

Summary
A large scale bioinformatics and molecular analysis of a 34 Mb interval on chromosome 6q12 was undertaken as part of our ongoing study to identify the gene responsible for an autosomal recessive retinitis pigmentosa (arRP) locus, RP25. Extensive bioinformatics analysis indicated in excess of 110 genes within the region and we also noted unfinished sequence on chromosome 6q in the Human Genome Database, between 58 and 61.2 Mb. Forty three genes within the RP25 interval were considered as good candidates for mutation screening. Direct sequence analysis of the selected genes in 7 Spanish families with arRP revealed a total of 244 sequence variants, of which 67 were novel but none were pathogenic. This, together with previous reports, excludes 60 genes within the interval (~55%) as disease causing for RP. To investigate if copy number variation (CNV) exists within RP25, a comparative genomic hybridization (CGH) analysis was performed on a consanguineous family. A clone from the tiling path array, chr6tp-19C7, spanning ~100-Kb was found to be deleted in all affected members of the family, leading to a major refinement of the interval. This will eventually have a significant impact on cloning of the RP25 gene.
Keywords: retinitis pigmentosa, mutation screening, gene expression
Introduction
Retinal dystrophies represent the most common inherited form of human visual handicap, with an estimated prevalence of 1 in 3000. They can be classified according to which photoreceptor system, rods or cones, is primarily affected (Bird, 1995). Disorders primarily affecting the rod system such as retinitis pigmentosa (RP) present initially with night blindness and may progress to involve the peripheral visual field, with relative preservation of central vision. On the other hand, cone dystrophies manifest initially with loss of central visual acuity and defects in colour vision without significant loss of peripheral vision.
Several loci with retinal dystrophy phenotypes have been mapped to the pericentromeric region of chromosome 6 (6q14-q21) (Abd El-Aziz et al. 2005). The genes for most of these loci have been identified. For example, ELOVL4 is the gene for autosomal dominant Stargardt-like disease (STGD3) as well as for autosomal dominant macular dystrophy (adMD) (Zhang et al. 2001). RIM1 and IMPG1 have also been reported as the causative genes for cone-rod dystrophy (CORD7) and benign concentric annular macular dystrophy (BCAMD), respectively (Johnson et al. 2003; van Lith-Verboeven et al. 2004). Recently, the gene for Leber congenital amaurosis type 5 (LCA5), Lebercillin, was also identified (den Hollander et al. 2007). It is interesting to note that the above retinal genes overlap with the autosomal recessive RP locus, RP25, for which the gene still remains to be identified.
The locus for RP25 was originally established by homozygosity mapping through targeting functional candidate genes. This resulted in the mapping of 4 Spanish families with arRP between microsatellite markers D6S257 and D6S1644 (Ruiz et al. 1998). The region flanked by these markers is ~34 Mb in size and contains approximately 110 known and predicted genes. Subsequently, evidence of linkage to the same region has been reported in three additional Spanish families with arRP as well as in other ethnic groups (Khaliq et al. 1999; Barragan et al. 2005a; Abd El-Aziz et al. 2007). Based on these findings, the isolation of the RP25 gene would represent a major achievement in RP research.
Previously we and others reported the exclusion of 17 genes from the RP25 interval (Marcos et al. 2000; Li et al. 2001; Marcos et al. 2002, 2003; Barragan et al. 2005a, 2005b; Abd El-Aziz et al. 2005, 2006; Barragan et al. 2008. Herein we report extensive molecular analysis of the original RP25 interval (~34 Mb) and mutation screening of a further 43 genes; therefore along with previous reports, 55% of the 110 genes have been evaluated as to their role in disease-causation for RP25. In addition, a comparative genomic hybridization (CGH) analysis was performed on one of the RP25 families to investigate whether copy number variations (CNVs) exist and if this could have an impact on the RP25 phenotype.
Materials and Methods
Families and DNA
Seven Spanish families, two consanguineous (RP5 and RP167) and five non-consanguineous (RP73, RP214, RP299, RP260 and RP235), were included in the study (Fig. 1). Informed consent was obtained from all participants for clinical and molecular genetic studies. The study conformed to the tenets of the Declaration of Helsinki.
Figure 1.
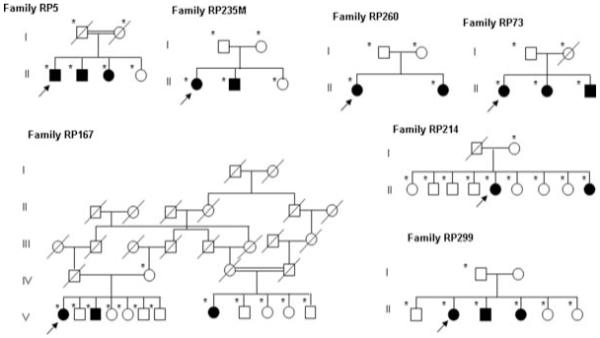
RP25 families which participated in the study. Open and closed symbols denote unaffected and affected individuals, respectively. Deceased family members are denoted by diagonal slashes, asterisks indicate individuals examined both clinically and genetically and arrows indicate probands in each family.
Bioinformatics Analysis
Genomic sequence of the screened genes within the interval was accessed through the National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/), the ENSEMBL database (http://www.ensembl.org) and the UCSC human genome browser (http://genome.ucsc.edu/). Information on expression pattern and expressed sequence tags (ESTs) were obtained from the NCBI UniGene database. In cases where several alternatively spliced transcripts were documented in ENSEMBL, or the exonic structure was uncharacterised, the BLAST tool (available through the NCBI) and the 6 Frame Translation tool from the Baylor College of Medicine (BCM) Search Launcher (http://searchlauncher.bcm.tmc.edu/seq-util/seq-util.html) were used to compare the sequences from these transcripts and the human genome sequence databases and to define the coding and protein sequences.
In order to evaluate the pathogenicity of the novel variants, we employed various softwares which analyse the potential role of a given variant on the function or structure of the encoded protein based on conservation and homology, physical properties of the amino acids, and prediction of the protein disorder (Conseq: http://conseq.tau.ac.il/; PolyPhen: http://coot.embl.de/PolyPhen/; SIFT: http://blocks.fhcrc.org/sift/SIFT.html; Disopred: http://bioinf.cs.ucl.ac.uk/disopred/disopred.html) (Thusberg & Vihinen 2006). In addition, intronic variants were evaluated for affecting any regulatory process at the transcriptional or splicing levels (TESS Transcription Element Search System: http://www.cbil.upenn.edu/cgi-bin/tess/tess; http://www.fruitfly.org/seq_tools/splice.html Splice Signal Analysis: http://www.ebi.ac.uk/asd-srv/wb.cgi; Alternative Splicing DataBase: http://hazelton.lbl.gov/~teplitski/alt/; http:/www.fruitfly.org/cgi-bin/seq_tools/splice.pl; Splicing Element Annotation: http://genes.mit.edu/acescan2/index.html; http://www.ensembl.org/Homo_sapiens/generegulationview; http://www.cisred.org/content/software; http://regrna.mbc.nctu.edu.tw/html/about.html) (Yeo et al. 2004; Matlin et al. 2005; Wang & Marin 2006).
