

Abstract
Sandhoff disease is a neurodegenerative disorder resulting from the autosomal recessive inheritance of mutations in the HEXB gene, which encodes the β-subunit of β-hexosaminidase. GM2 ganglioside fails to be degraded and accumulates within lysosomes in cells of the periphery and the central nervous system (CNS). There are currently no therapies for the glycosphingolipid lysosomal storage diseases that involve CNS pathology, including the GM2 gangliosidoses. One strategy for treating this and related diseases is substrate deprivation. This would utilize an inhibitor of glycosphingolipid biosynthesis to balance synthesis with the impaired rate of catabolism, thus preventing storage. One such inhibitor is N-butyldeoxynojirimycin, which currently is in clinical trials for the potential treatment of type 1 Gaucher disease, a related disease that involves glycosphingolipid storage in peripheral tissues, but not in the CNS. In this study, we have evaluated whether this drug also could be applied to the treatment of diseases with CNS storage and pathology. We therefore have treated a mouse model of Sandhoff disease with the inhibitor N-butyldeoxynojirimycin. The treated mice have delayed symptom onset, reduced storage in the brain and peripheral tissues, and increased life expectancy. Substrate deprivation therefore offers a potentially general therapy for this family of lysosomal storage diseases, including those with CNS disease.
The GM2 gangliosidoses are a group of glycosphingolipid (GSL) lysosomal storage diseases that includes Tay-Sachs disease, Sandhoff disease, and GM2 activator deficiency (1). They result from mutations in the genes encoding the hexosaminidase α-subunit, β-subunit and GM2 activator protein, respectively. They are characterized by progressive neurodegeneration in response to high levels of lysosomal storage of GM2 and related GSLs in neurons of the central nervous system (CNS) (1). There currently are no therapies for these diseases. Potential therapeutic strategies for Tay-Sachs and Sandhoff disease include enzyme augmentation and substrate deprivation (2, 3). Augmenting the level of enzyme can be achieved by using three clinical strategies: enzyme replacement, bone marrow transplantation, or gene therapy.
Intravenous administration of mannose-terminated glucocerebrosidase (β-d-glucosyl-N-acylsphingosine glucohydrolase, EC 3.2.1.45) is an effective therapy for type 1 Gaucher disease, which is a nonneurological GSL storage disease (4, 5). Because glycoprotein enzymes fail to cross the blood–brain barrier, this is not a suitable approach for diseases involving GSL storage in the CNS. Bone marrow transplantation has the potential to increase enzyme levels in the periphery and, to a limited extent, in the CNS because of secretion of enzyme from cells of bone marrow origin, including microglia (6). Results of bone marrow transplantation in GSL lysosomal storage diseases involving storage in the CNS have been mixed (7). Partial success was reported recently in a mouse model of Sandhoff disease given syngeneic wild-type bone marrow (8). This led to increased survival of the mice and improved neurological function. Gene therapy also has promise for treating these diseases, although this is currently experimental (9).
Substrate deprivation is a potentially generic pharmacological approach for treating the GSL storage diseases (3), including the GM2 gangliosidoses. This strategy is based on partial inhibition of the ceramide-specific glucosyltransferase (glucosylceramide synthase, UDP-glucose:N-acylsphingosine d-glucosyltransferase, EC 2.4.1.80), which catalyses the first step in GSL biosynthesis (10). This would reduce the levels of GSLs synthesized so they could be catabolized fully by the residual enzyme activity present in the cells.
Substrate deprivation, utilizing the GSL biosynthesis inhibitor N-butyldeoxynojirimycin (NB-DNJ), has been tested previously in an in vitro model of Gaucher disease and shown to prevent storage (11). NB-DNJ also has been evaluated in an asymptomatic mouse model of Tay-Sachs disease and shown to reduce GM2 accumulation in the brain and prevent the neuropathology associated with its storage (12). NB-DNJ currently is under clinical evaluation in type 1 Gaucher disease. In this study, we have extended the evaluation of substrate deprivation to the symptomatic mouse model of Sandhoff disease, which mimics the infantile onset variant (13). The Sandhoff disease mouse was generated through the targeted disruption of the Hexb gene and lacks hexosaminidase A and B isoenzymes resulting in GM2 and GA2 storage in the CNS and periphery (13). The mice undergo rapid, progressive neurodegeneration and die at 4–5 months of age (13). In this study, Sandhoff disease mice have been treated from 3 or 6 weeks of age with NB-DNJ. Substrate deprivation slows symptom onset, reduces GSL storage, and extends life expectancy.
