

Abstract
The UniProt knowledgebase, UniProtKB, is the main product of the UniProt consortium. It consists of two sections, UniProtKB/Swiss-Prot, the manually curated section, and UniProtKB/TrEMBL, the computer translation of the EMBL/GenBank/DDBJ nucleotide sequence database. Taken together, these two sections cover all the proteins characterized or inferred from all publicly available nucleotide sequences. The Plant Proteome Annotation Program (PPAP) of UniProtKB/Swiss-Prot focuses on the manual annotation of plant-specific proteins and protein families. Our major effort is currently directed towards the two model plants Arabidopsis thaliana and Oryza sativa. In UniProtKB/Swiss-Prot, redundancy is minimized by merging all data from different sources in a single entry. The proposed protein sequence is frequently modified after comparison with ESTs, full length transcripts or homologous proteins from other species. The information present in manually curated entries allows the reconstruction of all described isoforms. The annotation also includes proteomics data such as PTM and protein identification MS experimental results. UniProtKB and the other products of the UniProt consortium are accessible online at www.uniprot.org.
Keywords: Database, UniProt, Manual annotation, Plant, Proteomics, PTM
Introduction
The UniProt consortium was created in 2002 by the joining of forces between the Swiss Institute of Bioinformatics (SIB), the European Bioinformatics Institute (EBI) and the Protein Information Resource (PIR) group at the Georgetown University Medical Center and National Biomedical Research Foundation.
The main goal of the consortium is to provide the scientific community with a single, stable, high quality, comprehensive and authoritative protein knowledgebase, UniProtKB (www.uniprot.org). This knowledgebase consists of two sections: UniProtKB/Swiss-Prot, which contains all the fully manually annotated, non-redundant records, and UniProtKB/TrEMBL, the computer-annotated section that contains the translation of all the coding sequences (CDS) deposited in the EMBL/GenBank/DDBJ nucleotide sequence database. Taken together, the two sections cover all the proteins characterized or inferred from all publicly available nucleotide sequences.
Besides this centerpiece, the UniProt consortium also produces and maintains several other products such as UniRef, which consists of clusters of sequences sharing 100%, 90% or 50% of identity, UniParc, a highly redundant archive that contains original protein sequences retrieved from several different sources, or UniMES, a collection of metagenomic and environmental sequences (fig. 1). For a detailed description of UniProt and its various products, see [1].
Fig. 1.
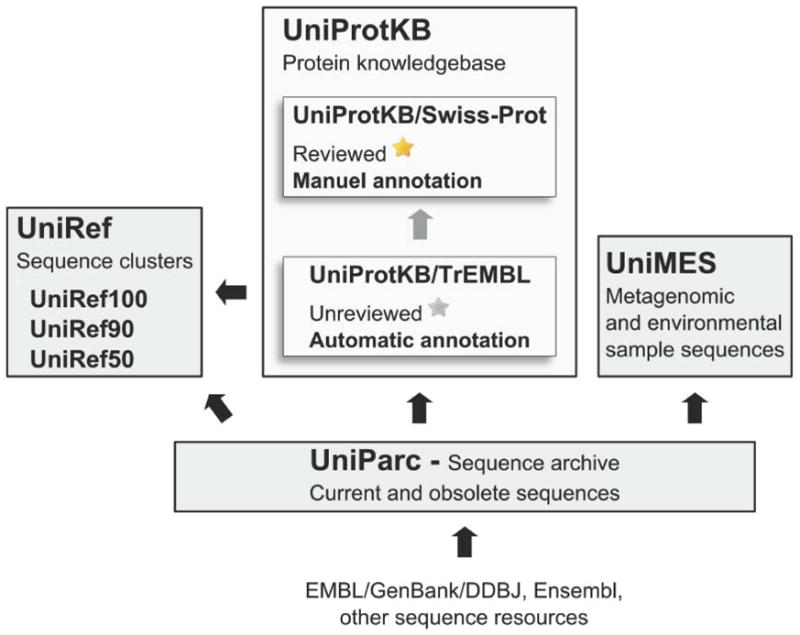
Sources and flow of data for UniProt component databases.