PCR Amplification of Candidate Genes
In total 530 primers pairs were designed using Primer 3 Output (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) in order to ensure a total coverage of the entire coding region, the intronic flanking sequences, the regulatory factors binding sites and the 5′ untranslated region (UTR) of the major transcript as well as the additional exons contributed by other published alternatively spliced isoforms (details of primers are available on request). Polymerase chain reaction (PCR) was carried out as previously reported (Barragan et al. 2005a).
Mutation Screening
The PCR products were purified by adding Exosap [1 U shrimp alkaline phosphatase (SAP, Amersham LifeScience, Buckinghamshire, UK; ExoSAP-IT®, USB Corporation) and 1 U Exonuclease I (United States Biochemicals, Ohio, USA) to 1 μl of the PCR product and incubated at 37 °C for 15 minutes then at 80 °C for another 15 minutes to deactivate the enzyme. Sequencing reaction was then performed as previously described (Abd El-Aziz et al. 2005). Direct sequence analysis of one affected and one unaffected individual from each of the Spanish families was performed on the automated fluorescence DNA sequencer (ABI 3730, Applied Biosystems, Warrington, UK), according to manufacture’s instructions. Subsequently, the data were analysed using SeqManTM 4.03 software (DNAStar, Wisconsin, USA).
Evaluation of the Identified Changes
Novel variants were tested in a matching control population by direct sequencing using the same protocol as for the mutation detection. In some other cases the changes were genotyped by fragment analysis using primers labelled at the 5′ end with 6-FAM fluorocrome. PCR products were then injected in an ABI3730 automated genotyper. Subsequently, data collection and allele identification were performed using GeneMapper software (Applied Biosystems).
Comparative Genomic Hybridization
Comparative genomic hybridization (CGH) was performed on 6 DNA samples (parents, 3 affected and 1 unaffected members) from one of the consanguineous families (RP5) using a whole genome tiling path (WGTP) array. The methods employed were as previously published (Redon et al. 2006). Two pairs of primers, Fragment 1 F and R (ACAATCCCCTGTCATGGCTA and CCTTTCCACCTGTATATTAGGAGA) and Fragment 2 F and R (GATCCACTGACAGCTTGCAC and TCGCAAATCTCATGTACTTCTCA), were designed at the beginning and end of a deleted clone. PCR amplification was then carried out in order to test the existence of this clone in all members of the family.
Results
Organization of the RP25 Interval: Distribution of Known/Predicted Genes
According to the genetic data obtained from the RP25 linked families, we have divided the RP25 interval into four regions (A, B, C and D; Fig. 2) which as stated earlier contains in excess of 110 genes. Region A, between microsatellite markers D6S257 and D6S1053, spans approximately 8 Mb and contains 15 genes. It is important to note that within this region a part of the human genome (Ensembl release 48), ~50 Kb between 58 and 59 Mb and ~3 Mb between 59 and 61.2 Mb on chromosome 6q, remains to be sequenced. A ~6 Mb interval between microsatellite markers D6S1053 and D6S1557 constitutes the second region (B) and contains 11 genes. The third region (C) is localised between D6S1557 and D6S421 spans only ~1 Mb and contains 7 genes. The last region, D, is the largest since it spans the rest of the RP25 locus (~18 Mb) and contains 79 genes (Fig. 2). Even though a large number of genes are included within the region, some of the sequenced intervals are devoid of genes. These regions could either represent gene desert areas in the human genome or regions where newly predicted genes might soon appear.
Figure 2.
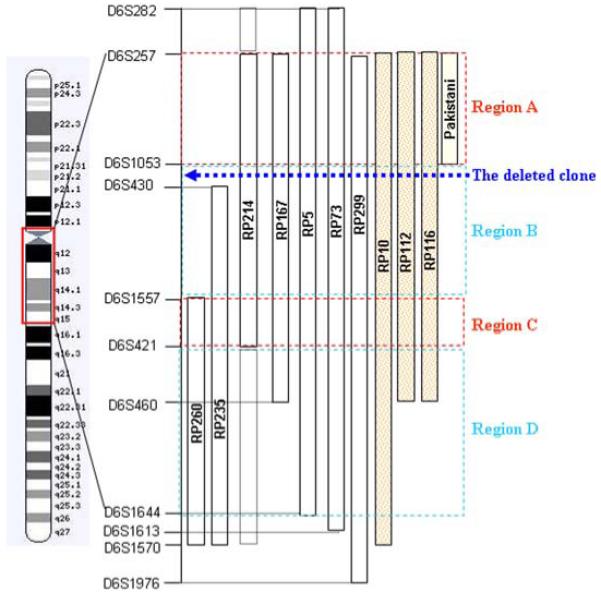
Schematic representation of chromosome 6, the RP25 locus (between microsatellite markers D6S257 and D6S1644). The genetic intervals for the Spanish (RP299, RP73, RP5, RP167, RP214, RP235 and RP260), Chinese (RP10, RP112 and RP116) and Pakistani families are represented by vertical bars. The dashed boxes in family RP214 represent the non homozygous linked intervals in this family. The interval is divided into four regions (A, B, C and D) according to the genetic data. The dotted arrow represents the location of the deleted clone in relation to the larger map.
Strategies for Selecting Candidate Genes
A comprehensive bioinformatics analysis of the RP25 interval indicated approximately 110 genes with many of them showing retinal expression. Information about gene function, expression pattern and/or the genetic data was among the criteria for selecting candidate genes from this interval. Here we present data on 43 genes, and a description of the adopted strategies for selecting these genes is detailed below.
Based on the organization of the RP25 interval as described above, region A was evaluated because of the possibility that the Pakistani family, the 3 Chinese and 5 of the Spanish families may have a common gene. Hence, 12 genes from this region were considered as good candidates for mutation screening (Table 1). Secondly, 5 genes, from region B where all families except the Pakistani and one of the Spanish (RP260) families shared linkage, were also considered as high priority for screening (Fig. 2 and Table 1). Following the assumption that apart from the Pakistani family all families could have a common gene; 3 genes from region C were evaluated (Table 1).
Table 1.
