

Abstract
IgA nephropathy is a chronic kidney disease defined by deposition of IgA in the glomeruli. An abnormality in the glycosylation of the hinge region of the IgA1 isotype of IgA is fundamental to the origins of this very common form of glomerulonephritis. In this issue of the JCI, Suzuki and coworkers describe the characteristics of IgG autoantibodies to the abnormally glycosylated IgA1 secreted by immortalized B cells derived from patients with sporadic forms of IgA nephropathy (see the related article beginning on page 1668). These IgG autoantibodies displayed remarkably restricted heterogeneity. These observations offer new insights into disease pathogenesis and may lead to new methods of diagnosis, monitoring, and therapy for patients with IgA nephropathy.
Just over four decades ago, a brief report appearing in a French-language journal, for which the authors utilized the new technique of immunofluorescence microscopy, described the observation of mesangial deposits of IgA and IgG in renal biopsies from a small number of patients with recurrent hematuria (1). Further descriptions of this disorder, which came to be known as Berger disease (after the first author of this now famous paper), quickly spread throughout the globe. In less than 20 years after its first description, this disorder was declared by Giuseppe D’Amico, one of the renowned early students of the disease, to be the “commonest glomerulonephritis in the world” (2). The extraordinary diversity of its clinical as well as light microscopic pathological findings has been amply demonstrated, and its progression to end-stage renal failure over a protracted period of 10–30 years in a substantial fraction of patients (25%–50%) is now widely appreciated (3).
IgA nephropathy, as this disorder is now generally called, is characterized by the predominant deposition of IgA in a granular fashion diffusely in the mesangial zones of glomeruli often, but not universally, accompanied by deposits of IgG in the same pattern (Figure 1) (4). IgA nephropathy was the first kidney disease to derive its name from the character of its immunopathological findings. Its exact origins in history are not well known, but it likely appeared much earlier than its first description in 1968. Indeed, a recent study suggested that Royal Prince Joseph Habsburg died with (or of) IgA nephropathy (and gouty nephropathy as well) in 1847 (5).
Figure 1. IgA nephropathy.
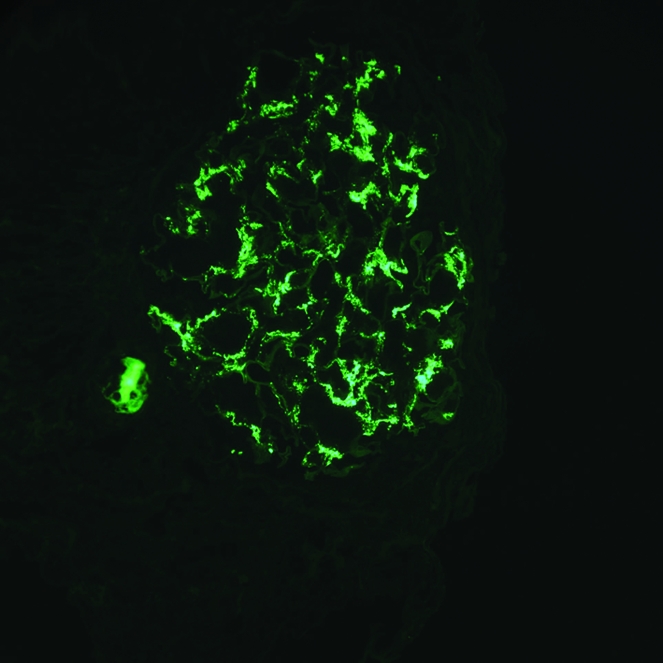
Immunofluorescence microscopy reveals granular deposits of IgA in the glomerular mesangium. Original magnification, ×400. Courtesy of Arthur Cohen, David Geffen School of Medicine at UCLA, Los Angeles, California, USA.