The Plant Proteome Annotation Program
Shortly after the publication of the first complete plant genome sequence in 2000 [2], the Swiss-Prot group initiated the Plant Proteome Annotation Program (PPAP). The main goal of this program is the manual annotation of plant-specific proteins or protein families, with a specific emphasis on the proteomes of two fully sequenced model organisms, Arabidopsis thaliana [2] and Oryza sativa [3].
We are currently working on the establishment and annotation of a comprehensive, non-redundant complete proteome of Arabidopsis. As a first step towards achieving this goal we have compared the content of our database with the list of proteins produced by alternative splicing published by The Arabidopsis Information Resource (TAIR) [4]. In several cases this has led us to complement the sequence information that was already present in UniProtKB with data available at TAIR.
Current status of the plant proteome annotation
By mid October 2008, UniProtKB/Swiss-Prot (Rel. 14.3) contained 399’749 manually curated entries, including 23’951 plant proteins (Table 1). Of these, 7’064 are from Arabidopsis thaliana, with 999 having one or more splice variant, while 1’865 originate from Oryza sativa, spp japonica, with 124 having one or more splice variant.
Table 1.
Content of UniProtKB release 14.3 (14-Oct-2008) sites.
| All species combined | Viridiplantae | |
|---|---|---|
| UniProtKB/TrEMBL (computer annotated entries, waiting for manual curation) | 6,212,793 | 488,400 |
| UniProtKB/Swiss-Prot (Manually annotated proteins) | 397,539 | 23,668 |
| Total in UniProtKB | 6,610,332 | 512,068 |
1’894 different plant species are currently represented in the manually annotated section of UniProtKB, and 12’205 proteins, half of all the entries from Viridiplantae, originate from the 10 most highly represented species (Table 2).
Table 2.
The 10 most highly represented plant species in UniProtKB/Swiss-Prot (Rel. 14.3)
| Number | Frequency | Species | |
|---|---|---|---|
| 1 | 6970 | ARATH | Arabidopsis thaliana (Mouse-ear cress) |
| 2 | 1786 | ORYSJ | Oryza sativa subsp. japonica (Rice) |
| 3 | 609 | MAIZE | Zea mays (Maize) |
| 4 | 438 | ORYSI | Oryza sativa subsp. indica (Rice) |
| 5 | 428 | TOBAC | Nicotiana tabacum (Common tobacco) |
| 6 | 400 | SOLLC | Solanum lycopersicum (Tomato) (Lycopersicon esculentum) |
| 7 | 376 | SOLTU | Solanum tuberosum (Potato) |
| 8 | 351 | PEA | Pisum sativum (Garden pea) |
| 9 | 344 | SOYBN | Glycine max (Soybean) |
| 10 | 318 | WHEAT | Triticum aestivum (Wheat) |
Structure and content of a UniProtKB/Swiss-Prot entry
Database redundancy is minimized by merging all submitted sequence data from different sources about a given protein in a given organism into a single entry. This implies the detection and correction of potential frameshifts, sequencing errors and erroneous gene model predictions. The sequence displayed in the entry is the most correct sequence version according to annotator judgment. If protein sequences are only derived from a computer gene prediction program running on a genomic sequence, the proposed gene model is validated, whenever possible, by multiple alignments with paralogs (other members of the same protein family) or orthologs (proteins having the same function in related species). These comparisons allow not only the correction of a great number of predicted gene models, but may also permit the inference of certain biological properties for as yet uncharacterized proteins.
1) Core structure
The minimal information contained in each entry, be it UniProtKB/TrEMBL or UniProtKB/Swiss-Prot, consists of an entry identifier, an accession number, a description including a recommended name, taxonomic classification of the organism in which the protein is present, bibliographical reference(s) and the protein sequence. In fully annotated UniProtKB/Swiss-Prot entries additional information is found in 11 different sections:
Names and origin
Protein attributes
General annotation (Comments)
Ontologies
Alternative products
Sequence annotation (Features)
Sequences
References
Cross-references
Entry information
Relevant documents
The level of confidence associated with each item of information is indicated by the presence or otherwise of one or more non-experimental qualifiers. “Potential” indicates that the feature is predicted by computer analysis. “Probable” means that the information is not explicitly given in the literature, but can be inferred from other sources (other proteins, obvious targeting signals, common knowledge, etc.). “By similarity” indicates that the information is not proven for this particular protein, but that it has been demonstrated for a homologous protein.
