

Abstract
Phrenic long-term facilitation (pLTF) following acute intermittent hypoxia (AIH) is a form of spinal, serotonin-dependent synaptic plasticity that requires reactive oxygen species (ROS) formation. We tested the hypothesis that spinal NADPH oxidase activity is a necessary source of ROS for pLTF. Sixty minutes post-AIH (three 5-min episodes of 11% O2, 5 min intervals), integrated phrenic and hypoglossal (XII) nerve burst amplitudes were increased from baseline, indicative of phrenic and XII LTF. Intrathecal injections (∼C4) of apocynin or diphenyleneiodonium chloride (DPI), two structurally and functionally distinct inhibitors of the NADPH oxidase complex, attenuated phrenic, but not XII, LTF. Immunoblots from soluble (cytosolic) and particulate (membrane) fractions of ventral C4 spinal segments revealed predominantly membrane localization of the NADPH oxidase catalytic subunit, gp91phox, whereas membrane and cytosolic expression were both observed for the regulatory subunits, p47phox and RAC1. Immunohistochemical analysis of fixed tissues revealed these same subunits in presumptive phrenic motoneurons of the C4 ventral horn, but not in neighbouring astrocytes or microglia. Collectively, these data demonstrate that NADPH oxidase subunits localized within presumptive phrenic motoneurons are a major source of ROS necessary for AIH-induced pLTF. Thus, NADPH oxidase activity is a key regulator of spinal synaptic plasticity, and may be a useful pharmaceutical target in developing therapeutic strategies for respiratory insufficiency in patients with, for example, cervical spinal injury.
Plasticity is an important property of neural systems, including the neural network controlling breathing (Mitchell & Johnson, 2003). One of the most comprehensively studied models of respiratory plasticity is long-term facilitation (LTF) of hypoglossal (XII) and phrenic motor output following acute intermittent hypoxia (AIH) (Mitchell et al. 2001; Feldman et al. 2003; Mahamed & Mitchell, 2007). In brief, LTF in phrenic nerve activity (pLTF) is a central neural (spinal) mechanism manifested as a progressive and sustained increase in phrenic motor output lasting more than 1 h (Mitchell et al. 2001). pLTF requires spinal serotonin receptor activation (Baker-Herman & Mitchell, 2002), new synthesis of brain-derived neurotrophic factor (BNDF) (Baker-Herman et al. 2004), and is constrained by spinal okadaic acid-sensitive protein phosphatases (Wilkerson et al. 2007, 2008). Although factors regulating this phosphatase constraint are not well known, we have hypothesized that increased reactive oxygen species (ROS) formation during and following AIH inhibits relevant phosphatases, thereby enabling pLTF (Wilkerson et al. 2007, 2008; MacFarlane et al. 2008). Consistent with this hypothesis, both phrenic and hypoglossal (XII) LTF are blocked by intravenous administration of a superoxide dismutase mimetic (MacFarlane & Mitchell, 2008), whereas phrenic (but not XII) LTF was rescued in rats that were also pre-treated with okadaic acid applied intrathecally to the cervical (∼C4) spinal cord (MacFarlane et al. 2008). Further, phrenic, but not XII LTF was blocked following a localized intrathecal (i.t.) injection of the same superoxide dismutase (SOD) mimetic near the phrenic motor nucleus suggesting a spinal mechanism of ROS formation (MacFarlane & Mitchell, 2008). Although these data demonstrate that spinal ROS formation near the phrenic motor nucleus is necessary for pLTF, the cellular source of the relevant ROS is unknown.
Hypoxia and re-oxygenation each independently stimulate ROS formation from multiple sources, including xanthine/xanthine oxidase, mitochondria and the NADPH oxidase complex (Abramov et al. 2007). Mitochondrial ROS formation has been implicated in carotid chemosensory plasticity following chronic intermittent hypoxia (Peng et al. 2003). On the other hand, NADPH oxidase activity is necessary for carotid chemosensory plasticity induced by episodic 5-HT2A-receptor activation (Peng et al. 2006). The NADPH oxidase complex has been implicated in other forms of neuroplasticity, including hippocampal long-term potentiation (Kishida et al. 2006; Kishida & Klann, 2007). Further, NADPH oxidase activity and ROS formation are increased by a protein kinase C (PKC)-dependent mechanism in renal mesangial cells following 5-HT2A receptor activation (Grewal et al. 1999). PKC (Neverova et al. 2007; McGuire & Ling, 2004) and 5-HT2 receptor activation (Kinkead & Mitchell, 1999; Fuller et al. 2001b) have both been implicated in AIH-induced LTF.
Since many forms of neuroplasticity share (multiple) common features in their underlying cellular/synaptic mechanisms, spinal NADPH oxidase activity could be a relevant source of ROS in AIH-induced pLTF. Thus, we tested the specific hypotheses that (1) spinal NADPH oxidase activity is necessary for AIH-induced pLTF, and (2) NADPH oxidase subunits are expressed in the phrenic motor nucleus.
Methods
Animals
All experiments were performed on 3- to 4-month-old male Sprague–Dawley rats (Sasco, colony 236, Harlan, Indianapolis, IN) and approved by The Animal Care and Use Committee at the School of Veterinary Medicine, University of Wisconsin–Madison.
Surgical preparation and measurements
Rats were anaesthetized with isoflurane, trachaeotomized and pump ventilated (tidal volume, 2.5 ml; Rodent Ventilator, model 683; Harvard Apparatus, South Natick, MA, USA). Anaesthesia was induced with isoflurane and maintained (3.5% in 50% O2, balance N2) for the duration of the surgical procedures, and then the rats were slowly converted to urethane anaesthesia (1.8 mg kg−1) via a tail vein catheter. Prior to beginning surgeries, the adequacy of isoflurane anaesthesia was confirmed by the lack of response to a toe-pinch. Surgery was performed on a temperature-controlled stainless-steel surgical table. Rectal temperature was monitored continuously with a temperature sensor (Fisher Scientific, Pittsburgh, PA, USA), and maintained constant by adjusting the temperature of the surgical table. The concentration of inspired O2 was monitored throughout experiments using a fuel-cell O2 sensor (TED 60T, Teledyne Analytical Instruments, City of Industry, CA, USA). A slow, continuous infusion of a 1 : 1 solution of heta-starch and Standard Lactated Ringer solution was achieved via a tail vein to maintain blood pressure and acid–base balance throughout the experiment.