Information about the genes screened in this study
| Gene/ Description of Gene | Gene Size (Kb)/ No. of Exons /No. Primers/ Position (Mb) | UniGene | Published Information About the Gene |
|---|---|---|---|
| Genes in-between microsatellite markers D6S257 and D6S1053 | |||
| 1. BMP5 Bone morphogenetic protein 5 | 120.1/4/7/ 55,72-55,84 |
Hs.296648 | Part of the transforming growth factor-beta superfamily; signaling molecule within the trabecular meshwork and optic nerve head; potential role in glaucoma pathogenesis. |
| 2. C6orf65/ Chromosome 6 open reading frame 65 | 72.05/8/10 56.92-57.00 |
Hs.582993 | Sugar porter activity; regulation of transcription, DNA-dependent. |
| 3. KIAA1586/ KIAA1586 protein | 8.68/4/7 57.01-57.02 |
Unknown | Hypothetical protein |
| 4. ZNF451/ Zinc finger protein 451 | 80.27/14/19 57.06-57.14 |
Hs.485628 | Coactivator for steroid receptors |
| 5. BAG2/ BCL2-associated athanogene 2 | 12.61/3/4/ 57.14-57.15 |
HS.55220 | Was reported along with BAG1 and BAG3 to interact with heat shock 70 KD protein 8 ATPase domain and inhibit its chaperone activity (Takayama et al. 1999). |
| 6. PRIM2A/ Primase, polypeptide 2A, 58kDa | 329.44/14/11/ 57.28-57.62 |
HS.485640 | Plays a role in both the initiation of DNA replication and the synthesis of Okazaki fragments (Shiratori et al. 1995). |
| 7.GUSBL2/ Glucuronidase, beta-like 2 | 41.64/8/9 58.35-58.39 |
HS.561539 | Plays an important role in the degradation of dermatan and keratan sulfates. |
| 8. LOC442225 | 2.86/1/3 63.960-63.963 |
Similar to KRT8 protein | |
| 9. FKBP1C/Q5VVH2_human/ FK506 binding protein 1C | 1.58/1/1 63.97-63.98 |
Unknown | Paralog to FKBP1A on chromosome 20p13. |
| 10. LOC442226 | 2.86/1/3 63.99-64.00 |
Unknown | Similar to chromosome 15 open reading frame 2 |
| 11. PHF3/ PHD finger protein 3 | 79.64/16/16 64.40-64.48 |
HS.348921 | PHF3 expression was reported to be significantly reduced in glioma (Fischer et al. 2001). |
| 12. Q9NQ15/ Novel protein with EGF-like and laminin G domains - Fragment | 1.2/1/3 64.488-64.489 |
HS.25067 | Has EGF-like and laminin G domains. |
| Genes in-between microsatellite markers D6S1053 and D6S1557 | |||
| 13. Q5T669_human | 8.87/2/2 65.38-65.39 |
Unknown | Unknown |
| 14. EGFL11/ EGF-like-domain, multiple 11 | 161.09/9/11 66.10-66.26 |
HS.454341 | Unknown |
| 15. BAI3/ Brain-specific angiogenesis inhibitor 3 | 750.22/30/31 69.40-70.15 |
HS.13261 | Plays an important role in suppression of glioblastoma supported by its markedly reduced expression in glioblastoma cell lines (Shiratsuchi et al. 1997). |
| 16. LMPRD1/C6orf209/LMBR1 domain containing 1 | 21.03/16/15 70.44-70.56 |
Unknown | Unknown |
| 17. COL19A1/ NM 001858/Collagen, type XIX, alpha 1 | 345.71/51/41 70.63-70.97 |
HS.444842 | COL19A1 and COL9A1, were duplicated from the same ancestral gene of the FACIT family (Khaleduzzaman et al. 1997). |
| Genes in-between microsatellite markers D6S1557 and D6S421 | |||
| 18. COL9A1/ Collagen, type IX, alpha 1 | 87.03/38/39/ 70.98-71.06 |
HS.590892 | A mutation in the COL9A1 gene was reported in a family with multiple epiphyseal dysplasia (Czarny-Ratajczak et al. 2001). A homozygous mutation was also reported in a recessive form of Stickler syndrome (Van Camp et al. 2006). |
| 19. C6orf57/ Chromosome 6 open reading frame 57 | 22.65/3/4 71.33-71.35 |
HS.418495 | Unknown |
| 20. C6orf155/ Chromosome 6 open reading frame 155 | 6.30/4/3 72.180-72.187 |
Unknown | Unknown |
| Genes in-between microsatellite markers D6S421 and D6S1644 | |||
| 21. KCNQ5/ Potassium voltage-gated channel, KQT-like subfamily, member 5 | 573.12/14/16 73.38-73.96 |
HS.98129 | A KCNQ5 splice variant was identified in skeletal muscle displayed altered gating kinetics (Schroeder et al. 2000). |
| 22. DPPA5 / Developmental pluripotency associated 5 | 1.23/3 74.11-74.12 |
Unknown | Reported to have an important role in stemness in human embryonic stem (Kim et al. 2005). |
| 23. C6orf150/ Chromosome 6 open reading frame 150 | 27.41/6/6 74.17-74.21 |
HS.14577 | Unknown |
| 24. MTO1/ Mitochondrial translation optimisation 1 homolog | 39.69/13 74.22-74.26 |
HS.347614 | Reported to be involved in the process of mitochondrial RNA modification; an important regulatory pathway in the phenotypic expression of the deafness-associated mitochondrial A1555G mutation (Bykhovskaya et al. 2004). |
| 25. SLC17A5/ Solute carrier family 17 (anion/sugar transporter), member 5 | 60.78/11/11 74.35-74.42 |
HS.597422 | Defects in SLC17A5 are the cause of Salla disease (SD); an ar neurodegenerative disorders (Verheijen et al. 1999; Aula et al. 2000; Martin et al. 2003). |
| 26. CD109/ CD109 molecule | 128.96/33 74.46-74.59 |
HS.399891 | A glycosylphosphatidyl -inositol (GPI)-linked cell surface antigen expressed by CD34+ acute myeloid leukemia cell lines, T-cell lines, activated T lymphoblasts and activated platelets (Lin et al. 2002). The platelet-specific antigen system, implicated in neonatal allo-immune thrombocytopenia and post-transfusion purpura, is carried by CD109 (Kelton et al. 1990). |
| 27. FILIP1/ Filamin A interacting protein 1 | 185.69/6 76.07-76.26 |
HS.526972 | The C terminus of the long form of rat Filip interacted with mouse filamin A (FLNA), an actin-binding protein required for cell motility (Nagano et al. 2002). |
| 28. SENP6 / SUMO1/sentrin specific peptidase 6 | 114.37/24 76.36-76.48 |
HS.485784 | Reported to be highly expressed in reproductive organs, such as testis, ovary and prostate suggesting that it may play a role in reproduction (Kim et al. 2000). |
| 29. MYO6/ Myosin VI | 167.28/35/34 76.51-76.68 |
HS.149387 | Defects in MYO6 are the cause of both ad nonsyndromic sensorineural deafness type 22 (DFNA22) (Melchionda et al. 2001) and ar congenital neurosensory deafness type 37 (DFNB37) (Ahmed et al. 2003). |
| 30. HTR1B/ 5-hydroxytryptamine (serotonin) receptor 1B | 1.17/1/1 78.228-78.229 |
HS.123016 | A genetic variation (SNP; G861C) in the HTR1B gene was associated with attention deficit/hyperactivity disorder (ADHD) (Smoller et al. 2006). |
| 31. HMGN3/ High mobility group nucleosomal binding domain 3 | 33.49/6/6 79.96-80.00 |
HS.77558 | Unique within the HMGN family, the HMGN3 transcript undergoes alternative splicing and generates two different variants, HMGN3a and HMGN3b. (West et al. 2001). |
| 32. Q9H8D1_human/ CDNA FLJ13744 fis | 0.534/1/1 80.076-80.077 |
Unknown | Unknown |
| 33. C6orf152/ Chromosome 6 open reading frame 152 | 52.41/8/8 80.25-80.30 |
HS.21945 | Reported as the causative gene for Leber congenital amaurosis type 5 (LCA5) (den Hollander et al. 2007). |
| 34. SH3BGRL2/ SH3 domain binding glutamic acid-rich protein like 2 | 72.37/4/4 80.39-80.47 |
HS.232772 | Reported as a novel human homologue of the SH3 binding glutamic acid-rich establishing a new family of highly conserved proteins related to Thioredoxin Superfamily (Mazzocco et al. 2002). |
| 35. TTK/ TTK protein kinase Dual specificity protein kinase | 37.88/22/18 80.77-80.80 |
HS.169840 | Has dual specificity protein kinase (tyrosine and serine/threonine residues) (Lindberg et al. 1993). Is required for centrosome duplication and for the normal progression of mitosis (Fisk et al. 2003). |
| 36. Q9H3A8/ | 0.231/1/1 82.319-82.319.2 |
Unknown | Unknown |
| 37. IBTK/ Inhibitor of Bruton agammaglobulinemia tyrosine kinase | 77.49/29/29 82.93-83.00 |
HS.306425 | Btk deficiency causes Xl agammaglobulinemia (XLA) in humans and X-linked immunodeficiency in mice (Liu et al. 2001). |