The content of each entry is continuously evolving as the information is regularly updated and completed. While for reasons of consistency it is sometimes necessary to change the entry name (identifier), the primary accession number is never altered and therefore should always be used to unambiguously identify and cite UniProtKB entries.
Differences between the displayed sequence and those inferred from various sequencing reports, including differences due to splice variants, polymorphisms, mutagenesis or sequencing errors, are listed in the “Sequence annotation (Features)” section of the entry (fig. 2).
Fig. 2.
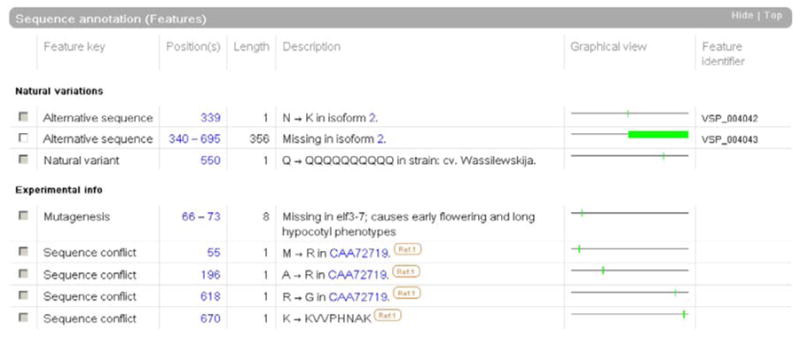
Part of the “Sequence annotation” section of an entry (extracted from O82804).
This allows all alternative sequences described in UniProtKB/Swiss-Prot to be easily recreated, and UniProtKB provides several tools which utilize this feature. For example, a BLAST search launched from the UniProt web site against UniProtKB or a subsection thereof will automatically include all described isoforms. It should be noted that a file containing the sequence of all the splice variants present in UniProtKB/Swiss-Prot is available for download under the FASTA format from our FTP server (ftp.uniprot.org/pub/databases/uniprot/knowledgebase/uniprot_sprot_varsplic.fasta.gz)
When the submitted sequence extensively differs from the displayed sequence, as in the case of an erroneous gene prediction, individual conflicts are not described in the “Sequence annotation (Features)” section; instead, this fact is indicated in the “Sequence Caution” part of the “General annotation (Comments)” section (fig. 3).
Fig. 3.

Example of a “Sequence caution” warning (extracted from Q3E6Y4).
2) Scientific literature as a source of validated information
Most of the data included in a UniProtKB/Swiss-Prot entry are extracted from the scientific literature by specially trained life scientists. If the information relates to defined regions of the protein then the precise positions of these regions are indicated in the “Sequence annotation (Features)” section. Otherwise, data are stored as “General annotation (Comments)”. This section is divided into subsections, each containing information pertinent to particular properties of the protein such as its function, catalytic activity, subcellular location, subunit structure, mass spectrometry or biophysicochemical properties. Whenever possible, individual fields are structured and controlled vocabularies are used in order to facilitate text searches and database interoperability. Currently, Gene Ontology (GO) terms are manually mapped to the UniProtKB keyword and subcellular location controlled vocabularies, to EC number, to InterPro [5] matches or to HAMAP family rules [6] and then transferred automatically to the matching entries. Mapping to Plant Ontology terms or between UniProtKB pathways and GO terms is underway. UniProt will integrate the GO consortium in the forthcoming months and active GO annotation will be implemented in UniProtKB by mid 2009. Keywords and GO terms are both stored in the “Ontologies” section of the entries.
A description of the various line types and their format is available at http://www.uniprot.org/manual.
The sources from which the data have been extracted are listed in the “References” section, with an indication of the nature of the information retrieved, and its source or scope (for example the tissue or cultivar used in the experiment). When available, direct links to the abstract in PubMed and to the full text version through its Digital Object Identifier (DOI) are also provided (fig. 4). The bibliographic list presented in an entry is not exhaustive but consists of a selection of the most relevant articles used for the current annotation.
Fig. 4.
