in the gyrA or rnpA mRNAs.
The rEGS assay for the gyrA and rnpA mRNAs was performed as described (15). The prEGS in 10-, 100-, or 1,000-fold molar excess was incubated with 10 nM 5′ end-labeled rnpA mRNAs in PA buffer [50 mM Tris·Cl (pH 7.5), 10 mM MgCl2, 100 mM NH4Cl] for 5 min at room temperature. Subsequently, 10 nM M1 RNA and 100 nM C5 protein were added to the mixture and further incubated for 30 min at 37 °C. Reactions were terminated by adding 10 μL of 10 M urea/10% phenol solution. Partial RNase T1 reaction was performed as described (8). After the reaction, the mRNA was separated on a 5% or 8% polyacrylamide sequencing gel that contained 7 M urea. Partial alkaline ladder digests were carried out as described (15). Cleavage sites were determined by comparing rEGS-mediated and partial RNase T1 cleavage products to partial alkaline ladder results. Primer extension was also carried out for gyrA prEGS313 as described (19) to confirm the identity of the cleavage site.
Preparation of the Target rnpA mRNAs and prEGSs.
The templates for the rnpA mRNAs were prepared as described (15). The PCR fragments were digested with the restriction enzymes KpnI and EcoRI, and then inserted into the vector pBluscript II SK (+) digested with the same enzymes. The resulting plasmids pBSKC5BAf, pBSKC5BMf, pBSKC5YPf, and pBSKC5FTf were digested with the restriction enzymes PvuII and EcoRI, ethanol-precipitated and dissolved in distilled water, and used as the template for in vitro transcription using T7 RNA polymerase (15). For construction of the plasmids carrying partial rnpA fragments of 4 bacteria the primers used were: C5BAK, 5′- Bsubt-F (5′-GACTGGTACCCGTGTCGGGCTTTCCGTCAG-3′), Bsubt-R (5′-GACTGGATCCGACTTTCTGAATAGATGCTGCAG-3′), Efaec-F (5′- GACTGGTACCGTGGGGATTTCTGTTGGGAAG-3′), Efaec-R (5′-GACTGGATCCCTACTCAATTCCCTCTCTTACA-3′).
The construction of the template for the prEGS targeting the rnpA was performed as described (15). Primers of C5EGS (5′ - CGCTAATACGACTCACTATAGGCKNYTNMWNMGRTTNCACCAGGGATTGTTCAGTAT-3′), map F, and map R were used for PCR amplification to yield a prEGS library for transcription in vitro with T7 RNA polymerase (14).
PPMO-EGSs in Growth Medium.
PPMO-EGSs were received from Avi Biopharma. The peptide composition covalently linked to either the 5′ or 3′ termini of the oligonucleotide is (RXR)4XB-AcpP-PMO (1), where X is 6-aminohexanoic acid and B is β-alanine. These compounds were stored and diluted according to the manufacturer's instructions. Bacterial cells were grown overnight in LB broth and diluted to start fresh cultures. PPMO-EGSs were added to certain cultures at time 0 and the cultures were allowed to grow at 37 °C in a shaking incubator. Aliquots were taken as indicated and plated out to measure cell growth. The position of the peptide, 5′ or 3′ ends, makes no difference in our experiments.
Supplementary Material
Acknowledgments.
We thank R. Kole for comments on a draft of this manuscript; R. Kole and Patrick Iversen (both of Avi Biopharma) for the supply of PPMO-EGSs; and our colleagues for useful discussions. S.A. thanks Dr. Tim Nilsen for discussions concerning EGSs several years ago. S.A. and M.I. were supported by Defense Threat Reduction Agency Grant W81XWH-06-2-0066.
Footnotes
Conflict of interest statement: B.G. is an employee of Avi Biopharma. The other authors have no competing interests.
This article contains supporting information online at www.pnas.org/cgi/content/full/0903491106/DCSupplemental.