| 38. ME1/ Malic enzyme 1 | 220.68/14 83.97-84.19 |
HS.21160 | Computer analysis revealed the presence of additional putative recognition motifs suggesting that ME1 gene is under complex regulatory control rather than on T3 (Gonzalez-Manchon et al. 1997). |
| 39. CYB5R4/ Cytochrome b5 reductase 4 | 100.74/16 84.62-84.72 |
HS.5741 | Targeted inactivation of the NCB5OR gene in mice lead to severe hyperglycemia with markedly decreased serum insulin levels and nearly normal insulin tolerance at 7 weeks of age and EM showed degranulation of beta cells and hypertrophic and hyperplastic mitochondria (Xie et al. 2004). |
| 40. SNX14 / Sorting nexin 14 | 88.41/29 86.27-86.36 |
HS.485871 | Cloned by a gene-trap strategy which set up in embryonic stem (ES) cells aiming to trap genes expressed in restricted neuronal lineages (Carroll et al. 2001). |
| 41. C6orf165/ Chromosome 6 open reading frame 165 | 56.46/13 88.17-88.20 |
HS.82921 | Unknown |
| 42. SLC35A1/ Solute carrier family 35 (CMP-sialic acid transporter), member A1 | 39.36/8/8 88.23-88.27 |
HS.423163 | By 5 prime RACE, a cDNA encoding SLC35A1, the human homolog of the murine cytidine monophosphate (CMP)-sialic acid transporter was obtained (Ishida et al. 1996). |
| 43. CNR1/ Cannabinoid receptor 1 | 5.5/2/5 88.90-88.93 |
HS.75110 | A triplet repeat polymorphism was significantly associated with schizophrenia especially the hebephrenic subtype (Ujike et al. 2002). A homozygous genotype within CNR1 1359A/A was associated with the vulnerability to alcohol withdrawal delirium (Schmidt et al. 2002). |
Genes from 2-7, 9, 11 and 14 were considered as good candidates due to confirmed expression in eye tissues by the EyeSAGE database. Genes’ numbers 41 and 42 were expressed in the cilia of the Trypanosome proteome. Genes’ numbers 17, 33 and 43 were considered as good candidates based on the expression pattern in the retinal degeneration (rd) mouse in comparison to the wild type mouse.
Lastly, the possibility of the disease gene lying within region D from which the remaining 23 genes were chosen cannot be ruled out. These genes were selected according to their functional relevance to RP; for example homologous genes from the ciliated Trypanosome mapping to retinal disease loci were used to identify functional candidates (Table 1) (Broadhead et al. 2006). This was based on the hypothesis that successful transmission of several proteins from the cell body to the outer segment of the photoreceptor cells depends on their transport along a modified cilium and that defective passage of certain molecules results in retinal degeneration associated with RP (Marszalek & Goldstein, 2000). Also, the study of novel genes whose expression can be modulated by a primary defect in a retina-specific gene was also used as an approach to identify functional candidates. For this reason the genes highlighted by the retinal degeneration (rd) mouse model, caused by a defect in the β subunit of rod cGMP-phosphodiesterase, were considered as good candidates (Table 1) (Bowes et al. 1990). Finally, EyeSAGE (Serial Analysis of Gene Expression) and the National Eye Institute Bank (NEIBank) were used as excellent databases comparing the pattern of gene expression in the retina and retinal pigment epithelium (RPE) to those in different tissues and hence highlighting important genes for different eye diseases including RP25 (Table 1).
Bioinformatics of the Genes within the RP25 Interval
The results of bioinformatics analyses for exon-intron structure, gene size and number of transcripts of all the screened genes apart from 3 genes, PRIM2A and LOC442225 and LOC442226, confirmed the previously published data (Table 1). Both LOC442225 and LOC442226 were uncharacterised and hence the Blast and the 6 Frame Translation tools were used to identify their exonic structure. Additionally, at the time of screening the PRIM2A gene, only 10 exons were reported in the Ensembl database, the initiation codon (ATG) was predicted but the stop codon had not been identified. Recently 4 additional exons have been predicted; however the stop codon still remains to be identified and hence the open reading frame (ORF) needs to be fully characterised.
Mutation Screening
Mutation screening of the selected 43 genes led to the identification of 244 sequence variants of which 76 were novel (Table 2). All changes were assigned a nucleotide number starting at the first translation base of all genes studied according to the GenBank entries summarised in table 2.
Table 2.
Novel changes identified in the screened genes
| Gene/GenBank Entry | Sequence Variation | AA Change | Allele Frequency§ |
|---|---|---|---|
| C6orf65/ NM_152731 | c.68-106_68-105insCA | - | - |
| c.520-122A>G | - | - | |
| KIAA1586 / NM_020931.1 | c.2146A>G | p.I716V | 12.0% |
| ZNF451/NM_001031623 | c.29-64_-65delAA | - | - |
| c.*1569_1573delGTGGT | - | - | |
| c.*166G>A | - | - | |
| c.106-85C>T | - | - | |
| BAG2/ NM_004282 | c.114-24G>A | - | 1.0% |
| c.596-69_75delGAGAT | - | 0.0% | |
| c.*398G>A | - | - | |
| PRIM2A/ NM_000947.2 | c.762-20T>A | - | - |
| c.762-38A>G | - | - | |
| c.810G>A | p.K270K | - | |
| c.834+38G>T | - | - | |
| c.834+44G>T | - | - | |
| LOC442225/ | c.105-35_29delCTCCTTG | - | - |
| FKBP1C | -20G>A | - | - |
| -6G>A | - | - | |
| EGFL11/ NM_198283.1 | c.281C>A | p. P93P | 5.0% |
| c.863-21insT | - | 20.0% | |
| c.1712A>G | p.Q570R | 40.0% | |
| C6orf209/ NM_018368.2 | c.916-48delA | - | - |
| COL19A1/ NM_001858.4 | c.166 + 12A>G | - | - |
| COL9A1/ NM_001851 | c.1090 -78T>C | - | - |
| c.2034 +3C>G | - | - | |
| C6orf57 /NM_145267 | c.-38T>C | - | - |
| c.*121AT(6_18) | - | - | |
| DPPA5/ NM_001025290 | c.297-30 G>A | - | - |
| C6orf150/ NM_138441 | c.300G>A | - | - |
| c.1145C>T | - | - | |
| MTO1/ NM_133645 | c.879-46A>G | - | - |
| c.1201-59 C>G | - | - | |
| c.1409 + 14 T>A | - | - | |
| SLC17A5/NM_012434.3 | c.979-87A>G | - | - |
| CD109/NM_133493 | c.248-51C>T | - | 47.0% |
| c.508-121 G>A | - | 33.0% | |
| c.673 + 190_673 + 191 ins A | - | 9.0% | |
| c.674 -93 G>A | - | 21.0% | |
| c.758 +16 C>A | - | 0.0% | |
| c.758 +22 A>G | - | 1.0% | |
| c.105A>G | p.S335S | 0.0% | |
| c.1435 -4_1435 -5 insT | - | 9.0% | |
| c.1498-3 T>C | - | 0.0% | |
| c.2556 +68_2556 + 69 insT | - | - | |
| c.2701 +102delA | - | - | |
| c.2991C>T | p.H997H | - | |
| c.3703-86G>A | - | - | |
| FILIP1/ NM_015687 | c.834A>G | p.E278E | 2.0% |
| c.844C>T | p.L282F | 7.0% | |
| SENP6/ NM_001100409.1 | c. 1129 A>G | - | - |
| c. 420 + 453G>T | - | - | |
| c. 637-135 G>C | - | - | |
| MYO6/ NM_004999.3 | -49C>G | - | - |
| c.1224-4A>G | - | - | |
| C6orf152/ NM_181714. | c.77A>C | p.D26A | - |
| c.858 del+126_129 ATC | - | - | |
| TTK/ NM_003318.3 | g.520C>T | - | - |
| c.471-39 del CATTIns TAAAGA | - | - | |
| c.1521 + 25G>T | - | - | |
| c.2050-75_80delAATAAT | - | - | |
| IBTK /NM_015525 | -1531 A>G | - | - |
| c.418 +237 G>A | - | - | |
| c.654 +84 654 + 85 ins T | - | - | |
| c.798G>A | p.P266P | - | |
| c.2509 +88_2509 +91 del AAAC | - | 1.0% | |
| c.2520-101_2520 -102insT | - | 0% | |
| c.3025-35A>G | - | - | |
| c.3432-144_140 del AAAT | - | 5.0% | |
| c.3432 -133A>C | - | 5.0% | |
| c.4967-68_70delTC | - | - | |
| c.3830C>A | p.S1277Y | 0% | |
| ME1/NM_002395 | c.302 + 21A>C | - | - |
| C6orf165/ NM_001031743.1 | c.353A>G | p.D118G | 0% |
| c.473-15del T | - | - | |
| c.921C >T | p.T307T | - | |
| SLC35A1/ NM_006416.3 | c.887-14T>C | - | - |
denotes allele frequency in Spanish population only.