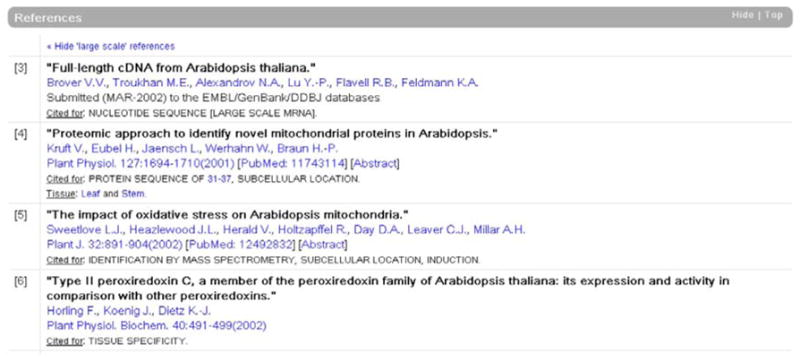
Example of bibliographical references (extracted from Q9M7T0).
3) Evidence of the existence of a protein
Some protein sequences may be mere predictions derived from the hypothetical translation of a nucleotide sequence, while others may be from well characterized proteins whose existence is proven, for example by mass spectrometry. We therefore provide an indication of the available evidence for the existence of each protein in the form of the Protein Existence (PE) line included in the “Protein attributes” section of every UniProtKB entry.
The PE line may take one of the following values:
1:Evidence at protein level;
2:Evidence at transcript level;
3:Inferred from homology;
4:Predicted;
5:Uncertain;
- Level 1 (Evidence at protein level) is attributed to any protein whose existence is supported by clear experimental evidence, such as Edman sequencing, unambiguous identification by mass spectrometry, X-ray or NMR structure, detection by antibodies, etc.
- Level 2 (Evidence at transcript level) is used to indicate that the existence of a protein has not been strictly proven, but is supported by transcription data such as cDNAs, RT-PCR, Northern blots or micro-array data extracted from the ArrayExpress or CleanEx databases.
- Level 3 (Inferred from homology) is used to indicate that the existence of the protein is probable since clear orthologs exist in closely related species or because the protein is the member of a family including multiple paralogs in the same species.
- Level 4 (Predicted) is attributed to entries without evidence at protein, transcript, or homology levels.
- Level 5 (Uncertain) indicates that the existence of the protein is unsure and that we consider that the proposed protein sequence may represent the translation of a pseudogene or an erroneously assigned ORF (to a non-coding RNA for example).
Criteria used to assign a PE level to entries are described in a document file available on the UniProt web site (www.uniprot.org/docs/pe_criteria). The distribution of the UniProtKB/Swiss-Prot plant entries according to their protein existence level is shown in fig. 5.
Fig. 5.
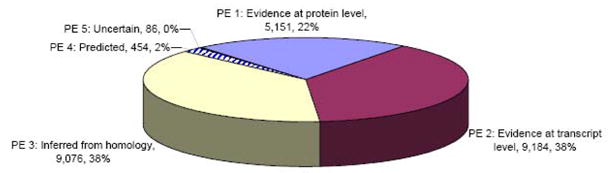
Distribution of the manually annotated plant entries according to their PE level.
For each type of evidence supporting the existence of a protein, the absolute number and proportion of the 23’951 plant entries present in UniProtKB/Swiss-Prot (Rel. 14.3) is indicated.
It should be pointed out that the PE line does not describe the accuracy or correctness of the sequence displayed but only the evidence for the existence of the protein. Sequences derived from gene predictions from genomic sequences in particular are prone to errors and may well be not entirely accurate.
4) Proteomics information
Two specific topics of the “General annotation” section about mass spectrometry and PTM might be of special interest for scientists working in the field of proteomics.
4.1) Mass spectrometry data
A comment “Mass spectrometry” is added when the entire protein or a biologically active peptide has been specifically studied by MS. It includes the range of the peptide submitted to MS, its determined molecular weight in Daltons with the error range of the machine if available, and the MS method used. When the protein studied carries a PTM, this fact is indicated in a note (fig. 6).
Fig. 6.

Example of annotation of mass spectrometry data (extracted from P0AE67).
When proteomic identification has been made by MS or MS/MS on tryptic fragments instead of biologically relevant peptides, the comment “Mass spectrometry” is not used, but the corresponding bibliographic reference is tagged as having provided identification of the protein concerned (cf. ref. 5 in fig. 4).