Rats were vagotomized and a catheter was inserted into the right femoral artery to monitor blood pressure using a calibrated pressure transducer (Gould pressure transducer, P23, USA). Blood samples were analysed for O2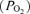 and CO2
and CO2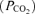 partial pressures and pH with a blood gas analyser (ABL 500, Radiometer, Copenhagen, Denmark); base excess, calculated by the analyser, was used as an indicator of metabolic acid–base disturbances.
partial pressures and pH with a blood gas analyser (ABL 500, Radiometer, Copenhagen, Denmark); base excess, calculated by the analyser, was used as an indicator of metabolic acid–base disturbances.
The left phrenic and hypoglossal (XII) nerves were dissected and exposed via a dorsal approach, cut distally, and de-sheathed. Nerves were submerged in mineral oil and placed on bipolar silver recording electrodes to record their activity; once electrical signals were detected, the rats were then assessed for adequate depth of anaesthesia by checking for an immediate increase in blood pressure and/or respiratory neural output; after confirming adequate anaesthesia, the rats were then subjected to neuromuscular block with pancuronium bromide (∼1.2 ml i.v., 1 mg ml−1). The rats received another toe-pinch to test for anaesthetic depth immediately prior to and at the end of the experimental protocol. We did not observe increased blood pressure or respiratory nerve activity in any of the rats, consistent with previous studies demonstrating the efficacy of urethane anaesthesia for many hours longer than the duration of our experimental protocols (Maggi & Meli, 1986). End tidal CO2 was monitored using a flow-through capnograph (Novametrix, model 1265, Wallingford, CT, USA) with sufficient response time, and maintained ∼40 mmHg for 1 h to allow stabilization of the preparation and nerve signals. Nerve activity was amplified (gain, 10 000; A-M systems, Everett, WA, USA), bandpass-filtered (100 Hz to 10 kHz), rectified and integrated (CWE 821 filter; Paynter, Ardmore, PA, USA; time constant, 50 ms). The signal was then digitized and recorded using WINDAQ data acquisition system (DATAQ Instruments, Akron, OH, USA). Data analysis was performed using custom designed software based on a Labview platform (LabVIEW, National Instruments, Austin, TX, USA).
An intrathecal catheter enable localized injections of NADPH oxidase inhibitors (apocynin or diphenyleneiodonium chloride, DPI), into the cerebral spinal fluid (CSF) of the cervical spinal cord. In brief, after dorsal laminectomy at C2, an incision was made in the dura and a small silicone catheter was inserted and advanced ∼2 mm caudally (2 French; Access Technologies, Skokie, IL, USA). Thus, the catheter tip was placed proximal to the region just rostral to the cervical sections containing the phrenic motor nucleus (∼C4–C5). The catheter was primed with either apocynin, DPI or their vehicles prior to positioning through the dura (doses listed below). All drugs were purchased from Sigma (Sigma, USA); apocynin was prepared fresh prior to each experiment, whereas aliquots of DPI in dimethyl sulphoxide (DMSO) were stored in a freezer and then diluted in artificial cerebrospinal fluid (aCSF) on the day of experiments.
Neurophysiological experiments
Stable nerve activity was established while the rat was ventilated with a hyperoxic inspired gas mixture (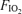 ∼0.5;
∼0.5; 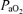 , >200 mmHg), with sufficient levels of inspired CO2, to maintain arterial
, >200 mmHg), with sufficient levels of inspired CO2, to maintain arterial 
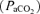 constant and at a level necessary to prevent the rat from becoming apnoeic (typically between 40–45 mmHg). After the preparation had stabilized, the apnoeic threshold for rhythmic phrenic and XII activity was determined by progressively lowering the inspired (and arterial) CO2 until rhythmic phrenic activity ceased. From apnoea, the end-tidal CO2 was progressively increased in 1 mmHg increments every 2–3 min until nerve activity resumed (i.e. the recruitment threshold). Baseline nerve activity was established with
constant and at a level necessary to prevent the rat from becoming apnoeic (typically between 40–45 mmHg). After the preparation had stabilized, the apnoeic threshold for rhythmic phrenic and XII activity was determined by progressively lowering the inspired (and arterial) CO2 until rhythmic phrenic activity ceased. From apnoea, the end-tidal CO2 was progressively increased in 1 mmHg increments every 2–3 min until nerve activity resumed (i.e. the recruitment threshold). Baseline nerve activity was established with 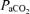 set 2–3 mmHg above the CO2 recruitment threshold. This procedure allows a standardized level of respiratory drive during baseline conditions in different rats (i.e. different rats have intrinsically different apnoeic and recruitment thresholds).
set 2–3 mmHg above the CO2 recruitment threshold. This procedure allows a standardized level of respiratory drive during baseline conditions in different rats (i.e. different rats have intrinsically different apnoeic and recruitment thresholds).
In different experimental treatment groups, AIH-induced phrenic and XII LTF were determined. The groups included (1) rats without treatment, (2) rats with intrathecal vehicle injections, (3) varied doses of intrathecal apocynin to determine dose–response characteristics on AIH-induced pLTF, and (4) intrathecal injections of DPI (1 mm). Additional rats were used as time controls (i.e. without hypoxia) to test for effects of vehicle and drugs. Fifteen minutes after establishing the CO2 recruitment threshold, rats received an intrathecal injection of apocynin, DPI or their vehicles into the CSF at a rate of 3 μl per 30 s until a total volume of 12 μl had been injected. The same volumes were used for vehicle treated rats and at different apocynin concentrations or with DPI (see below), thus standardizing convective drug spread between injections.