A considerable number of novel variants were excluded as pathogenic, based on their non-segregation with the disease phenotype or their presence in the control population.
Of the 76 identified sequence variants, only 5 showed segregation with the disease phenotype and were considered as potentially pathogenic. The first change was a heterozygous amino acid substitution of an aspartic acid into glycine (D118G) within the C6orf165 gene and was observed in the proband of a non-consanguineous family (RP73). Even though the D118G genetic variant was not detected in 192 control chromosomes it could not be considered as a pathogenic alteration due to the absence of a second change that is necessary to explain the phenotype in the case of a recessive disorder.
Mutation screening of BAG2 revealed 2 heterozygous novel variants, c.596-69_75delGAGAT and c.114-24G>A, in the RP299 and RP73 families, respectively. The deletion in family RP299 has not been detected in 192 control chromosomes, whereas the G>A transition identified in family RP73 was detected in a control individual. Nevertheless, in silico information has not provided enough support to consider these changes as pathogenic in the absence of a second change in either family.
The last 2 changes, C>A transversion at c.758 + 16 C>A and T>C transition in intron 14 at c.1498-3T>C, were identified within the CD109 gene in the RP214 and RP299 families, respectively. Both changes were segregating with the disease phenotype and were not detected in 192 control chromosomes. However, as we could not find a second change in either family, these changes are likely to be rare polymorphisms.
Heterozygous Changes within Homozygous Intervals
Interestingly, all the changes within one of the screened genes, PRIM2A, were found to be heterozygous in all family members. This does not fit with the genetic data for the RP5 and RP214 families, since affected individuals in both should be homozygous for any changes. By blast search no additional copy of this gene was detected elsewhere in the human genome; however by searching the database for genome variants (TCAG), CNV was reported for the PRIM2A gene (Redon et al. 2006). Additionally, CNV was reported for other genes within this interval, such as EGFL11, FKBP1C, GUSBL2, and LOC442226; in all cases heterozygous SNPs were observed in affected samples tested in both RP5 and RP214 families. CGH analysis was undertaken to look for CNV as an explanation for the above finding.
Comparative Genomic Hybridization
The data obtained from the WGTP analysis revealed an inappropriate signal compatible with a deletion of a single clone in affected members of the RP5 family. The clone, chr6tp-19C7, spans ~100-Kb and is located at 65.7 Mb. A 95-Kb region proximal to this clone was not covered by the array used in this experiment. Therefore, it is possible that the deleted clone may extend over this gap. Additionally, a normal signal was observed from the Chr6tp-10D10 clone upstream of the gap. On the other hand, the deleted clone overlaps by 40-Kb with its distal neighbour, Chr6tp-10G7, which also shows a normal signal (Fig. 3). Based on this data it is possible that the deleted clone could be as much as 200-Kb in size.
Figure 3.
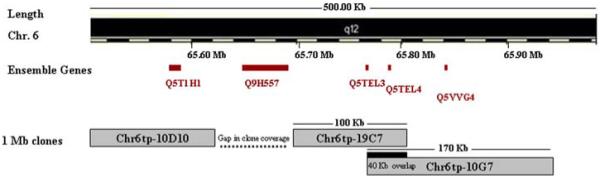
Schematic representation showing part of chromosome 6 along with the deleted clone, the gap in the clone coverage and the overlap between the deleted clone and the distal clone (Ch6tp-10G7). The genes within the region of the deleted clone are depicted.
The identification of at least a 100-Kb deletion in family RP5 is interesting and indicates that one of the genes, Q5T1H1, Q9H557, Q5TEL3, Q5TEL4, Q5VVG4 and Q5T3C8, within this interval might be responsible for the RP25 phenotype (Fig. 3). However, we cannot rule out the possibility that this deletion might represent a rare non-pathogenic CNV. The existence of the deletion was further validated by non-amplification of two PCR amplicons designed within the deleted clone in all affected members of the family (Fig. 4). It is noteworthy that the genes within the deleted clone interval share a common epidermal growth factor (EGF-like) domain, however they represent incomplete transcripts that range between 1 to 4 exons, without an initiation or stop codon. Assuming that one of the genes from the deleted interval would be responsible for the RP25 phenotype, the families whose linked regions do not overlap with the deletion, such as RP235 and RP260 (Fig. 2), should not possess mutations in this gene. Linkage to the RP25 interval in these two families could therefore be coincidental, since many regions in the genome are expected to show suggestive evidence of linkage given the small size of these families.
Figure 4.