When MS/MS spectra are studied manually and provided along with each amino acid and ion series, the results are considered as direct protein sequencing and annotated as such.
4.2) PTM
When a protein is modified post-translationally but the precise site or sites of modification are not known, this fact is indicated in the “Post-translational modification” subsection of the “General annotation” section. The same subsection is also used to indicate proteolytic cleavage or N-terminal blockage when the precise identity of the N-terminal residue is unknown. If the biological role of a particular PTM is known, the corresponding detailed information is also given in the same “General annotation” section (fig. 7).
Fig. 7.

Example of annotation of PTMs in the “General annotation” section (extracted from O24591).
When the precise positions of individual modifications are known these are given in the “Sequence annotation (Features)” section of the UniProtKB entry (fig. 8).
Fig. 8.
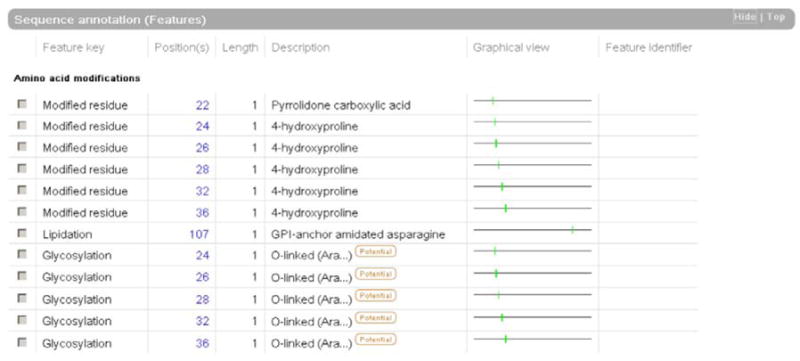
Example of annotation of PTMs in the “Sequence annotation” section, with a precise indication of the position of the modifications (extracted from Q9M0S4).
Currently, 336 different forms of PTM (not including the various types of glycosylation) are annotated in UniProtKB, and a full list is available at www.uniprot.org/docs/ptmlist. Included in this list are also the average mass differences caused by the various modifications.
In release 14.3 of UniProtKB, 4’225 manually annotated plant proteins contained one or more defined PTM sites. The frequency of occurrence of some of the most commonly encountered PTMs in plants is given in Table 3.
Table 3.
Number of plant entries containing some of the most frequent PTM sites.
| PTM | Number of occurrence |
|---|---|
| Disulfide bonds | 1985 |
| Modified residues | 1806 |
| phosphorylations | 653 |
| acetylations | 633 |
| methylations | 602 |
| pyrrolidone carboxylic acid | 147 |
| Glycosylations | 1319 |
| Cross-links | 290 |
| Lipidation | 268 |
| GPI-anchor | 109 |
| geranylation | 76 |
| myristoylation | 47 |
| palmitoylation | 19 |
| farnesylation | 13 |
4.3) Large scale proteomic experiments
We also incorporate information extracted from large scale proteomic experiments, mostly dealing with PTM identification or organelle profiling. Due to a high level of genome duplication, proteomic studies in plants are particularly difficult. Often fragments cannot be assigned unequivocally to a single protein as they match several members of large protein families. We pay particular attention to the quality of the data to be integrated, first at the level of sample preparation and purity, then at the level of MS protocols and apparatus, and finally at the level of data analysis and filtering. We often change the threshold of acceptable identification scores, or manually remove potential chemical artifacts from biologically relevant PTM.
Curation of proteomics experiments, performed on a variety of biological materials, using different apparatus, software and scoring system is not an easy task. Some initiatives such as PRIDE [7] have been launched in order to create repositories displaying all the technical details needed for evaluating and comparing proteomic experiments. We are working in close collaboration with these groups to find adequate selection criteria to facilitate the automatic retrieval of high quality data from public repositories.
Therefore raw proteomic identification data should ideally be submitted first to specialized databases such as PRIDE. Specific cross-references to those databases could then be inserted in the corresponding UniProtKB entries.