MATERIALS AND METHODS
Animals and Treatment Procedures.
Sandhoff mice (13) were housed and drug-treated as described previously (14). The mice were bred as homozygotes. Females only produce one litter before symptom onset, and their disease course is accelerated if bred. The experimental females used in this study, therefore, were not bred and had a life expectancy similar to that of males. A humane end point was applied when the mice became moribund and unable to right themselves when laid on their side. NB-DNJ was a gift from Searle/Monsanto and Oxford Glycosciences.
Behavioral Tests.
Mice were tested (between 2:00 and 6:00 p.m.) individually with randomizing of subjects with respect to treatment, test sequence, and sex.
Horizontal Bar Test.
The apparatus used was modified from ref. 15. Briefly, a metal bar (1.2 mm × 26 cm) was suspended horizontally between two wooden supports 30 cm high (see Fig. 1) over a cushioned surface. The animal was held by the tail and allowed to grasp the center of the bar with forepaws only, the tail was released, and the clock was started. Latency to cross or fall from the bar was scored, with 120 sec, maximum, to termination of each trial. A score was given to each animal between +120 and −120. If crossing time (CT) was greater than 0, then score = 120 − CT; if falling time (FT) was greater than 0, the score = FT − 120.
Figure 1.

Representative behavioral analysis of Sandhoff mice treated from 6 weeks of age with 2,400 mg/kg per day of NB-DNJ. The data are from male siblings (n = 3 per group). The data are summarized as the means ± SD at each time point. (A) Performance of the mice in the bar-crossing test. (B) Performance of the mice in the inverted screen. (C) Activity levels on the inverted screen. ○, Untreated wild-type mice; ▵, untreated Sandhoff mice; and ▴, NB-DNJ-treated Sandhoff mice. Comparable data were obtained from female mice (data not shown).
Inverted Screen Test.
The mouse was placed in the center of a screen (30-cm2 square-wire mesh, 25-mm2 holes), and the screen was inverted over a 2-sec period with the mouse’s head declining. The screen was held steadily 20–30 cm above a solid, cushioned surface. Latency to falling from the screen was noted, with a 120-sec time limit for each trial. The percentage of time spent upside down on the screen was scored, as was the frequency of hindpaw grasping and release, as an indicator of activity levels of the mice.
Statistical Analysis.
Data from untreated and NB-DNJ treated mice were analyzed by using a nonlinear mixed-effects model (16) and s-plus 3.4 software (MathSoft, Seattle). The equation used was
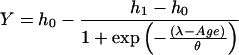 |
where ho and h1 are the lower and upper asymptotes, respectively, λ is the location parameter, and θ is the slope. The GSL levels were analyzed by using Student’s t test.
Biochemical Analysis.
The biochemical analysis was performed either on liver whole brain or on dissected brain regions according to published methods (12). Densitometry was performed by using nih image 1.49 software. Mass spectrometry analysis was performed to confirm the purity of the TLC bands that comigrated with the GM2 and GA2 standard. Briefly, glycolipids were separated by TLC (as above). The plate was sprayed with 1% iodine in 100% methanol (wt/vol). On the basis of detection of an authentic standard, the band in the experimental lanes was scraped and Folsch extracted. The glycolipids were permethylated and analyzed on a Micromass Tofspec 2E mass spectrometer (Micromass, Manchester, U.K.) operated in reflectron mode by using 2,5-dihydroxybenzoic acid as the matrix.
Histology.