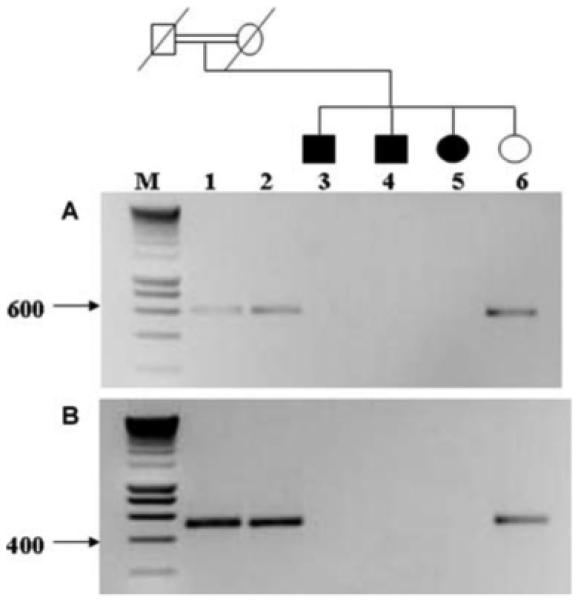
Cosegregation study of family RP5 using 2 pairs of primers within the deleted clone (A and B). Heterozygous parents and the unaffected daughter (lanes 1, 2 and 6) showed PCR products corresponding to the expected size of the amplified fragments. Lanes 3, 4 and 5 (affected members) showed absent PCR products confirming the data obtained from the CGH array analysis. M is a 10-Kb smart ladder.
Discussion
Herein, we report the molecular analysis of a 34 Mb interval on chromosome 6q which resulted in the exclusion of forty three genes as disease-causing for RP25 in 7 Spanish families. In addition, we have identified a ~100 Kb deletion within this interval in one of the RP25 families leading to a major refinement of the locus that should facilitate the subsequent identification of the disease gene (Fig. 2 and 3).
The RP25 interval spans approximately 34 Mb containing more than 110 genes, and bioinformatics analysis has revealed a sequence gap of ~3 Mb close to the centromere of chromosome 6. Moreover, some of the sequenced intervals within the RP25 region are devoid of any genes, representing either gene desert areas in the human genome or regions containing unpredicted genes that are yet to be characterised. Therefore, the number of genes within the RP25 interval should be considered as a rough estimate. However, given the large number of genes, it was impractical to screen all of them without adopting a specific strategy. As a result 43 genes were selected as good candidates for mutation screening in the Spanish families.
Both positional and functional candidate gene approaches were employed in order to identify the RP25 gene. The two strategies were overlapping throughout the study. The positional approach was considered as an essential part of the strategy for selection of candidate genes. This is due to fact that within the linkage interval there was no shared haplotype between each of the Spanish and/or other families originating from diverse geographical regions (Fig. 2). Different mutations within one gene are therefore expected to be responsible for the RP phenotype in all the families. However, the possibility of two genes responsible for the RP25 phenotype cannot be ruled out based on the presence of different crossovers from different families.
The functional candidate gene approach was also used to select genes for mutation screening. This is based on the fact that a gene could demonstrate a functional relationship with the underlying defect (RP) or there could be human homologs of genes responsible for a similar disease phenotype in other species. Alternatively, some genes might have been identified as members of a gene family of which other members have been implicated in a related disorder. For example, C6orf165 and SLC35A1 were considered as the human orthologs for the protein that was shown to be expressed in the cilia of the Trypanosome proteome, which made these genes excellent candidates (Broadhead et al. 2006).
Highly efficient genetic databases as well as information about genes expressed in the rd mouse were also used in order to identify functional candidates (Table 1). Interestingly, our strategy of selecting genes from the RP25 interval has been further validated by the identification of 2 genes within this locus, Lebercillin and COL9A1, as responsible for the retinal degeneration phenotype in both LCA5 (Leber Congenital Amaurosis), and Stickler syndrome, respectively (den Hollander et al. 2007; Van Camp et al. 2006).
Using the above described candidate gene approaches, 43 genes underwent mutation screening in 7 Spanish families. In total 244 genetic variants were identified, of which 76 were novel. Out of the 76 SNPs only 5 were initially considered as significant changes in terms of either segregation with the disease phenotype and/or absence in 192 matching control chromosomes. However, by further investigations they were ruled out as pathogenic since they have been observed in non-consanguineous families where a second change is necessary to explain their role in disease causation.
In any of the studied genes all the identified SNPs segregated with the genetic data apart from 5 genes, PRIM2A, EGFL11, FKBP1C, GUSBL2 and LOC442226, where heterozygous SNPs were observed within an interval of homozygosity. Even though this is in contradiction to the linkage data it was only acceptable in this case since a second copy of the gene was discovered to be existing within chromosome 6q12. It has been reported that CNVs could have a direct effect on transcription regulation which in turn may influence disease susceptibility and phenotypic variation (Redon et al. 2006). Similarly, we have postulated that CNVs existing within the RP25 locus could have an impact on the phenotype of the RP25 families. For this purpose a WGTP array was carried out on one family (RP5) and the data obtained revealed that a clone within the RP25 locus was deleted in all of the affected members of the investigated family. It is interesting to note that recently 5 new Spanish families showed suggestive evidence of linkage to the RP25 locus. Moreover a crossover in one of the families has probably refined the interval from 34 to 16 Mb between microsatellite markers D6S257 and D6S1557 (published in this issue). This complements our current findings presented here.
In summary no pathogenic changes were observed in the screened genes; hence their role in the causation of the RP25 phenotype is excluded. Nevertheless, the exclusion of this significant number of genes from the interval would help in prioritising the remaining genes for mutation screening and hence in identifying the causative gene for RP25. The identification of a deleted clone from the tiling path array has narrowed down the RP25 interval from 34 Mb to only 100 Kb. It is very likely that one of the genes within the deleted clone interval could be responsible for the RP phenotype in the studied families. This confirms our initial assumption of the presence of more than one gene as responsible for RP25. If that is the case we should expect the mutations in this gene to be responsible for the RP phenotype in all RP25 linked families apart from two families, (RP235 and RP260). Future work will therefore involve full characterisation and mutation screening of the current and/or novel genes within the deleted interval in the RP25 families.
Acknowledgments
We would like to thank the families who participated in the study. This study was funded by Fondo de Investigación Sanitaria, Spain (PI050857), Consejería de Salud, Junta de Andalucía, Spain(PI-0334/2007), British Retinitis Pigmentosa Society (Grant ref.GR556), NIHR Biomedical Research Centre for Ophthalmology (BMRC) and Special Trustees of Moorfields Eye Hospital.