Deposition and abnormal glycosylation of IgA1
The pathogenesis of IgA nephropathy was initially a profound mystery. Although the pattern of glomerular IgA/IgG deposits suggested an immune complex–mediated mechanism, this remained largely an unproven assertion. Several discoveries have considerably clarified this mystery. Two of the three most important prior breakthroughs concerning the pathogenesis of IgA nephropathy have been published in the JCI (6, 7). The first of these appeared in 1980, when Conley and coworkers described the selective deposition of the IgA1 subclass of IgA in the mesangial zones of patients with IgA nephropathy (6). IgA1 is the dominant circulating form of IgA, and it is prominently synthesized in the bone marrow (and the tonsil as well) of hominid primates but not other species (8). IgA1 is unique, as it contains a number of O-linked glycan side chains (containing N-acetylgalactosamine, galactose, and sialic acid) in the hinge region of the molecule (unlike other immunoglobulins or IgA2) (8). Immunopathological findings identical to those of IgA nephropathy are also found in the related condition of Henoch-Schönlein purpura nephritis (4). At an international symposium held in Nancy, France, in August/September 1992 to commemorate the 25th anniversary of the discovery of IgA nephropathy, Mestecky and coworkers (9) and Hiki and colleagues (10, 11) first noted that the IgA1 molecule was aberrantly glycosylated in IgA nephropathy. By 1995, this finding had been confirmed by Allen and coworkers (12), and the glycosylation abnormality of IgA1 in IgA nephropathy is now very firmly established. This observation had been suggested by an earlier study using lectin (jacalin) binding to circulating IgA by Andre et al. (13).
It is now apparent that levels of this defective form of IgA1 are commonly elevated in the serum of patients with IgA nephropathy (14). Immune complexes were found in the serum (and urine) of patients with IgA nephropathy containing both the aberrantly glycosylated IgA1 and IgG autoantibodies to the abnormal IgA1 (15). The glomerular immune deposits also contain the abnormal IgA1 (15).
Immortalization of B cells from IgA nephropathy patients
The final series of breakthroughs in our understanding of the pathogenesis of IgA nephropathy occurred very recently as a consequence of the cloning and immortalization of B cells from subjects with the disorder (7), making it possible to study the fine details and the possible mechanisms responsible for the defective glycosylation of IgA1 and also to examine in greater depth the character of the autoantibodies developing against the glycan-associated neoepitopes on IgA1. In 2008, Suzuki and colleagues studied such immortalized human B cell lines and clearly demonstrated that unique abnormalities involving premature enzymatic sialylation and/or reduced galactosylation of the O-linked serine residues at the hinge region of IgA1 were the basis for the aberrant glycosylation of circulating and glomerular deposits of IgA1 in individuals with IgA nephropathy (7). These observations set the stage for their current study, appearing in this issue of the JCI, in which the authors undertook the molecular characterization of IgG autoantibodies to the abnormal IgA1 molecule secreted by immortalized B cells from subjects with IgA nephropathy and compared these autoantibodies to those secreted by B cells from normal subjects (16). The IgG autoantibodies generated by these immortalized B cell clones reacted specifically with the N-acetylgalactosamine (GalNAc) epitopes on the IgA1 molecule. Thus, B cells in IgA nephropathy are programmed to manufacture both the autoantigen and the autoantibodies to the autoantigen (a situation believed to be unique in autoimmune disease). Presumably, the individual B cell sources for these two components of immune complexes in IgA nephropathy are distinct.
Most strikingly, Suzuki and coworkers found that these IgG autoantibodies contained a common A to S amino acid substitution in a region of the IgG VH molecule arising from mutations of the variable region of the IGH gene (IgG VH). These autoantibody-specific alterations greatly enhanced the binding characteristics of the IgG autoantibodies to the galactose-deficient IgA1. This study also suggests that these mutations arose because of positive selection during the immune response. However, the design of the study could not differentiate whether the unique molecular “signature” of IgG autoantibodies that react to galactose-deficient IgA1 develops because of underlying genetic variation or by somatic mutation. In this regard, it is important to note that none of the subjects serving as donors of the lymphocytes for B cell immortalization were identified as having a familial form of IgA nephropathy.
In any case, the description of a unique molecular “signature” for IgG autoantibodies in IgA nephropathy offers a novel approach to both the diagnosis and monitoring of this disorder, as suggested from the preliminary observations of Suzuki et al. (16). Perhaps more importantly, it also opens up a new avenue for the development of “designer” therapy directed at inhibition of the interaction between the “mutant” IgG autoantibodies and their relevant epitopes on the aberrantly glycosylated IgA1 molecule.