Mice were sacrificed, the brains were removed, and one hemisphere of each was freeze-embedded in OCT compound (BDH). Cryostat sections (10 μm, Lauda 1720 Leitz) were collected onto poly-l-lysine-coated slides, air-dried for 4–6 hr, and stored at −70°C. For periodic acid/Schiff reagent (PAS) staining, frozen sections were warmed to room temperature, fixed in formalin-ethanol fixative (5 min, 3.7% formaldehyde in 95% ethanol), stained with PAS according to the manufacturer’s instructions (Sigma), counterstained with hematoxylin (Gill No. 3), and mounted in DePeX (BDH). Apoptotic cells were detected in the thalamus on frozen brain sections according to ref. 17 by using the ApoTag Plus in situ apoptosis detection kit (Oncor). The sections were counterstained with methyl green.
Electron Microscopy.
Mice were anesthetized (Hypnorm/Hypnovel i.p; Janssen/Roche Products, Welwyn Garden City, Hertfordshire, U.K.), perfused through the left cardiac ventricle with heparinized saline and then 2% paraformaldehyde/2% glutaraldehyde in PBS, and fixed at 4°C for 2–3 days. For brain and liver samples, the fixed tissue was sectioned (100 μm, vibrotome), washed three times in 0.1 M phosphate buffer, postfixed with 1% OsO4 in 0.1 M phosphate buffer for 35–40 min, dehydrated through an alcohol series and propylene oxide, placed in Durcupan-ACM resin (Fluka) overnight at room temperature, transferred to glass slides, and kept at 60°C for 48 hr. Selected areas of the tissues were cut and processed for ultra-thin sectioning, stained with uranyl acetate/lead citrate, and examined (Philips EM410 electron microscope; Philips Electron Optics, Cambridge, U.K.).
RESULTS
Symptom Onset and Life Expectancy.
Mice were tested for their ability to cross a horizontal bar or hang from an inverted screen as a measure of hind-limb strength (both tests) and motor coordination (horizontal bar). As neurodegeneration progressed, the mice became impaired in their performance in these tests because of hind limb paralysis and impaired coordination. The disease can be subdivided operationally into four discreet stages: the presymptomatic phase; early symptomatic phase, characterized by tremor but maintenance of muscle strength and coordination; the late symptomatic phase, characterized by progressive muscle wastage and reduced coordination; and the terminal phase, when the animals are moribund.
Sandhoff mice were treated from 3 or 6 weeks of age with 4,800 or 2,400 mg/kg of NB-DNJ per day, respectively. Serum levels were approximately 50 μM (data not shown), in keeping with previous studies in healthy mice (14). Approximately 10% of the serum level (5 μM) is reflected in the cerebrospinal fluid (12), which, in HL-60 cells in tissue culture, gives approximately 30% inhibition of GSL biosynthesis (18) and, against the ceramide glucosyltransferase in vitro, gives almost complete inhibition (Ki = 7 μM) (3, 12). The data were similar with both treatment groups, and a representative data set from mice treated from 6 weeks of age with 2,400 mg/kg NB-DNJ per day is shown (Fig. 1). Normal wild-type mice (or NB-DNJ-treated wild-type mice, data not shown) crossed the horizontal bar in less than the 120-sec duration of the test (Fig. 1A) (average crossing time, 7.8 sec ± 2.8). The early symptomatic phase began at 12 weeks of age and was characterized by a fine tremor. This aspect of the disease was not affected by NB-DNJ treatment. Untreated Sandhoff mice performed comparably to wild-type mice until approximately 104 days of age, when the late symptomatic phase began, their performance rapidly deteriorated, and they fell from the apparatus within 20 sec. The mice reached the humane end point at approximately 125 days of age. In contrast, NB-DNJ-treated mice crossed the bar at up to 136 days of age (approximately 30 days after the untreated mice reached the late symptomatic phase; P < 0.001), at which time they underwent rapid deterioration (Fig. 1A). When the slopes of the curves were compared, the rate of decline in the treated mice was significantly slower than in the untreated controls (P < 0.001). The terminal phase of the disease persisted for approximately 30 days, in contrast with approximately 15 days in the untreated controls. The NB-DNJ-treated mice were sacrificed at the humane end point at approximately 170 days of age as opposed to 125 days in the untreated controls, an increase in life expectancy of approximately 40%. The ability of the mice to hang from an inverted screen without falling from the apparatus during the test period of 120 sec also was determined (Fig. 1B). Similar observations were made of the data in Fig. 1A, with the untreated Sandhoff mice falling from the inverted screen within 2 min by 111 days (Fig. 1B). The mice steadily declined because of muscle wastage until they were killed at 125 days. The NB-DNJ-treated mice showed a significantly slower rate of decline (P < 0.001) in their performance until the humane end point at 170 days (Fig. 1). The untreated and NB-DNJ-treated mice were significantly different in terms of the age at which they exhibited reduced hind-limb movement (Fig. 1C, P < 0.001), but the rate of decline was comparable between the two groups (P = 0.1). The data were very similar irrespective of the sex of the animals (data not shown).