References
- Abd El-Aziz MM, El-Ashry MF, Barragan I, Marcos I, Borrego S, Antinolo G, Bhattacharya SS. Molecular genetic analysis of two functional candidate genes in the autosomal recessive retinitis pigmentosa, RP25, locus. Curr Eye Res. 2005;30:1081–1087. doi: 10.1080/02713680500351039. [DOI] [PubMed] [Google Scholar]
- Abd El-Aziz MM, El-Ashry MF, Chan WM, Chong KL, Barragan I, Antinolo G, Pang CP, Bhattacharya SS. A Novel Genetic Study of Chinese Families with Autosomal Recessive Retinitis Pigmentosa. Ann Hum Genet. 2007;70:1–14. doi: 10.1111/j.1469-1809.2006.00333.x. [DOI] [PubMed] [Google Scholar]
- Abd El-Aziz MM, Patel RJ, El-Ashry MF, Barragan I, Marcos I, Borrego S, Antinolo G, Bhattacharya SS. Exclusion of four candidate genes, KHDRBS2, PTP4A1, KIAA1411 and OGFRL1, as causative of autosomal recessive retinitis pigmentosa. Ophthalmic Res. 2006;38:19–23. doi: 10.1159/000088493. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Riazuddin S, Griffith AJ, Friedman TB, Wilcox ER. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet. 2003;72:1315–1322. doi: 10.1086/375122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aula N, Salomaki P, Timonen R, Verheijen F, Mancini G, Mansson JE, Aula P, Peltonen L. The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet. 2000;67:832–840. doi: 10.1086/303077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragan I, Borrego S, Abd El-Aziz MM, El-Ashry MF, Abu-Safieh L, Bhattacharya SS, Antinolo G. Genetic Analysis of FAM46A in Spanish Families with Autosomal Recessive Retinitis Pigmentosa: Characterisation of novel VNTRs. Ann Hum Genet. 2008;71:281–294. doi: 10.1111/j.1469-1809.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- Barragan I, Marcos I, Borrego S, Antinolo G. Molecular analysis of RIMI in autosomal recessive retinitis pigmentosa. Ophthalmic Res. 2005a;37:89–93. doi: 10.1159/000084250. [DOI] [PubMed] [Google Scholar]
- Barragan I, Marcos I, Borrego S, Antinolo G. Mutation screening of three candidate genes, ELOVL5, SMAP1 and GLULD1 in autosomal recessive retinitis pigmentosa. Int J Mol Med. 2005b;16:1163–1167. [PubMed] [Google Scholar]
- Bird AC. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119:543–562. doi: 10.1016/s0002-9394(14)70212-0. [DOI] [PubMed] [Google Scholar]
- Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N, Shaw MK, Ginger ML, Gaskell SJ, McKean PG, Gull K. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Mengesha E, Wang D, Yang H, Estivill X, Shohat M, Fischel-Ghodsian N. Phenotype of non-syndromic deafness associated with the mitochondrial A1555G mutation is modulated by mitochondrial RNA modifying enzymes MTO1 and GTPBP3. Mol Genet Metab. 2004;83:199–206. doi: 10.1016/j.ymgme.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Carroll P, Renoncourt Y, Gayet O, De Bovis B, Alonso S. Sorting nexin-14, a gene expressed in motoneurons trapped by an in vitro preselection method. Dev Dyn. 2001;221:431–442. doi: 10.1002/dvdy.1163. [DOI] [PubMed] [Google Scholar]
- Czarny-Ratajczak M, Lohiniva J, Rogala P, Kozlowski K, Perala M, Carter L, Spector TD, Kolodziej L, Seppanen U, Glazar R, Krolewski J, Latos-Bielenska A, Ala-Kokko L. A mutation in COL9A1 causes multiple epiphyseal dysplasia: further evidence for locus heterogeneity. Am J Hum Genet. 2001;69:969–980. doi: 10.1086/324023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Koenekoop RK, Mohamed MD, Arts HH, Boldt K, Towns KV, Sedmak T, Beer M, Nagel-Wolfrum K, McKibbin M, Dharmaraj S, Lopez I, Ivings L, Williams GA, Springell K, Woods CG, Jafri H, Rashid Y, Strom TM, van der Zwaag B, Gosens I, Kersten FF, van Wijk E, Veltman JA, Zonneveld MN, van Beersum SE, Maumenee IH, Wolfrum U, Cheetham ME, Ueffing M, Cremers FP, Inglehearn CF, Roepman R. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Manchon C, Butta N, Ferrer M, Ayuso MS, Parrilla R. Molecular cloning and functional characterization of the human cytosolic malic enzyme promoter: thyroid hormone responsiveness. DNA Cell Biol. 1997;16:533–544. doi: 10.1089/dna.1997.16.533. [DOI] [PubMed] [Google Scholar]
- Fischer U, Struss AK, Hemmer D, Michel A, Henn W, Steudel WI, Meese E. PHF3 expression is frequently reduced in glioma. Cytogenet Cell Genet. 2001;94:131–136. doi: 10.1159/000048804. [DOI] [PubMed] [Google Scholar]
- Fisk HA, Mattison CP, Winey M. Human Mps1 protein kinase is required for centrosome duplication and normal mitotic progression. Proc Nat Acad Sci. 2003;100:14875–14880. doi: 10.1073/pnas.2434156100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Miura N, Yoshioka S, Kawakita M. Molecular cloning and characterization of a novel isoform of the human UDP-galactose transporter, and of related complementary DNAs belonging to the nucleotide-sugar transporter gene family. J Biochem. 1996;120:1074–1078. doi: 10.1093/oxfordjournals.jbchem.a021523. [DOI] [PubMed] [Google Scholar]
- Johnson S, Halford S, Morris AG, Patel RJ, Wilkie SE, Hardcastle AJ, Moore AT, Zhang K, Hunt DM. Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7) Genomics. 2003;81:304–314. doi: 10.1016/s0888-7543(03)00010-7. [DOI] [PubMed] [Google Scholar]
- Kelton JG, Smith JW, Horsewood P, Humbert JR, Hayward CPM, Warkentin TE. Gov(a/b) alloantigen system on human platelets. Blood. 1990;75:2172–2176. [PubMed] [Google Scholar]
- Khaliq S, Hameed A, Ismail M, Mehdi SQ, Bessant DA, Payne AM, Bhattacharya SS. Refinement of the locus for autosomal recessive Retinitis pigmentosa (RP25) linked to chromosome 6q in a family of Pakistani origin. Am J Hum Genet. 1999;65:571–574. doi: 10.1086/302493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaleduzzaman M, Sumiyoshi H, Ueki Y, Inoguchi K, Ninomiya Y, Yoshioka H. Structure of the human type XIX collagen (COL19A1) gene, which suggests it has arisen from an ancestor gene of the FACIT family. Genomics. 1997;45:304–312. doi: 10.1006/geno.1997.4921. [DOI] [PubMed] [Google Scholar]
- Kim KI, Baek SH, Jeon YJ, Nishimori S, Suzuki T, Uchida S, Shimbara N, Saitoh H, Tanaka K, Chung CH. A new SUMO-1-specific protease, SUSP1, that is highly expressed in reproductive organs. J Biol Chem. 2000;275:14102–14106. doi: 10.1074/jbc.275.19.14102. [DOI] [PubMed] [Google Scholar]
- Kim SK, Suh MR, Yoon HS, Lee JB, Oh SK, Moon SY, Moon SH, Lee JY, Hwang JH, Cho WJ, Kim KS. Identification of developmental pluripotency associated 5 expression in human pluripotent stem cells. Stem Cells. 2005;23:458–462. doi: 10.1634/stemcells.2004-0245. [DOI] [PubMed] [Google Scholar]
- Li Y, Marcos I, Borrego S, Yu Z, Zhang K, Antinolo G. Evaluation of the ELOVL4 gene in families with retinitis pigmentosa linked to the RP25 locus. J Med Genet. 2001;38:478–480. doi: 10.1136/jmg.38.7.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Sutherland DR, Horsfall W, Totty N, Yeo E, Nayar R, Wu XF, Schuh AC. Cell surface antigen CD109 is a novel member of the alpha-2 macroglobulin/C3, C4, C5 family of thioester-containing proteins. Blood. 2002;99:1683–1691. doi: 10.1182/blood.v99.5.1683. [DOI] [PubMed] [Google Scholar]