Cautions
Several cautions are in order as one attempts to interpret and integrate the findings of Suzuki et al. (7, 16) in the context of other observations regarding IgA nephropathy. First, IgG is not universally deposited in the glomeruli of subjects with IgA nephropathy. The percentage of renal biopsies showing concomitant IgG deposition shows considerable variation, from approximately 15% to 80% (4). If these IgG autoantibodies to aberrantly glycosylated IgA1 are crucial to disease pathogenesis, why are they not universally found in clinically affected subjects? Perhaps glomerular deposition of IgG autoantibodies modifies the severity of the disease but is not itself the “prime mover.” Suzuki et al. have attempted to address this issue and suggest that the variation in codeposition of IgG observed in IgA nephropathy might be due to differences in methods, timing of renal biopsy, or the sensitivity of the immunofluorescence microscopy utilized for the detection of IgG. Alternatively, there may be a predominance of IgA autoantibodies to the abnormally glycosylated IgA1 in some patients with IgA nephropathy. Clearly, further studies will be necessary to sort out this conundrum.
Under-galactosylated IgA1 also tends to spontaneously aggregate and may bind nonspecifically to constituents of extracellular matrix (“sticky IgA”) (17). Theoretically, it is possible that these aberrant IgA1 molecules deposit as “planted” antigens in glomeruli and then the circulating IgG autoantibodies react with the predeposited under-galactosylated IgA1. In this formulation, circulating IgA1/IgG immune complexes would not play a necessary role in disease pathogenesis. However, there is little doubt that immune complexes composed of aberrantly glycosylated IgA1 and IgG autoantibodies to this abnormal IgA1 molecule as well as free (uncomplexed) IgG autoantibodies to the abnormal IgA1 are present in the circulation of subjects with IgA nephropathy (15). Further, in IgA nephropathy, serum C3 complement levels are typically normal, and extensive glomerular mesangial deposition of C3 is very common, but prominent C1q deposition is seldom observed, indicating conspicuous local activation via the alternate and/or mannose-binding lectin pathways for complement activation (18). The subclass of the IgG produced by the immortalized B cells was IgG1 in the studies reported by Suzuki et al. (16). According to Aucouturier et al. (19), the IgG1 and IgG3 subclasses are present in 81% and 63%, respectively, of renal biopsies showing IgA nephropathy. This observation provides good concurrence between the subclass of IgG autoantibodies from B cells and actual glomerular deposits. IgG1 antibodies are known to activate the classical complement cascade (18). The discrepancy between the pattern of glomerular complement component deposition (prominent C3, scanty C1q) and the subclass of IgG glomerular deposition (IgG1 and IgG3) deserves some explanation. Finally, it has also been noted that apparently healthy individuals (dying of suicide or trauma or serving as donors for kidney transplantation) have an appreciable prevalence (4%–16%) of “latent” IgA deposits in the glomeruli, as determined by immunofluorescence (20). It is not known whether these individuals also demonstrate aberrantly glycosylated IgA1 in these deposits, but the codeposition of IgG (and C3 as well) is much less common in these circumstances than in subjects affected with clinically apparent IgA nephropathy. In the studies of Moldoveanu et al. (14) and Suzuki et al. (16), it is noteworthy that some control subjects have slightly elevated serum levels of under-galactosylated IgA1 or an IgG VH amino acid sequence similar to the “signature sequence” of the IgG from patients with IgA nephropathy. A few control subjects showed binding of IgG to under-galactosylated IgA1 similar to that found in patients with IgA nephropathy. One might speculate that these are the same individuals that might show “latent” IgA deposits in their glomeruli.
Conclusions
Despite these cautionary statements, little doubt exists that the elegant and breakthrough studies reported by Suzuki et al. (7, 16) have brought us dramatically closer to a comprehensive understanding of the pathogenesis of IgA nephropathy. One can only hope that the promise of newer, sensitive and specific, noninvasive diagnostic tests, better means of monitoring disease activity, and novel disease-specific treatments for this disease will quickly ensue from these cutting-edge investigations. The hundreds of thousands of patients with already diagnosed IgA nephropathy and the many more who will be diagnosed with this very common disorder in the future eagerly await these developments.
Footnotes
Conflict of interest: The author has received consulting fees from Aspreva (Vifor Pharma), Genentech, Novartis, FibroGen, and Quest Diagnostics/Nichols Institute.
Nonstandard abbreviations used: GalNAc, N-acetylgalactosamine.
Citation for this article: J. Clin. Invest. 119:1450–1452 (2009). doi:10.1172/JCI39189.
See the related article beginning on page 1668.