Glycosphingolipid Analysis.
Brain and liver GM2 and GA2 levels were determined by quantitative high-performance (HPTLC) from untreated or NB-DNJ-treated 112-day-old mice (Fig. 2A). Brain GM2 and GA2 levels were reduced significantly by 41% (P < 0.05) and 35% (P < 0.01), respectively, in NB-DNJ-treated mice. In liver, GM2 and GA2 levels were reduced significantly by 86% (P < 0.001) and 38% (P < 0.05), respectively (Fig. 2A). When brain regions were dissected and the GSL composition was analyzed it was apparent that the storage of GSLs was not significantly different in untreated and NB-DNJ-treated Sandhoff mice at the terminal stage of disease (125 and 170 days, respectively, Fig. 2B). The average storage levels of GM2 and GA2 were determined in whole brain and liver at the end stage of disease in the untreated and NB-DNJ-treated mice (125 and 170 days, respectively) (Fig. 2C). In brain, there were equivalent levels of GM2 and GA2 at the respective end points (in agreement with the regional analysis shown in Fig. 2B). In liver there were comparable levels of GA2 at the respective end points whereas GM2 was reduced significantly (41% lower in NB-DNJ-treated mice, P < 0.01). The data were similar irrespective of the sex of the animals used (data not shown).
Figure 2.

(A) Graphical representation of HPTLC analysis of brain and liver GM2 and GA2 in Sandhoff mice in the presence or absence of NB-DNJ. Mice were treated from weaning at 3 weeks with 4,800 mg/kg per day of NB-DNJ, and GSL profiles were compared at 16 weeks of age relative to untreated age-matched controls. The HPTLC plates were analyzed by densitometry in relation to authentic GSL standards. (B) Graphical representation of HPTLC analysis of brain region GM2 and GA2 content at the end stage of untreated controls (125 days) and NB-DNJ-treated Sandhoff mice (170 days). CC, cerebral cortex; THP, thalamus/hypothalamus; MPM, midbrain, pons, and medulla; CBL, cerebellum; SPN, spinal cord. (C) Comparison of GM2 and GA2 levels in whole brain and liver at the end stage of diseases in untreated (125 days) and NB-DNJ-treated mice (170 days). The data are presented as the mean ± SD for three mice per group. Solid bars depict data from untreated mice, and open bars depict data from NB-DNJ-treated mice. Representative data are shown. The data set shown is from male mice, and comparable data were obtained from females (data not shown).
Light Microscopy.
Brain sections were stained with PAS to visualize the storage GSL within different regions of the brain at 112 days. Untreated Sandhoff mouse brain showed high levels of storage in several areas, including the hippocampus, in which the granular cells and the polymorph layer of the dentate gyrus exhibited extensive storage (Fig. 3 A and C). The brains from Sandhoff mice treated with NB-DNJ had lower levels of storage compared with the untreated mice when the same hippocampal region was examined (Fig. 3 B and D). This was in keeping with the reduction in GM2 and GA2 storage levels observed by HPTLC analysis (Fig. 2A).
Figure 3.

(A) PAS staining of Sandhoff brain sections in the hippocampus (dentate gyrus). (A and C) Untreated brain. (B and D) NB-DNJ-treated brain. The images shown are representative of at least three animals per group. (Bar = 50 μm.) The reduction in PAS staining in NB-DNJ-treated mouse brains also was observed in other storage regions of the brain (data not shown). The data are from female mice, and comparable data were obtained from males (data not shown).