5) Cross-references
Information related to the protein described in an entry but stored in a database external to UniProt can be accessed by following the links provided in the “Cross-references” section. Among many, this concerns the underlying DNA sequences stored in the EMBL/GenBank/DDBJ nucleotide sequence database, the 3D-protein structures from PDB [8], HSSP [9] or ModBase [10], the protein domains and family characterizations of PROSITE [11], Pfam [12], InterPro [5], ProDom [13], etc., the proteomic data from PeptideAtlas [14] and ProMEX [15], or all the various information stored in Model Organism Databases (MOD) such as Gramene [16], MaizeGDB [17], and TAIR [4] to cite only a few.
Currently UniProtKB is cross-linked to more than 100 external databases and a complete list can be downloaded from our web site (www.uniprot.org/docs/dbxref).
UniProtKB is synchronized with several of those external databases. For example, all the gene models proposed by TAIR are also included in UniProtKB and all the different 806 plant proteins with a determined 3D-structure are cross-linked to the corresponding PDB entries, while 672 of them (83%) are manually annotated in UniProtKB/Swiss-Prot.
Concluding remarks
UniProtKB provides the scientific community with a comprehensive, high-quality and freely accessible resource of protein sequence and functional information. It contains all the protein sequences inferred from publicly available nucleotides sequences, but only UniProtKB/Swiss-Prot, the fully manually annotated section, is non redundant. UniProtKB provides access to more than one hundred external resources and as such acts as a central hub for biomolecular information.
The “Protein Evidence” tag allows the discrimination between proteins whose existence has been experimentally proven and those whose existence has been inferred computationally. This is important since computational gene predictions can be prone to error: approximately one third of the initial gene predictions in Arabidopsis were revised following incorporation of information from expressed transcripts. However it is important to bear in mind that the “Protein Evidence” tag does not constitute a measure of the correctness of the protein sequence displayed.
As protein isoforms may differ considerably from the displayed protein sequence, with potentially less than 50% similarity, it may be important to include all the splice variants when the database is used to identify new proteins. All the information needed to recreate the various isoforms is provided in the original UniProtKB entries. In this way, an increased number of theoretical peptides can be produced for a potentially better identification of proteins after a MS experiment for example. By the same token the mass of the theoretical peptides can easily be corrected according to the post-transcriptional modifications described in the entries. Tools such as Phenyx (www.genebio.com/products/phenyx), take full advantage of the annotated sequence information found in the UniProtKB databases such as PTMs, binding sites, variants, alternative splicing events, etc. During a search for peptides Phenyx examines the difference between an experimental peptide mass and a theoretical peptide mass in order to determine modifications to the protein sequence. If a mass difference corresponds to a known PTM that is annotated in UniProtKB/Swiss-Prot (even if the modification was not selected by the user), then the peptide sequence is considered modified and reported in the results.
Measuring the false discovery rate of a MS/MS experiment through the use of decoy databases is now a requirement for most proteomic studies. To comply with these new standards, the UniProt consortium now provides decoy versions of UniProtKB and UniRef100, which ensure that no tryptic peptide is shared between the decoy and the initial databases. The various decoy databases can be retrieved in FASTA format from our public FTP site (ftp.uniprot.org/pub/databases/uniprot/current_release/decoy).
The major effort of the plant group is currently directed focused on the extensive annotation of the proteomes of both a monocot (Oryza sativa) and a dicot (Arabidopsis thaliana). In a later stage, we will then propagate the relevant annotation to orthologous proteins from other plant species. Once the sequences and the gene predictions are reliable enough, we will also annotate all the proteins involved in specific pathways not present in our two model plants, such as the nitrogen fixation pathway of Medicago truncatula.
As we are willing to keep up-to-date with the most recent research results, we are seeking active collaboration from the scientific community. We urge researchers to deposit their results in public databases such as the EMBL/GenBank/DDBJ database for nucleotide sequences (even if it is only a sequence produced by PCR) or PRIDE for proteomics data for example. Researchers should also favor the use of clear, unique and non-ambiguous identifiers in their publications. Usage of well recognized ordered locus names such as the AGI numbers in Arabidopsis, for example, is strongly recommended. Following those simple guidelines will already considerably facilitate the daily work of curators. Researchers are welcome to help us maintain a high-quality database by sending us feedback and corrections, or submitting relevant findings.