References
- 1.Berger J., Hinglais N. Les depots intercapillaries d’IgA-IgG (Intercapillary deposits of IgA-IgG). J. Urol. Nephrol. (Paris). 1968;74:694–695. [PubMed] [Google Scholar]
- 2.D’Amico G. The commonest glomerulonephritis in the world: IgA nephropathy. Q. J. Med. 1987;64:709–727. [PubMed] [Google Scholar]
- 3.D’Amico G., et al. Idiopathic IgA mesangial nephropathy. Clinical and histiological study of 374 patients. Medicine (Baltimore). 1985;64:49–60. [PubMed] [Google Scholar]
- 4. Habib, R., Niaudet, P., and Levy, M. 1994. Schonlein-Henoch purpura nephritis and IgA nephropathy. InRenal pathology with clinical and functional correlations. C.C. Tisher and B.M. Brenner, editors. J.B. Lippincott & Co. Philadelphia, Pennsylvania, USA. 472–523. [Google Scholar]
- 5.Jozsa L.G. Histologic diagnosis of tissue from two nineteenth century Habsburgs. Paleopathol. Newsl. 2008;141:12–18. [PubMed] [Google Scholar]
- 6.Conley M.E., Cooper M.D., Michael A.F. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura and systemic lupus erythematosus. . J. Clin. Invest. 1980;66:1432–1436. doi: 10.1172/JCI109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki H., et al. IgA1 secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Invest. 2008;118:629–639. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feehally J., Allen A.C. Structural features of IgA molecules which contribute to IgA nephropathy. J. Nephrol. 1999;12:59–65. [PubMed] [Google Scholar]
- 9.Mestecky J., et al. Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib. Nephrol. 1993;104:172–182. doi: 10.1159/000422410. [DOI] [PubMed] [Google Scholar]
- 10. Hiki, Y., Iwase, H., and Saitoh, M. 1992. O-linked oligosacchardie (Gal β1,3 GalNAc) on IgA hinge region in IgA nephropathy [abstract]. InIgA nephropathy. The 25th year International Symposium. M.C. Bene, G.C. Faure, and M. Kessler, editors. Karger Medical and Scientific Publishers. Nancy, France/Basel, Switzerland. 222 pp. [Google Scholar]
- 11.Hiki Y., et al. O-linked oligosaccharide on IgA1 hinge region in IgA nephropathy. Fundamental study for precise structure and possible role. Contrib. Nephrol. 1995;111:73–84. [PubMed] [Google Scholar]
- 12.Allen A.C., Harper S.J., Fehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin. Exp. Immunol. 1995;100:470–474. doi: 10.1111/j.1365-2249.1995.tb03724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andre P.M., Le Pogamp P., Chevet D.1990Impairment of jacalin binding to serum IgA in IgA nephropath y . J. Clin. Lab. Anal. 4115–119. 10.1002/jcla.1860040208 [DOI] [PubMed] [Google Scholar]
- 14.Moldoveanu Z., et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007;71:1148–1154. doi: 10.1038/sj.ki.5002185. [DOI] [PubMed] [Google Scholar]
- 15.Novak J., Julian B.A., Tomana M., Mestecky J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin. Nephrol. 2008;28:78–87. doi: 10.1016/j.semnephrol.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suzuki H., et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Invest. 2009;119:1668–1677. doi: 10.1172/JCI38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kokubo T., et al. Protective role of IgA1 glycan against IgA1 self-aggregation and adhesion to intracellular matrix proteins. J. Am. Soc. Nephrol. 1998;9:2048–2054. doi: 10.1681/ASN.V9112048. [DOI] [PubMed] [Google Scholar]
- 18. Hebert, L.A., Cosio, F.G., and Birmingham, D.J. 2001. Complement and complement regulatory proteins in renal disease. InImmunologic renal diseases. 2nd edition. E.G. Neilson and W.G. Couser, editors. Lippincott Williams & Wilkins. Philadelphia, Pennsylvania, USA. 367–394. [Google Scholar]
- 19.Aucouturier P., Monteiro R.C., Noel L.H., Preud’homme J.L., Lesavre P. Glomerular and serum immunoglobulin G subclasses in IgA nephropathy. Clin. Immunol. Immunopathol. 1989;51:338–347. doi: 10.1016/0090-1229(89)90032-9. [DOI] [PubMed] [Google Scholar]
- 20. Glassock, R.J. Future prospects for IgA nephropathy. 2009. InAdvances in IgA nephropathy. K.N. Lai, editor. World Scientific Publishing Co. Singapore. 403–411 [Google Scholar]