Approximately 20 min post-injection, a baseline blood sample was taken and the rat was exposed to AIH, consisting of 3 × 5 min hypoxic episodes (∼11% O2) with 5 min intervals of baseline conditions (i.e. 50% O2). Nerve activity was monitored for 60 min after the last hypoxic episode while maintaining baseline levels of arterial blood gases. Blood samples (0.3 ml in a heparinized syringe) were drawn and analysed before (baseline), during the first hypoxic episode, and at 15, 30 and 60 min post-hypoxia to ensure that blood gases met the criteria outlined below. During hypoxia (∼11% O2) 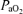 decreased to 38.8 ± 0.6 mmHg compared to baseline (∼50% O2, 278.2 ± 4.0 mmHg) in association with a 29.2 ± 3.0 mmHg decrease in MAP.
decreased to 38.8 ± 0.6 mmHg compared to baseline (∼50% O2, 278.2 ± 4.0 mmHg) in association with a 29.2 ± 3.0 mmHg decrease in MAP. 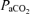 was maintained constant throughout the experimental (baseline, 42.5 ± 0.4; hypoxia, 42.4 ± 0.5; 15 min, 42.5 ± 0.5; 30 min 42.5 ± 0.4; and 60 min, 42.3 ± 0.4 mmHg).
was maintained constant throughout the experimental (baseline, 42.5 ± 0.4; hypoxia, 42.4 ± 0.5; 15 min, 42.5 ± 0.5; 30 min 42.5 ± 0.4; and 60 min, 42.3 ± 0.4 mmHg).
At the end of the experiment the rat was humanely killed by urethane overdose administered via the tail vein, followed by discontinuation of pump ventilation. In one group (n= 20; BW = 370 ± 7 g), rats received a 12 μl i.t. injection of vehicle prior to AIH. Other rat groups received 12 μl i.t. injections of apocynin at the following concentrations: 250 μm (n= 4), 450 μm (n= 3), 600 μm (n= 14) or 1 and 2.5 mm (n= 1 each). Since 600 μm was the lowest, most effective dose that blocked pLTF, a separate group of rats (n= 5) were treated with a 600 μm i.t. injection, but without AIH exposure to serve as a drug time control (TC). To further test if NADPH oxidase activity is necessary for AIH-induced phrenic LTF, additional rats (n= 3) were given a 1 mm i.t. injection of DPI (12 μl), a drug that inhibits NADPH oxidase by a distinct mechanism. Neither drug nor their vehicles affected baseline respiratory burst frequency, nerve burst amplitude or blood pressure. Apocynin was dissolved in DMSO and then diluted in aCSF for a final vehicle with 0.1% DMSO/aCSF. DPI was dissolved in the same way, except a 3% DMSO/aCSF mixture was required because of its lower solubility in aCSF.
Throughout the experimental protocol, arterial blood gases and pressure were monitored and corrected as necessary to ensure that they were maintained near baseline levels. 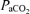 was adjusted by manipulating inspired CO2. Decreases in base excess of more than 3 mEq l−1 were corrected with intravenous sodium bicarbonate infusions. Progressive reductions in blood pressure were offset with intravenous injections of lactated Ringer solution (∼1–2 ml as needed). Data from rats were included in the analysis only if they complied with the following criteria: (1)
was adjusted by manipulating inspired CO2. Decreases in base excess of more than 3 mEq l−1 were corrected with intravenous sodium bicarbonate infusions. Progressive reductions in blood pressure were offset with intravenous injections of lactated Ringer solution (∼1–2 ml as needed). Data from rats were included in the analysis only if they complied with the following criteria: (1)  during hypoxia was between 35 and 45 mmHg; (2)
during hypoxia was between 35 and 45 mmHg; (2) 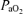 during the hyperoxic baseline and recovery periods was >180 mmHg; and (3)
during the hyperoxic baseline and recovery periods was >180 mmHg; and (3)  remained within 1 mmHg of baseline throughout the post-AIH recovery period.
remained within 1 mmHg of baseline throughout the post-AIH recovery period.
Immunohistochemical analysis of NADPH oxidase subunit distribution
Three rats were perfused with heparinized saline followed by ice-cold fixative (4% paraformaldehyde in 1× phosphate-buffered saline (PBS) (i.e. 1 : 9 ratio of PBS (10×): double distilled water) at the end of the experiment. The brain and cervical spinal cord were removed and placed in fixative overnight at 4°C, then transferred to a refrigerated cryoprotectant solution (30% sucrose in PBS) until it sank. Serial coronal sections from cervical regions containing phrenic motoneurons (C4–C5, 40 mm) were prepared using a freezing microtome and processed for the detection of NADPH oxidase subunits as well as markers for cell types in the phrenic motor nucleus (neuron: NeuN; astrocytes: GFAP; microglia: OX-42). Briefly, tissues were washed with Tris-buffered saline (TBS), 0.1% Triton X-100 in TBS (TBS-T), then incubated in 5% normal goat serum for 1 h. Serial sections were incubated overnight at 4°C in TBS-T containing primary antibodies of p47phox (1/500, Upstate, Lake Placid, NY, USA) or gp91phox (1/500 dilution, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and mouse-monoclonal anti-NeuN (1 : 200; Chemicon, Temecula, CA, USA), mouse-monoclonal anti-glial fibrillary acidic protein (GFAP; 1:1000; Chemicon), and mouse polyclonal anti-OX-42 (CD11b; 1 : 500, Serotec, Oxford, UK) at 4°C overnight. After washing with TBS-Tx (3 × 5 min), tissues were incubated in a mixture of Alexa Fluor 594 conjugated to goat anti-rabbit IgG and Alexa Fluor 488 conjugated to goat anti-mouse IgG (1:200, Molecular Probes, Eugene, OR, USA) at RT for 60 min. Stained tissue were mounted in glass using anti-fade solution (Prolong Gold antifade reagent, Invitrogen, OR, USA) and examined using an epifluorescence microscope (Nikon, Japan) with Spot Imaging software (Diagnostic Instruments, Sterling Heights, MI, USA).