Acknowledgments
The authors would like to thank Alan Bridge and Sylvain Poux for critical reading and correction of the manuscript. The Swiss-Prot group is part of the Swiss Institute of Bioinformatics (SIB) and of the UniProt consortium. Its activities are supported by the Swiss Federal Government through the Federal Office of Education and Science and by the National Institutes of Health (NIH) grant 2 U01 HG02712-04. Additional support comes from the European Commission contract FELICS (021902RII3).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.The UniProt Consortium. The universal protein resource (UniProt) Nucleic Acids Res. 2008;36:D190–95. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 3.International rice genome sequencing project (IRGSP) The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 4.Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L, Radenbaugh A, Singh S, Swing V, Tissier C, Zhang P, Huala E. The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 2008;36:D1009–14. doi: 10.1093/nar/gkm965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Buillard V, Cerutti L, Copley R, Courcelle E, Das U, Daugherty L, Dibley M, Finn R, Fleischmann W, Gough J, Haft D, Hulo N, Hunter S, Kahn D, Kanapin A, Kejariwal A, Labarga A, Langendijk-Genevaux PS, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McAnulla C, McDowall J, Mistry J, Mitchell A, Nikolskaya AN, Orchard S, Orengo C, Petryszak R, Selengut JD, Sigrist CJ, Thomas PD, Valentin F, Wilson D, Wu CH, Yeats C. New developments in the InterPro database. Nucleic Acids Res. 2007;35:D224–28. doi: 10.1093/nar/gkl841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lima T, Auchincloss AH, Coudert E, Keller G, Michoud K, Rivoire C, Bulliard V, de Castro E, Lachaize C, Baratin D, Phan I, Bougueleret L, Bairoch A. HAMAP: a database of completely sequenced microbial proteome sets and manually curated microbial protein families in UniProtKB/Swiss-Prot. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkn661. in Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones P, Cote RG, Cho SY, Klie S, Martens L, Quinn AF, Thorneycroft D, Hermjakob H. PRIDE: new developments and new datasets. Nucleic Acids Res. 2008;36:D878–D883. doi: 10.1093/nar/gkm1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sander C, Schneider R. The HSSP data base of protein structure-sequence alignments. Nucleic Acids Res. 1993;21:3105–09. doi: 10.1093/nar/21.13.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pieper U, Eswar N, Davis F, Madhusudhan MS, Rossi A, Marti-Renom MA, Karchin R, Webb B, Eramian D, Shen MY, Kelly L, Melo F, Sali A. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2006;34:D291–95. doi: 10.1093/nar/gkj059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJ. The 20 years of PROSITE. Nucleic Acids Res. 2008;36:D245–49. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn RD, Tate J, Mistry J, Coggill PC, Sammut SJ, Hotz HR, Ceric G, Forslund K, Eddy SR, Sonnhammer EL, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–88. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bru C, Courcelle E, Carrere S, Beausse Y, Dalmar S, Kahn D. The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 2005;33:D212–15. doi: 10.1093/nar/gki034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deutsch EW, Lam H, Aebersold R. PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO Rep. 2008;9:429–34. doi: 10.1038/embor.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hummel J, Niemann M, Wienkoop S, Schulze W, Steinhauser D, Selbig J, Walther D, Weckwerth W. ProMEX: a mass spectral reference database for proteins and protein phosphorylation sites. BMC Bioinformatics. 2007;8:216. doi: 10.1186/1471-2105-8-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang C, Jaiswal P, Hebbard C, Avraham S, Buckler ES, Casstevens T, Hurwitz B, McCouch S, Ni J, Pujar A, Ravenscroft D, Ren L, Spooner W, Tecle I, Thomason J, Tung CW, Wei X, Yap I, Youens-Clark K, Ware D, Stein L. Gramene: a growing plant comparative genomics resource. Nucleic Acids Res. 2008;36:D947–D953. doi: 10.1093/nar/gkm968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lawrence CJ, Schaeffer ML, Seigfried TE, Campbell DA, Harper LC. MaizeGDB’s new data types, resources and activities. Nucleic Acids Res. 2007;35:D895–D900. doi: 10.1093/nar/gkl1048. [DOI] [PMC free article] [PubMed] [Google Scholar]