- Lindberg RA, Fischer WH, Hunter T. Characterization of a human protein threonine kinase isolated by screening an expression library with antibodies to phosphotyrosine. Oncogene. 1993;8:351–359. [PubMed] [Google Scholar]
- Liu W, Quinto I, Chen X, Palmieri C, Rabin RL, Schwartz OM, Nelson DL, Scala G. Direct inhibition of Bruton’s tyrosine kinase by IBtk, a Btk-binding protein. Nature Immun. 2001;2:939–946. doi: 10.1038/ni1001-939. [DOI] [PubMed] [Google Scholar]
- Marcos I, Borrego S, Antinolo G. Molecular cloning and characterization of human RAB23, a member of the group of Rab GTPases. Int J Mol Med. 2003;12:983–987. [PubMed] [Google Scholar]
- Marcos I, Galan JJ, Borrego S, Antinolo G. Cloning, characterization, and chromosome mapping of the human GlcATS gene. J Hum Genet. 2002;47:677–680. doi: 10.1007/s100380200103. [DOI] [PubMed] [Google Scholar]
- Marcos I, Ruiz A, Blaschak CJ, Borrego S, Cutting GR, Antinolo G. Mutation analysis of GABRR1 and GABRR2 in autosomal recessive retinitis pigmentosa (RP25) J Med Genet. 2000;37:E5. doi: 10.1136/jmg.37.6.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Goldstein LS. Understanding the functions of kinesin-II. Biochim Biophys Acta. 2000;1496:142–150. doi: 10.1016/s0167-4889(00)00015-x. [DOI] [PubMed] [Google Scholar]
- Martin RA, Slaugh R, Natowicz M, Pearlman K, Orvisky E, Krasnewich D, Kleta R, Huizing M, Gahl WA. Sialic acid storage disease of the Salla phenotype in American monozygous twin female sibs. Am J Med Genet. 2003;120:23–27. doi: 10.1002/ajmg.a.10246. [DOI] [PubMed] [Google Scholar]
- Matlin AJ, Clark F, Smith CW. Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol. 2005;6:386–398. doi: 10.1038/nrm1645. [DOI] [PubMed] [Google Scholar]
- Mazzocco M, Maffei M, Egeo A, Vergano A, Arrigo P, Di Lisi R, Ghiotto F, Scartezzini P. The identification of a novel human homologue of the SH3 binding glutamic acid-rich (SH3BGR) gene establishes a new family of highly conserved small proteins related to Thioredoxin Superfamily. Gene. 2002;29:233–239. doi: 10.1016/s0378-1119(02)00602-9. [DOI] [PubMed] [Google Scholar]
- Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, Arbones ML, Notarangelo A, Di Iorio E, Carella M, Zelante L, Estivill X, Avraham KB, Gasparini P. MYO6, the human homologue of the gene responsible for deafness in Snell’s Waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. A J Hum Genet. 2001;69:635–640. doi: 10.1086/323156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Yoneda T, Hatanaka Y, Kubota C, Murakami F, Sato M. Filamin A-interacting protein (FILIP) regulates cortical cell migration out of the ventricular zone. Nature Cell Biol. 2002;4:495–501. doi: 10.1038/ncb808. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Borrego S, Marcos I, Antinolo G. A major locus for autosomal recessive retinitis pigmentosa on 6q, determined by homozygosity mapping of chromosomal regions that contain gamma-aminobutyric acid-receptor clusters. Am J Hum Genet. 1998;62:1452–1459. doi: 10.1086/301866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt LG, Samochowiec J, Finckh U, Fiszer-Piosik E, Horodnicki J, Wendel B, Rommelspacher H, Hoehe MR. Association of a CB1 cannabinoid receptor gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend. 2002;65:221–224. doi: 10.1016/s0376-8716(01)00164-8. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Shiratori A, Okumura K, Nogami M, Taguchi H, Onozaki T, Inoue T, Ando T, Shibata T, Izumi M, Miyazawa H, Hanaoka F, Murakami Y, Eki T. Assignment of the 49-kDa (PRIM1) and 58-kDa (PRIM2A and PRIM2B) subunit genes of the human DNA primase to chromosome bands 1q44 and 6p11.1-p12. Genomics. 1995;28:350–353. doi: 10.1006/geno.1995.1155. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi T, Nishimori H, Ichise H, Nakamura Y, Tokino T. Cloning and characterization of BAI2 and BAI3, novel genes homologous to brain-specific angiogenesis inhibitor 1 (BAI1) Cytogenet Cell Genet. 1997;79:103–108. doi: 10.1159/000134693. [DOI] [PubMed] [Google Scholar]
- Smoller JW, Biederman J, Arbeitman L, Doyle AE, Fagerness J, Perlis RH, Sklar P, Faraone SV. Association between the 5HT1B receptor gene (HTR1B) and the inattentive subtype of ADHD. Biol Psychiat. 2006;59:460–467. doi: 10.1016/j.biopsych.2005.07.017. [DOI] [PubMed] [Google Scholar]
- Takayama S, Xie Z, Reed JC. An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J Biol Chem. 1999;274:781–786. doi: 10.1074/jbc.274.2.781. [DOI] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Bioinformatic analysis of protein structure-function relationships: case study of leukocyte elastase (ELA2) missense mutations. Hum Mutat. 2006;27:1230–1243. doi: 10.1002/humu.20407. [DOI] [PubMed] [Google Scholar]
- Ujike H, Takaki M, Nakata K, Tanaka Y, Takeda T, Kodama M, Fujiwara Y, Sakai A, Kuroda S. CNR1, central cannabinoid receptor gene, associated with susceptibility to hebephrenic schizophrenia. Mol. Psychiatry. 2002;7:515–518. doi: 10.1038/sj.mp.4001029. [DOI] [PubMed] [Google Scholar]
- Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, Fukuoka H, Wagatsuma M, Suzuki H, Smets RME, Vanhoenacker F, Declau F, Van De Heyning P, Usami S. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006;79:449–457. doi: 10.1086/506478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lith-Verboeven JJC, Hoyng CB, van den Helm B, Deutman AF, Brink HMA, Kemperman MH, de Jong WHM, Kremer H, Cremers FPM. The benign concentric annular macular dystrophy locus maps to 6p12.3-q16. Invest Ophthalmol Vis Sci. 2004;45:30–35. doi: 10.1167/iovs.03-0392. [DOI] [PubMed] [Google Scholar]
- Verheijen FW, Verbeek E, Aula N, Beerens CEMT, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, Van Der Spek PJ, Mancini GMS. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23:462–465. doi: 10.1038/70585. [DOI] [PubMed] [Google Scholar]
- Wang M, Marin A. Characterization and prediction of alternative splice sites. Gene. 2006;366:219–227. doi: 10.1016/j.gene.2005.07.015. [DOI] [PubMed] [Google Scholar]
- West KL, Ito Y, Birger Y, Postnikov Y, Shirakawa H, Bustin M. HMGN3a and HMGN3b, two protein isoforms with a tissue-specific expression pattern, expand the cellular repertoire of nucleosome-binding proteins. J Biol Chem. 2001;276:25959–25969. doi: 10.1074/jbc.M101692200. [DOI] [PubMed] [Google Scholar]
- Xie J, Zhu H, Larade K, Ladoux A, Seguritan A, Chu M, Ito S, Bronson RT, Leiter EH, Zhang CY, Rosen ED, Bunn HF. Absence of a reductase, NCB5OR, causes insulin-deficient diabetes. Proc Nat Acad Sci. 2004;101:10750–10755. doi: 10.1073/pnas.0404044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Hoon S, Venkatesh B, Burge CB. Variation in sequence and organization of splicing regulatory elements in vertebrate genes. Proc Natl Acad Sci U S A. 2004;101:15700–15705. doi: 10.1073/pnas.0404901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, MacDonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]