Immunoblot analysis for NADPH oxidase subunits
In additional rats (n= 4), fresh spinal segments (C4–C5) were harvested, immediately frozen and then stored at −80°C. The dorsal section of the spinal cord was removed and the tissue was homogenized using a Potter-Elvehjem homogenizer in 10 mm Hepes, pH 7.9, with 1.5 mm MgCl2, 10 mm KCl, 1 mm EDTA, protease inhibitor cocktail (Roche, Indianapolis, IN, USA) and phosphatase inhibitor cocktail (Pierce Biotechnology, Rockford, IL, USA). 4000 g supernatants were centrifuged at 100 000 g in a Beckman TLA55 rotor for 60 min at 4°C. The 100 000 g pellet contained enriched membrane components while the soluble fraction consisted mainly of cytosolic components (Schack et al. 2008). Fractions were diluted for immunoblotting to 30 mm Tris, 3 mm EDTA, 1.5 mm Na3VO4, 3 mm DTT, 3% SDS and 30% glycerol. Samples were boiled for 5 min before loading onto 12.5% polyacrylamide gels (4.5 μg per lane for membrane fractions and 16 μg per lane for cytosolic fractions). Protein was transferred to Immobilon-P polyvinylidene difluoride membrane (Millipore Corp., Bedford, MA, USA). After blocking for 2 h with 6% blotto, blots were incubated overnight at 4°C with primary antibodies raised against p47phox and gp91phox (both 1/100, Santa Cruz Biotechnology), or RAC1 (1/2000, Upstate). Blots were rinsed and proteins were visualized with horseradish peroxidase (HRP)-conjugated secondary antibodies and enhanced chemiluminescence (Pierce Biotechnology). Analysis was performed using LabWorks 4.6 (UVP, Inc., Upland, CA, USA). Blots were stripped with Restore Stripping Buffer (Pierce Biotechnology) and probed with antibodies raised against pan-actin (1 : 5000, Chemicon) or the sodium-potassium ATPase (1 : 1000, Abcam, Cambridge, MA, USA).
Statistical analysis
Peak amplitude and burst frequency of phrenic and XII nerve activity were averaged in 1 min bins at each recorded data point (baseline, at the end of the first hypoxic exposure, and 15, 30 and 60 min after the final hypoxic episode). Minute phrenic activity (expressed as a percentage change from baseline) was determined by multiplying burst frequency and integrated nerve burst amplitude. Changes in nerve burst amplitude were normalized as a percentage of the baseline value, whereas burst frequency was reported as an absolute change from baseline. We compared phrenic and hypoglossal responses of rats receiving vehicle injections (time control and AIH exposure) versus drug injections. Statistical comparisons were made for time and drug treatment effects using two-way, repeated measures ANOVA. Bonferroni's post hoc test was used to identify statistically significant individual comparisons. Differences were considered significant at P < 0.05. All values are expressed as the mean ± 1 s.e.m.
Results
Phrenic and XII LTF: effects of apocynin
AIH caused a progressive and sustained increase in phrenic nerve burst amplitude (Fig. 1), indicative of pLTF. Sixty minutes post-AIH, phrenic burst amplitude was significantly increased (46 ± 6%) from baseline and time control rats (Fig. 2A), confirming robust pLTF. XII burst amplitude was also significantly elevated above baseline 60 min post-AIH (13 ± 4%), indicating a small, but statistically significant LTF characteristic of some substrains of Sprague–Dawley rats (Fuller et al. 2001a); XII LTF was also significantly greater than TC rats at 60 min post-AIH (Fig. 2B).
Figure 1. Three representative neurograms recorded from the phrenic nerve of anaesthetized rats.

Top trace, a rat that did not receive AIH (i.e. time control, TC); middle trace, an AIH-treated rat that received an i.t. injection of vehicle (12 μl); bottom trace, an AIH-treated rat that received a prior injection of apocynin (600 μm; 12 μl). Arrows indicate time points that blood samples were analysed for blood gases. Injections were administered 20 min prior to AIH. Dashed line extrapolates baseline (i.e. pre-AIH) nerve burst amplitude. Note the gradual increase in phrenic nerve burst amplitude after AIH (i.e. LTF), which was inhibited by an i.t. injection of apocynin prior AIH.
Figure 2. Dose–response curve for i.t. injection of apocynin and its effects on phrenic (A) and XII (B) nerve burst amplitude at 60 min post-AIH.

Values are means ± 1 s.e.m. and are expressed as percentage change from baseline amplitude (i.e. 0%). †Significant difference from baseline value (i.e. pre-AIH); ‡significant difference from time control (TC) value at the same time point; §significant difference from AIH value at the same time point (P < 0.05).
Intrathecal apocynin prior to AIH caused a dose-dependent decrease in pLTF. pLTF was completely blocked at 600 μm (7.8 ± 3.3%; Fig. 2A). Both phrenic and XII LTF were abolished by 1–2 mm apocynin, with the latter reaching levels below baseline at 60 min (Fig. 2B). Since 600 μm apocynin was most effective at blocking pLTF (but not XII LTF), apocynin treatment effects are presented at this dose.
This method of intrathecal drug delivery into the CSF of spinal regions containing the phrenic motor nucleus has been used previously to distinguish spinal mechanisms in pLTF versus effects at distant sites that could be affected by unintended drug distribution (Baker-Herman & Mitchell, 2002). In this approach, the magnitude of XII LTF serves as an internal control, demonstrating that the injected drug reached effective concentrations only within the spinal cord, and that the drug did not reach the brainstem at effective concentrations. Although XII LTF was small, it was significant (P < 0.05; Fig. 2B), and was unaffected by intrathecal apocynin (600 μm; Fig. 2B).
AIH also caused a small, but significant frequency LTF (46.2 ± 1.7 breaths min−1versus 41.5 ± 1.5 breaths min−1; P < 0.05). Frequency LTF was unaffected by 600 μm apocynin (baseline, 40.6 ± 1.3 breaths min−1versus 46.5 ± 1.7 breaths min−1 at 60 min post-AIH) (Fig. 3A), strengthening the argument that apocynin reached effective concentrations only in the cervical spinal cord, and did not reach the brain at effective concentrations. With our experimental approach, however, we are not certain if NADPH oxidase activity is necessary for frequency LTF.
Figure 3. Frequency of respiratory motor output (A) and changes in minute neural activity (quantified as nerve burst amplitude (mV) × frequency (breathsmin−1)) in phrenic (B) and XII (C) nerve amplitude at 15, 30, and 60 min post-AIH.