Electron Microscopy.
Representative electron micrographs from mice treated with 4,800 mg/kg per day for 90 days are shown in Fig. 4. Neurons from the polymorph layer of the dentate gyrus showed inclusions in the cytoplasm, which had zebra body-like morphology (Fig. 4a). At higher power these storage bodies could be seen to consist of sheets of lamellae (Fig. 4b). Some storage bodies had a more diffuse morphology or included circular vesicular structures surrounded by single- or multilayered membranous structures (Fig. 4a). When comparable neurons were examined in NB-DNJ-treated mice, the storage bodies were considerably less electron-dense (Fig. 4 c and d). The overall electron density of the cytoplasm also was observed to be lower in NB-DNJ-treated mice.
Figure 4.

Electron microscopy (low and high magnification) on brain and liver from untreated and NB-DNJ-treated Sandhoff mice (4,800 mg/kg per day for 90 days). Neurons from the polymorph layer of the dentate gyrus: untreated neuron (a and b), NB-DNJ-treated neuron (c and d), untreated hepatocyte (e and f), NB-DNJ-treated hepatocyte (g and h), untreated Kupffer cell (i and j), and NB-DNJ-treated Kupffer cell (k and l). The images are representative of data obtained from at least three mice. The images shown are from female mice, and comparable data were obtained from males (data not shown). (Bar = 0.5 μm.)
In untreated Sandhoff mouse hepatocytes, the storage bodies had a relatively homogeneous morphology comprising multiple vesicular structures contained within a single membrane-enclosed structure (Fig. 4 e and f). In NB-DNJ-treated mice the hepatocytes contained large numbers of cytoplasmic vesicles containing diffuse contents and fewer circular vesicular inclusions (Fig. 4 g and h). Much of the material within the hepatocyte vesicles has an appearance similar to glycogen. This has been confirmed in healthy mice treated with NB-DNJ, but does not appear to cause progressive storage of glycogen or cause any overt pathology (U. Andersson and F.M.P., unpublished observation). The biochemical basis for this change in glycogen catabolism currently is under investigation. In Kupffer cells the storage material was morphologically diverse and comprised predominantly zebra body-like structures and, to a lesser extent, vesicular structures similar to those seen in hepatocytes (Fig. 4 i and j). At higher magnification the storage material could be seen to be organized into lamellae similar to those seen in neurons of the brain (Fig. 4 a and b). In NB-DNJ-treated mice the storage material within the Kupffer cell storage vesicles was diffuse because of reduced electron density (Fig. 4 k and l). Lamellal structures were infrequent and poorly defined (Fig. 4l).
Apoptotic Cell Death.
It has been reported previously (17) that a feature of Sandhoff disease is the apoptotic death of neurons. This has been determined in human autopsy samples and in Sandhoff mice at an advanced stage of disease (17). Therefore, we investigated whether NB-DNJ treatment of mice prevented apoptotic death of neurons. Animals at 17 weeks of age were compared. At this stage the untreated mice were at an advanced stage of disease whereas the NB-DNJ-treated mice were still performing well in the behavioral tests. When comparable sections of the thalamus were stained by using DNA end-labeling techniques, positive cells (dying neurons) were readily observed in the untreated mice but were absent in the NB-DNJ-treated mice (Fig. 5). Similar observations were made in other brain regions (data not shown).
Figure 5.

Detection of apoptotic cells in the thalamus of untreated Sandhoff mouse at 17 weeks of age (A) and an NB-DNJ-treated mouse at 17 weeks of age (B). The apoptotic cells appear brown (arrows). (Bar = 50 μm.) The sections were derived from male mice, and comparable data were obtained from females (data not shown).
DISCUSSION
Substrate deprivation is anticipated to be most effective in adult- and juvenile-onset GSL storage disease variants, where low to moderate levels of residual enzyme activity are present. In the infantile-onset variants it should be possible only to slow symptom onset and increase life expectancy, but not totally arrest the disease process. The limiting factor is the availability of sufficient residual enzyme to catabolize fully the reduced substrate levels entering the lysosome.