References
- 1.Mellbye BL, Puckett SE, Tilley LD, Iversen PL, Geller BL. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob Agents Chemother. 2009;53:525–530. doi: 10.1128/AAC.00917-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tilley LD, Mellbye BL, Puckett SE, Iversen PL, Geller BL. Antisense peptide-phosphorodiamidate morpholino oligomer conjugate: Dose–response in mice infected with Escherichia coli. J Antimicrob Chemother. 2007;59:66–73. doi: 10.1093/jac/dkl444. [DOI] [PubMed] [Google Scholar]
- 3.Palm C, Netzereab S, Hällbrink M. Quantitatively determined uptake of cell-penetrating peptides in nonmammalian cells with an evaluation of degradation and antimicrobial effects. Peptides. 2006;27:1710–1716. doi: 10.1016/j.peptides.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Patel PC, Giljohann DA, Seferos DS, Mirkin CA. Peptide antisense nanoparticles. Proc Natl Acad Sci USA. 2008;105:17222–17226. doi: 10.1073/pnas.0801609105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abes S, et al. Peptide-based delivery of steric-block PNA oligonucleotides. Methods Mol Biol. 2009;480:85–99. doi: 10.1007/978-1-59745-429-2_6. [DOI] [PubMed] [Google Scholar]
- 6.Nekhotieva N, Awasthi SK, Nielsen PE, Good L. Inhibition of Staphyloccocus aureus gene expression and growth using antisense peptide nucleic acids. Mol Ther. 2004;10:652–659. doi: 10.1016/j.ymthe.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Forster AC, Altman S. External guide sequences for an RNA enzyme. Science. 1990;249:783–786. doi: 10.1126/science.1697102. [DOI] [PubMed] [Google Scholar]
- 8.Guerrier-Takada C, Altman S. Inactivation of gene expression using Ribonuclease P and external guide sequences. Methods Enzymol. 2000;313:442–456. doi: 10.1016/s0076-6879(00)13028-9. [DOI] [PubMed] [Google Scholar]
- 9.Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130:101–112. doi: 10.1016/j.cell.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Altman S. Inhibition of the expression of the human RNase P protein subunits Rpp21, Rpp25, and Rpp29 by external guide sequences (EGSs) and siRNA. J Mol Biol. 2004;342:1077–1183. doi: 10.1016/j.jmb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Guerrier-Takada C, Salavati R, Altman S. Phenotypic conversion of drug-resistant bacteria to drug sensitivity. Proc Natl Acad Sci USA. 1997;94:8468–8472. doi: 10.1073/pnas.94.16.8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKinney J, Guerrier-Takada C, Wesolowski D, Altman S. Inhibition of Escherichia coli viability by external guide sequences complementary to two essential genes. Proc Natl Acad Sci USA. 2001;98:6605–6610. doi: 10.1073/pnas.121180398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKinney J, Zhang H, Kubori T, Galan JE, Altman S. Disruption of type III secretion in Salmonella enterica serovar typhimurium by external guide sequences. Nucleic Acids Res. 2004;32:848–854. doi: 10.1093/nar/gkh219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soler Bistué AJ, et al. External guide sequences targeting the aac(6′)-Ib mRNA induce inhibition of amikacin resistance. Antimicrob Agents Chemother. 2007;51:1918–1925. doi: 10.1128/AAC.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lundblad EW, Xiao G, Ko J-h, Altman S. Rapid selection of accessible and cleavable sites in RNA by E. coli RNase P and random external guide sequences. Proc Natl Acad Sci USA. 2008;105:2354–2357. doi: 10.1073/pnas.0711977105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;13:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko J-h, Izadjoo M, Altman S. Inhibition of expression of virulence genes of Yersinia pestis in Escherichia coli by external guide sequences and RNase P. RNA. 2008;14:1656–1662. doi: 10.1261/rna.1120508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao G, Lundblad EW, Izadjoo M, Altman S. Inhibition of expression in Escherichia coli of a virulence regulator MglB of Francisella tularensis. PLoS ONE. 2008;3:e3719–e3725. doi: 10.1371/journal.pone.0003719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ko J-h, Altman S. OLE RNA, an RNA motif that is highly conserved for several extremophilic bacteria, is a substrate for and can be regulated by RNase P RNA. Proc Natl Acad Sci USA. 2007;104:7815–7820. doi: 10.1073/pnas.0701715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.