Treatment groups are as indicated. Values are means ± 1 s.e.m. and, with the exception that burst frequency (A), values are expressed as a percentage change from baseline. Note apocynin inhibited phrenic minute activity (B) whereas XII was unaffected (C). †Significant difference from baseline value (i.e. pre-AIH); ‡significant difference from time control (TC) value at the same time point; §significant difference from AIH value at the same time point (P < 0.05).
Calculated values of minute activity (frequency × amplitude) for phrenic (Fig. 3B) and XII (Fig. 3C) nerves were also significantly elevated at 60 min post-AIH. Whereas minute phrenic activity was suppressed by 600 μm apocynin (Fig. 3B), XII was unaffected by drug administration (Fig. 3C), once again confirming the spinal site of action. However, minute phrenic activity was not completely blocked by 600 μm apocynin since frequency LTF remained (Fig. 3A).
Previous studies of pLTF have shown a positive correlation between the magnitudes of pLTF and the hypoxic phrenic response (Fuller et al. 2000) although no causal relationship between has been demonstrated. No significant differences were observed between the hypoxic phrenic responses during AIH + vehicle versus AIH + apocynin (600 μm) protocols. Thus, the increase in phrenic (Fig. 4A and B) and XII (Fig. 4C and D) activities during hypoxia was unaffected by apocynin, and variations in the hypoxic chemoreflex are unlikely to have affected the outcome.
Figure 4. Comparison between vehicle, apocynin (600 μm, 12 μl), and DPI (1 mm, 12 μl) injected rats on the magnitude of the hypoxic response.

A, phrenic and XII nerve burst amplitude for each treatment group during hypoxia. B, breath frequency during hypoxia for each treatment group. Note, apocynin or DPI had no effect on the phrenic nerve amplitude, XII nerve amplitude, or frequency response to hypoxia compared to AIH treated rats. Also note, hypoxia caused a significant increase in phrenic and XII amplitude and breath frequency compared to pre-AIH. Values are means ± 1 s.e.m.
Phrenic and XII LTF: effects of DPI
An intrathecal injection of DPI (1 mm), an agent that inhibits NADPH oxidase via a distinct mechanism from apocynin (O’Donnell et al. 1993), also attenuated, but did not completely abolish, pLTF (19.3 ± 2.5%; Fig. 5). XII LTF was also unaffected by DPI (19.3 ± 5.1%) compared to AIH and so was the magnitude of the hypoxic phrenic response (Fig. 4A and B).
Figure 5. Time domains for AIH-induced phrenic (top) and XII LTF (bottom) and the effects of pre-treatment with apocynin (600 μm) or DPI (1 mm).

AIH caused a progressive increase in nerve amplitude (i.e. LTF) in phrenic and XII nerves, which was attenuated or blocked by DPI and apocynin, respectively. Note that AIH-induced XII LTF was not affected by intrathecal DPI or apocynin. Values are averaged for each treatment group for 15, 30 and 60 min post-AIH. Values are means ± 1 s.e.m. † indicates significant difference from baseline value; ‡ significant difference from time control (TC) value at the same time point.
Localization of NADPH oxidase subunits in C4 spinal cord
Immunohistochemical analysis of cervical sections (∼C4) containing the phrenic motor nucleus revealed large NeuN positive cells in regions typically occupied by phrenic motor neurons (Fig. 6A–F). These presumptive phrenic motor neurons expressed both p47phox (Fig. 6A–C) and gp91phox (Fig. 6D–F) subunits of the NADPH oxidase complex. High power micrographs show a group of neurons with p47phox and gp91phox labelling in the soma (and possibly dendrites) of presumptive phrenic motor neurons. However, neither subunit was expressed at high levels in either GFAP or Ox42 positive cells, indicating that they are not constitutively expressed, or are expressed at relatively low levels, in astrocytes and microglia, respectively (Fig. 7A–L).
Figure 6. Photomicrographs of double immunofluorescence staining in ventral horn of cervical regions.

Motor neurons are stained with NeuN against NADPH subunits p47phox (upper panel) and gp91phox (lower panel). NADPH oxidase subunits p47phox (A–C) and gp91phox (D–F) subunits are co-localized in presumptive phrenic motor neurons. The western blot (G) showed that p47phox and Rac-1 were present in the membrane and cytosol fractions from homogenates of ventral spinal segments C4–C5, while gp91phox was detected only in membrane fractions. The purity of the preparations was assessed by probing for the Na+,K+-ATPase, present mainly in the membrane fractions, and for GAPDH, present mainly in the cytosolic fractions. Scale bar in all large panels = 200 μm, small panel = 50 μm.
Figure 7. Double immunoflourescence staining of NADPH subunits p47phox and gp91phox with astrocytes marker (GFAP) and microglia marker (OX-42).

Note, both NADPH subunits are not co-localized in these glial cells. Scale bar in all large panels = 200 μm, small panel = 50 μm.
In tissues harvested from ventral C4–C5 at the end of TC experiments, immunoblot analysis of particulate (i.e. ‘membrane’) and soluble (i.e. ‘cytosolic’) fractions revealed the presence of key NADPH oxidase subunits. Immunoblots demonstrated that the catalytic subunit, gp91phox, is predominantly localized to the membrane fraction (Fig. 6G), whereas the regulatory subunits, p47phox and RAC1, are expressed in both membrane and cytosolic fractions, with somewhat greater cytosolic levels.
Discussion
Here, we demonstrate that spinal NADPH oxidase activity (i.e. superoxide anion formation) is necessary for AIH-induced phrenic long-term facilitation (pLTF). Our conclusion is supported by the localization of key NADPH oxidase subunits within putative phrenic motor neurons, but with only minimal expression in adjacent GFAP positive (presumptive astrocytes) or OX-42 positive cells (presumptive microglia). These results provide important insights concerning the source of ROS relevant to spinal respiratory plasticity following acute intermittent hypoxia.