Having shown previously that substrate deprivation prevents GSL storage in the asymptomatic mouse model of Tay-Sachs disease (12), we have evaluated substrate deprivation in a disease model that closely mimics the most severe forms of these diseases, that is, the infantile-onset variants. The Sandhoff mouse is a progressive neurodegenerative model that has very low levels of residual enzyme activity conferred by the minor hexosaminidase S (αα) isoenzyme. The mice die at 4–5 months of age (13).
When Sandhoff mice were treated with NB-DNJ, either at 3 or 6 weeks of age, life expectancy was extended by approximately 40%. This correlated with reduced GSL storage both in peripheral tissues, such as liver, and in the CNS. If therapy was commenced during the late presymptomatic period (11 weeks of age), no increase in life expectancy could be achieved, suggesting that an irreversible disease process had commenced that could not be halted or reversed (data not shown). The first clinical signs of disease in the Sandhoff mice begin at 12 weeks of age and are characterized by tremor, which probably reflects CNS dysfunction. This aspect of the disease was not affected by NB-DNJ treatment. Very early postnatal or even in utero administration of drug may be required to impact this aspect of the pathology. After the onset of the later symptomatic phase, characterized by changes in motor coordination, the rate of decline was significantly different in untreated and NB-DNJ-treated mice, as was the age at which deterioration could be detected in the behavioral tests (approximately 100 days for untreated mice and approximately 135 days for NB-DNJ-treated mice). This probably results from a slower rate of GSL accumulation, leading to a prolongation of the presymptomatic phase and a slower rate of symptom progression. However, the terminal stage of the disease was also prolonged in NB-DNJ-treated mice. When GSL storage levels were measured in the untreated and NB-DNJ-treated Sandhoff mice at their end points (125 and 170 days, respectively), the GM2 and GA2 levels were comparable, indicating that death correlated with the same GSL storage levels in the brains of the two groups of mice. In liver, GA2 levels were comparable at the respective end points whereas GM2 levels were reduced by 41% in the NB-DNJ-treated mice. A more detailed understanding of the underlying mechanisms of pathogenesis is required before it can be determined whether peripheral or CNS storage is the critical component in the natural history of this disease. Histological examination of the mice at 120 days revealed lower levels of storage in the brain of NB-DNJ-treated mice, and at the ultrastructural level the neurons had greatly reduced storage burdens. This was true to an even greater extent in liver. The liver in common with other peripheral organs is exposed to higher levels of NB-DNJ, whereas only approximately 10% of the serum level is reflected in the cerebrospinal fluid (12). When the brains of untreated and NB-DNJ-treated mice were compared at 17 weeks of age, apoptotic cells were observed in the thalamus of the untreated mice but were absent from the NB-DNJ-treated mice. There is therefore a correlation between maintenance of performance in the behavioral tests and lack of neuronal apoptosis. The mechanism(s) through which apoptosis is triggered in response to GM2 and GA2 storage are not known (17). It is likely that NB-DNJ treatment keeps the storage levels below the critical threshold, which triggers neuronal death and, therefore, keeps the mice asymptomatic for longer relative to the untreated controls.
Substrate deprivation also has been evaluated by using a genetic approach in which Sandhoff mice have been crossed with mice null for β1,4-N-acetylgalactosaminyltransferase. These animals lack the substrate that would be catabolized by the defective enzyme and survive long term, indicating that balancing synthesis with degradation using this extreme genetic strategy is also effective (19).
It is interesting to compare our finding with those observed when Sandhoff mice were bone marrow transplanted (BMT) (8). The mice survived for up to 4 months longer than the untreated mice, and, in keeping with this study, the BMT-treated animals had a relatively normal phenotype at the time when the untreated controls were moribund. BMT transplantation reduced GSL storage in peripheral tissues but not in the CNS, and the CNS storage burden was actually higher in the BMT animals because of their enhanced survival. The overall levels of GM2 and GA2 storage in the CNS, therefore, is not the only factor involved in the onset of clinical disease, and the mechanisms of pathogenesis are clearly more complex than previously appreciated. In the NB-DNJ-treated Sandhoff mice, substrate deprivation had a profound effect on levels of peripheral storage (in keeping with BMT) but also reduced GSL storage in the CNS. NB-DNJ crosses the blood–brain barrier and therefore can impact neuronal storage in a way that is difficult to achieve with BMT. It is currently unclear why survival is more prolonged with BMT compared with NB-DNJ treatment even though NB-DNJ is achieving significant reduction in GSL storage in the CNS. It will be of interest to determine whether low-dose therapy with NB-DNJ, which primarily impacts the periphery rather than the CNS, will have the same effect as high-dose therapy, where NB-DNJ reaches relatively high levels in the CNS and inhibits GSL biosynthesis. Because augmentation of enzyme levels via BMT and substrate deprivation may act synergistically, it also will be of interest to treat BMT Sandhoff mice with NB-DNJ to see whether life expectancy can be enhanced beyond that of either therapy alone. We currently do not understand the precise mechanism(s) through which life expectancy is enhanced by either BMT or substrate deprivation. However, elucidation of these mechanisms should shed light on the underlying pathological process that results from the failure to degrade GM2 and GA2.
Slowing the disease course appears to be effective only if the therapy commences before symptom onset, which would limit the application of NB-DNJ to individuals genotyped in early infancy, who are presymptomatic at the time therapy commences. However, the ethics of treating simply to slow the course of the disease is questionable. It could be argued that slowing symptom onset could provide time to find a matched bone marrow donor, but this is offset by the fact that if substrate deprivation alone is used, the end stage of the disease, when the patient has advanced neurodegeneration, may be prolonged. What would seem more appropriate is evaluating this therapy for juvenile- and adult-onset variants of the CNS diseases or in infantile disease only if residual enzyme levels are augmented by BMT or gene therapy.
Testing of substrate deprivation in mouse models less extreme than those generated by “knock out” technology may be more appropriate for modeling the outcome of substrate deprivation in late-onset disease variants. To this end, a second generation of animal models has been reported recently in which the endogenous gene has been replaced by a gene encoding the human mutant gene, for instance, Gaucher disease models (20). Because of biological differences between mice and humans, the mice fail to form a ceramide barrier in the skin, dehydrate, and do not survive. However, it is anticipated that strategies could be found to overcome this problem. It is clear that this general approach should produce models that more closely mimic the human disease in which this and other therapies can be evaluated.
In summary, substrate deprivation using NB-DNJ or other GSL biosynthesis inhibitors (18, 21) holds promise as a general therapy for this family of lysosomal storage diseases, including those that exhibit CNS pathology. It also has the potential to be of benefit in severe infantile forms of these diseases if combined with an enzyme-augmenting therapy such as BMT, although this will await formal testing in animal models.
Acknowledgments
We thank Julia McAvoy and David Smith for excellent technical assistance, Mandy Townsend for help with electron microscopy, David Harvey for advice on mass spectrometry, Colin Beesley and Daniel Anthony for photography, John Freeman for graphics, and Rob Deacon for his advice on behavioral tests. We thank Searle/Monsanto and Oxford Glycosciences for NB-DNJ. F.M.P. is a Lister Institute Research Fellow, and M.J. is supported by a Biotechnology and Biological Sciences Research Council studentship.