NADPH oxidase activity is necessary for AIH-induced pLTF since two structurally and functionally distinct NADPH oxidase inhibitors (apocynin and DPI) blocked pLTF. Apocynin inhibits NADPH oxidase by preventing translocation and assembly of the cytosolic regulatory subunits (e.g. p47phox) with catalytic membrane subunits (e.g. gp91phox) (Valko et al. 2007). Some evidence suggests that apocynin requires activation by myeloperoxidase before it can effectively inhibit NADPH oxidase activity otherwise it functions as an antioxidant versus a specific inhibitor of the NADPH oxidase complex (Heumülleret al. 2008). Although myeloperoxidase is present in the spinal cord (Nakayama et al. 2007), in CNS neurons and in neuronal cell lines (Green et al. 2004), we are unable to completely exclude the possibility that apocynin acted via specific NADPH oxidase inhibition versus antioxidant effects. Therefore, we also used a functionally different compound, DPI, which interferes with NADPH oxidase activity by inhibiting gp91phox. DPI also blocked AIH-induced phrenic (but not XII) LTF, providing further reassurance that ROS generated by spinal NADPH oxidase activity are necessary for AIH-induced pLTF.
We controlled for unintended drug distribution to the brainstem by assessing XII LTF, a related form of respiratory plasticity in a distant motor pool (i.e. brainstem versus spinal cord). We used this strategy successfully in previous pharmacological studies of pLTF (Baker-Herman & Mitchell, 2002). Since systemic ROS scavengers block both phrenic and XII LTF following AIH (MacFarlane & Mitchell, 2008), the observation that phrenic, but not XII, LTF is blocked by cervical injections of apocynin and DPI confirms that effective drug concentrations were restricted to the cervical spinal regions of interest. NADPH oxidase activity also modulates carotid chemosensory responses to hypoxia (see Dinger et al. 2007). For example, hypoxic ventilatory chemoreflexes are augmented in mice lacking the NADPH oxidase regulatory subunit, p47phox, which renders NADPH oxidase incapable of generating ROS (Sanders et al. 2002). Since we did not observe changes in the magnitude of the hypoxic phrenic or XII responses during hypoxia (Fig. 4), intrathecal drug administration did not affect carotid chemoreceptor function. Collectively, our data provide strong evidence that NADPH oxidase activity is necessary for AIH-induced pLTF, and that the relevant NADPH oxidase is localized within cervical spinal segments associated with the phrenic motor nucleus.
Immunohistochemical localization of key NADPH oxidase subunits within the phrenic motor nucleus confirms that NADPH oxidase is located at key anatomical sites for pLTF (Baker-Herman & Mitchell, 2002; Mahamed & Mitchell, 2007; MacFarlane et al. 2008). NADPH oxidase subunits have been identified in other regions of the central nervous system, including brainstem regions associated with the central neural control of breathing (Tejada-Simon et al. 2005). Dendritic and synaptic NADPH oxidase in hippocampal neurons (Tejada-Simon et al. 2005) has been implicated in long-term potentiation, a form of activity-dependent synaptic plasticity thought to underlie learning and memory (Kishida et al. 2006; Kishida & Klann, 2007). The detailed, subcellular localization of NADPH oxidase subunits (p47phox, gp91phox and p22phox) has been described in neurons and astrocytes of the medial nucleus tractus solitarii of rats using electron microscopy (Glass et al. 2006). In contrast to their study, we did not observe significant labelling of NADPH oxidase subunits in GFAP-positive cells (i.e. presumptive astrocytes; Fig. 7), suggesting that constitutive ROS formation is small in astrocytes versus adjacent motor neurons. Here we provide the first evidence for NADPH oxidase subunit expression within putative motor neurons located in spinal regions associated with the phrenic motor nucleus. Although the precise role played by each subunit in pLTF requires further elaboration, they most likely assemble into an active NADPH oxidase complex, providing spinal ROS necessary for pLTF during and/or following AIH (MacFarlane & Mitchell, 2008; MacFarlane et al. 2008). These data are also supported by immunoblot analysis of subcellular fractions from the ventral cervical spinal cord. The catalytic subunit, gp91phox, was predominantly expressed in the particulate (membrane) fraction as expected (Tejada-Simon et al. 2005). On the other hand, mixed (particulate and soluble) localization of the regulatory subunits, p47phox and RAC1, suggest at least partial constitutive NADPH oxidase activity in the ventral cervical spinal cord. The NADPH oxidase complex is activated following phosphorylation of its cytosolic subunits (e.g. p47phox), which subsequently translocates to the membrane where it binds to membrane-bound subunits (i.e. gp91phox and p22phox) to enable superoxide formation (Valko et al. 2007). Consistent with such an effect, we have observed increased dihydroethidium fluorescence within presumptive phrenic motor neurons 60 min post-AIH, suggesting increased superoxide formation (MacFarlane et al. 2007). Although the source of ROS in that study is not known, it is consistent with increased NADPH oxidase activity and superoxide formation during and/or following AIH. This tentative conclusion does not rule out additional sources of ROS such as mitochondria and/or xanthine/xanthine oxidase. Regardless, our studies highlight the critical contribution of NADPH oxidase activity (whether constitutive or induced) to the necessary ROS formation for AIH-induced pLTF.
The cellular targets of the ROS necessary for pLTF have not been fully elucidated, although it could involve regulation of cellular phosphorylation events (Wilkerson et al. 2007, 2008; MacFarlane et al. 2008). For example, systemic administration of the SOD mimetic MnTMPyP blocked both phrenic and XII LTF (MacFarlane & Mitchell, 2008), whereas simultaneous pretreatment with an intrathecal injection of the serine/threonine protein phosphatase inhibitor, okadaic acid, restored only pLTF (MacFarlane et al. 2008). We propose that okadaic acid-sensitive protein phosphatases constrain pLTF (Wilkerson et al. 2007, 2008; MacFarlane et al. 2008), and the constraint is removed by ROS formed during and/or following AIH (Fig. 8). ROS-dependent inhibition of key phosphatases could favour pLTF mechanisms including new synthesis of BDNF, a molecule that is both necessary and sufficient for AIH-induced pLTF (Baker-Herman et al. 2004). Thus, sequestering ROS (with MnTMPyP) or inhibiting their formation (with apocynin/DPI) during AIH prevents removal of this phosphatase constraint and suppresses pLTF. Subsequent okadaic acid pre-treatment inhibits phosphatase activity and restores pLTF (MacFarlane et al. 2008). ROS also activates protein kinases (Klann et al. 1998; Klann & Thiels 1999; Kishida & Klann, 2007), some of which are relevant to pLTF including protein kinase C and ERK MAP kinases and receptor tyrosine kinases (Mahamed & Mitchell, 2007; Feldman et al. 2003). NAPDH oxidase activity is likely to provide the ROS necessary to regulate the kinase/phosphatase balance (Fig. 8).
Figure 8. Working cellular/synaptic model of the role played by NADPH oxidase (NOX) in pLTF.

We postulate that AIH stimulates episodic spinal serotonin (5-HT) release, activating serotonin type 2 (5-HT2) receptors on phrenic motor neurons (Baker-Herman and Mitchell, 2002). Subsequent activation of protein kinase C (PKC) is postulated to initiate new BDNF synthesis (Baker-Herman et al. 2004) and reactive oxygen species (ROS) formation from NOX activity. We suggest that the ROS inhibit serine/threonine protein phosphatases (PP) that normally constrain pLTF (Wilkerson et al. 2007, 2008; MacFarlane et al. 2008). By inhibiting these phosphatases, the PP constraint is relieved, and pLTF is expressed.
While NADPH oxidase activity is necessary for AIH-induced pLTF, the factors regulating NADPH oxidase activity are speculative. Hypoxia (Peng et al. 2006) and re-oxygenation (Abramov et al. 2007) are separate stimuli, each capable of increasing NADPH oxidase activity. Since pLTF is uniquely induced by intermittent, but not sustained, hypoxia (Baker & Mitchell, 2000), then repetitive re-oxygenation versus hypoxia per se may be the more important stimulus to NADPH oxidase activity. Likewise, 5-HT2A receptor activation (without hypoxia/re-oxygenation) increases NADPH oxidase activity and ROS formation in renal mesangial cells (Grewal et al. 1999) and a similar effect is observed in ex vivo carotid bodies (Peng et al. 2006). Since hypoxic activation of the carotid bodies stimulates raphe serotonergic neurons (Erickson & Millhorn, 1994; Morris et al. 1996; Teppema et al. 1997), subsequent release of serotonin on or near phrenic motor neurons (Kinkead et al. 2001) implicates that hypoxia may actually be only an indirect stimulus to NADPH oxidase activity, acting indirectly via serotonergic receptor activation. In support of this hypothesis, 5-HT2 receptor activation on or near phrenic motor neurons is necessary for pLTF (Baker-Herman & Mitchell, 2002) and we have recently observed that episodic spinal serotonin receptor activation elicits phrenic motor facilitation by an NADPH oxidase-dependent mechanism (MacFarlane & Mitchell, 2007). The relevant stimulus to NADPH oxidase activity and pLTF expression, however, requires further investigation.
In conclusion, we provide the first evidence that NADPH oxidase is expressed in cervical (respiratory) motor neurons, and that cervical spinal NADPH oxidase activity is necessary for AIH-induced pLTF. We suggest that NADPH oxidase is a critical regulator of spinal neuroplasticity, and that it is one factor that confers pattern sensitivity in the expression of respiratory plasticity (Wilkerson et al. 2007, 2008; MacFarlane et al. 2008). Since NADPH oxidase is an important regulator of respiratory plasticity, it is an attractive candidate molecule/complex in the development of novel therapeutic strategies for the treatment of devastating ventilatory control disorders associated with inadequate motor neuron output and/or activation (Mitchell, 2007). Examples include (1) obstructive sleep apnoea, where upper airway motor neuron activity is suspected to be inadequate for upper airway patency during sleep, (2) cervical spinal injury, where synaptic inputs to respiratory motor neurons have been disrupted and functional recovery may result from increased strength in spared synaptic pathways to those motor neurons, and (3) motor neuron disease, where loss of motor neurons must be compensated by greater activity in those that survive (Mitchell, 2007).
Acknowledgments
This work was supported by NIH HL80209. P.M.M. was the recipient of a Parker B Francis Fellowship. We thank Brad Hodgeman for excellent technical assistance and Safraaz Mahamed for use of his custom-designed computer program for analysis of neurograms.
Glossary
Abbreviations
- AIH
acute intermittent hypoxia
- DPI
diphenyleneiodonium chloride
- LTF
long-term facilitation
- pLTF
phrenic long-term facilitation
- ROS
reactive oxygen species
- CSF
cerebral spinal fluid
Author contributions
All authors contributed either to the conception and design of the experiments or analysis and interpretation of data as well as drafting the article and revising it critically for final approval of the version to be published. All experiments were conducted in the Department of Comparative Biosciences, School of Veterinary Medicine at the University of Wisconsin, Madison, USA.