ABBREVIATIONS
- CNS
central nervous system
- GSL
glycosphingolipid
- NB-DNJ
N-butyldeoxynojirimycin
- PAS
periodic acid/Schiff reagent
- HPTLC
high-performance TLC
References
- 1.Gravel R A, Clarke J T R, Kaback M M, Mahuran D, Sandhoff K, Suzuki K. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C R, Beadet A L, Sly W S, Valle D, editors. Vol. 2. New York: McGraw–Hill; 1995. pp. 2839–2879. [Google Scholar]
- 2.Radin N S. Glycoconj J. 1996;13:153–157. doi: 10.1007/BF00731489. [DOI] [PubMed] [Google Scholar]
- 3.Platt F M, Butters T D. Biochem Pharmacol. 1998;56:421– 430. doi: 10.1016/s0006-2952(98)00115-4. [DOI] [PubMed] [Google Scholar]
- 4.Grabowski G A, Barton N W, Pastores G, Dambrosia J M, Banerjee T K, McKee M A, Parker C, Schiffmann R, Hill S C, Brady R O. Ann Intern Med. 1995;122:33–39. doi: 10.7326/0003-4819-122-1-199501010-00005. [DOI] [PubMed] [Google Scholar]
- 5.Beutler E, Kay A, Saven A, Garver P, Thurston D, Dawson A, Rosenbloom B. Blood. 1991;78:1183–1189. [PubMed] [Google Scholar]
- 6.Krivit W, Sung J H, Shapiro E G, Lockman L A. Cell Transplant. 1995;4:385–392. doi: 10.1177/096368979500400409. [DOI] [PubMed] [Google Scholar]
- 7.Hoogerbrugge P M, Brouwer O F, Bordigoni P, Ringden O, Kapaun P, Ortega J J, O’Meara A, Cornu G, Souillet G, Frappaz D, et al. Lancet. 1995;345:1398–1402. doi: 10.1016/s0140-6736(95)92597-x. [DOI] [PubMed] [Google Scholar]
- 8.Norflus F, Tifft C J, McDonald M P, Goldstein G, Crawley J N, Hoffmann A, Sandhoff K, Suzuki K, Proia R L. J Clin Invest. 1998;101:1881–1888. doi: 10.1172/JCI2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salvetti A, Heard J M, Danos O. Br Med Bull. 1995;51:106–122. doi: 10.1093/oxfordjournals.bmb.a072940. [DOI] [PubMed] [Google Scholar]
- 10.Sandhoff K, van Echten G. Adv Lipid Res. 1993;26:119–142. [PubMed] [Google Scholar]
- 11.Platt F M, Neises G R, Dwek R A, Butters T D. J Biol Chem. 1994;269:8362–8365. [PubMed] [Google Scholar]
- 12.Platt F M, Neises G R, Reinkensmeier G, Townsend M J, Perry V H, Proia R L, Winchester B, Dwek R A, Butters T D. Science. 1997;276:428–431. doi: 10.1126/science.276.5311.428. [DOI] [PubMed] [Google Scholar]
- 13.Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald M P, Crawley J N, Sandhoff K, Suzuki K, Proia R L. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- 14.Platt F M, Reinkensmeier G, Dwek R A, Butters T D. J Biol Chem. 1997;272:19365–19372. doi: 10.1074/jbc.272.31.19365. [DOI] [PubMed] [Google Scholar]
- 15.Barclay L L, Gibson G E, Blass J P. Pharmacol Biochem Behav. 1981;14:153–157. doi: 10.1016/0091-3057(81)90236-7. [DOI] [PubMed] [Google Scholar]
- 16.Vonesh E F, Chinchilli V M. Linear and Nonlinear Models for the Analysis of Repeated Measurements. New York: Dekker; 1997. [Google Scholar]
- 17.Huang J Q, Trasler J M, Igdoura S, Michaud J, Hanai N, Gravel R A. Hum Mol Genet. 1997;6:1879–1885. doi: 10.1093/hmg/6.11.1879. [DOI] [PubMed] [Google Scholar]
- 18.Platt F M, Butters T D. Trends Glycosci Glycotechnol. 1995;7:495–511. [Google Scholar]
- 19.Liu Y, Wada R, Hiromichi K, Sango K, Deng C, Tai T, McDonald M P, Araujo K, Crawley J N, Bierfreund U, et al. J Clin Invest. 1999;103:497–505. doi: 10.1172/JCI5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y J, Suzuki K, Reed J D, Grinberg A, Westphal H, Hoffmann A, Doring T, Sandhoff K, Proia R L. Proc Natl Acad Sci USA. 1998;95:2503–2508. doi: 10.1073/pnas.95.5.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Platt F M, Neises G R, Karlsson G B, Dwek R A, Butters T D. J Biol Chem. 1994;269:27108–27114. [PubMed] [Google Scholar]