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol. 2000;15:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman T, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Dinger B, He L, Chen J, Liu X, Gonzalez C, Obeso A, Sanders K, Hoidal J, Stensaas L, Fidone S. The role of NADPH oxidase in carotid body arterial chemoreceptors. Respir Physiol Neurobiol. 2007;157:45–54. doi: 10.1016/j.resp.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Millhorn DE. Hypoxia and electrical stimulation of the carotid sinus nerve induce Fos-like immunoreactivity within catecholaminergic and serotoninergic neurons of the rat brainstem. J Comp Neurol. 1994;348:161–182. doi: 10.1002/cne.903480202. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic nerve output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Baker TL, Behan M, Mitchell GS. Expression of hypoglossal long-term facilitation differs between substrains of Sprague-Dawley rat. Physiol Genomics. 2001;4:175–181. doi: 10.1152/physiolgenomics.2001.4.3.175. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Selected contribution: phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Huang J, Oselkin M, Tarsitano MJ, Wang G, Iadecola C, Pickel VM. Subcellular localization of nicotinamide adenine dinucleotide phosphate oxidase subunits in neurons and astroglia of the rat medial nucleus tractus solitarius: Relationship with tyrosine hydroxylase immunoreactive neurons. Neurosci. 2006;143:547–564. doi: 10.1016/j.neuroscience.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green PS, Mendez AJ, Jacob JS, Crowley JR, Growdon W, Hyman BT, Heinecke JW. Neuronal expression of myeloperoxidase is increased in Alzheimers disease. J Neurochem. 2004;90:724–733. doi: 10.1111/j.1471-4159.2004.02527.x. [DOI] [PubMed] [Google Scholar]
- Grewal JS, Mukhin YV, Gamovskaya MN, Raymond JR, Greene EL. Serotonin 5-HT2A receptor induces TGF-β1 expression in mesangial cells via ERK: proliferative and fibrotic signals. Am J Physiol Renal Physiol. 1999;276:F922–F930. doi: 10.1152/ajprenal.1999.276.6.F922. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Mitchell GS. Time-dependent hypoxic ventilatory responses in rats: effects of ketanserin and 5-carboxyamidotryptamine. Am J Physiol Regul Integr Comp Physiol. 1999;277:R658–R666. doi: 10.1152/ajpregu.1999.277.3.R658. [DOI] [PubMed] [Google Scholar]
- Kinkead R, Bach KB, Johnson SM, Hodgeman BA, Mitchell GS. Plasticity in respiratory motor control: intermittent hypoxia and hypercapnia activate opposing serotonergic and noradrenergic modulatory systems. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:207–218. doi: 10.1016/s1095-6433(01)00393-2. [DOI] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antiox Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Klann E, Thiels E. Modulation of protein kinases and protein phosphatases by reactive oxygen species: implications for hippocampal synaptic plasticity. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:359–376. doi: 10.1016/s0278-5846(99)00002-0. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Serotonin-induced phrenic long-term facilitation requires reactive oxygen species signalling via the NADPH oxidase complex. Abstract SFN. 2007 [Google Scholar]
- MacFarlane PM, Satriotomo I, Mitchell GS. Reactive oxygen species generated by NADPH oxidase are necessary for phrenic long-term facilitation following acute intermittent hypoxia. FASEB J. 2007 [Google Scholar]
- MacFarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neurosci. 2008;152:189–197. doi: 10.1016/j.neuroscience.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane PM, Wilkerson JE, Lovett-Barr MR, Mitchell GS. Reactive oxygen species and respiratory plasticity following intermittent hypoxia. Respir Physiol Neurobiol. 2008;164:263–271. doi: 10.1016/j.resp.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi CA, Meli Suitability of urethane anesthesia for physiopharmacological investigations in various systems. Part 1: General considerations. Experientia. 1986;42:109–114. doi: 10.1007/BF01952426. [DOI] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- McGuire M, Ling L. Activation of protein kinase C near/in phrenic motoneurons is required for phrenic long-term facilitation in rats. Am J Respir Crit Care Med. 2004;169:A433. [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Johnson SM. Neuroplasticity in respiratory motor control. J Appl Physiol. 2003;94:358–374. doi: 10.1152/japplphysiol.00523.2002. [DOI] [PubMed] [Google Scholar]
- Mitchell GS. Respiratory plasticity following intermittent hypoxia: a guide for novel therapeutic approaches to ventilatory control disorders. In: Gaultier C, editor. Genetic Basis for Respiratory Control Disorders. New York: Springer; 2007. pp. 291–311. [Google Scholar]
- Morris KF, Arata A, Shannon R, Lindsey BG. Long-term facilitation of phrenic nerve activity in cats: responses and short time scale correlations of medullary neurons. J Physiol. 1996;490:463–480. doi: 10.1113/jphysiol.1996.sp021158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T, Harada N, Asano M, Nomura N, Saito T, Mishima A, Okajima K. Atrial natriuretic peptide reduces ischemia/reperfusion-induced spinal cord injury in rats by enhancing sensory neuron activation. J Pharmacol Exp Ther. 2007;322:582–590. doi: 10.1124/jpet.107.120725. [DOI] [PubMed] [Google Scholar]
- Neverova NV, Saywell SA, Nashold LJ, Mitchell GS, Feldman JL. Episodic stimulation of α1-adrenoreceptors induces protein kinase C-dependent persistent changes in motoneuronal excitability. J Neurosci. 2007;27:4435–4442. doi: 10.1523/JNEUROSCI.2803-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J. 1993;290:41–49. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body of intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Yuan G, Jacono FJ, Kumar GK, Prabhakar NR. 5-HT evokes sensory long-term facilitation of rodent carotid body via activation of NADPH oxidase. J Physiol. 2006;576:289–295. doi: 10.1113/jphysiol.2006.116020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KA, Sundar KM, He L, Dinger B, Fidone S, Hoidal JR. Role of components of the phagocytic NADPH oxidase in oxygen sensing. J Appl Physiol. 2002;93:357–364. doi: 10.1152/japplphysiol.00564.2001. [DOI] [PubMed] [Google Scholar]
- Schack VR, Morth JP, Toustrup-Jensen MS, Anthonisen AN, Nissen P, Andersen JP, Vilsen B. Identification and function of a cytoplasmic K+ site of the Na+,K+-ATPase. J Biol Chem. 2008;283:27982–27990. doi: 10.1074/jbc.M803506200. [DOI] [PubMed] [Google Scholar]
- Tejada-Simon MV, Serrano F, Villasana LE, Kanterewicz BI, Wu GY, Quinn MT, Klann E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol Cell Neurosci. 2005;29:97–106. doi: 10.1016/j.mcn.2005.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppema LJ, Veening JG, Kranenburg A, Dahan A, Berkenbosch A, Olievier C. Expression of c-fos in the rat brainstem after exposure to hypoxia and to normoxic and hyperoxic hypercapnia. J Comp Neurol. 1997;388:169–190. doi: 10.1002/(sici)1096-9861(19971117)388:2<169::aid-cne1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Wilkerson JER, MacFarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: roles of protein phosphatases and reactive oxygen species. Biochem Soc Trans. 2007;35:1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]
- Wilkerson JER, Satriotomo I, Baker-Herman TL, Watters TL, Mitchell GS. Okadaic acid-sensitive protein phosphatases constrain phrenic long-term facilitation after sustained hypoxia. J Neurosci. 2008;28:2949–2958. doi: 10.1523/JNEUROSCI.5539-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]