

Abstract
Reaction of the normal isomer of [B20H18]2− and the protected thiol anion, [SC(O)OC(CH3)3]−, produces an unexpected isomer of [B20H17SC(O)OC(CH3)3]4− directly and in good yield. The isomer produced under mild conditions is characterized by an apical–apical boron atom intercage connection as well as the location of the thiol substituent on an equatorial belt adjacent to the terminal boron apex. Although the formation of this isomer from nucleophilic attack of the normal isomer of [B20H18]2− has not been reported previously, the isomeric assignment has been unambiguously confirmed by one-dimensional and two-dimensional 11B NMR spectroscopy. Deprotection of the thiol substituent under acidic conditions produces a protonated intermediate, [B20H18SH]3−, which can be deprotonated with a suitable base to yield the desired product, [B20H17SH]4−. The sodium salt of the resulting [B20H17SH]4− ion has been encapsulated in small, unilamellar liposomes, which are capable of delivering their contents selectively to tumors in vivo, and investigated as a potential agent for boron neutron capture therapy. The biodistribution of boron was determined after intravenous injection of the liposomal suspension into BALB/c mice bearing EMT6 mammary adenocarcinoma. At low injected doses, the tumor boron concentration increased throughout the time-course experiment, resulting in a maximum observed boron concentration of 46.7 μg of B per g of tumor at 48 h and a tumor to blood boron ratio of 7.7. The boron concentration obtained in the tumor corresponds to 22.2% injected dose (i.d.) per g of tissue, a value analogous to the most promising polyhedral borane anions investigated for liposomal delivery and subsequent application in boron neutron capture therapy.
Boron neutron capture therapy (BNCT), a binary cancer therapy first proposed by Locher in 1936 (1), is based on the propensity of the boron-10 isotope to capture thermal neutrons. The neutron capture reaction yields an unstable boron-11 atom, which then undergoes fission to produce highly energetic lithium-7 and helium-4 species. The two fission products have an effective range of ≈10 μm in tissue, which essentially limits the fission event to a single cell or its immediate neighbors. Therefore, the selective concentration of boron-10 nuclei within tumor cells, followed by thermal neutron capture, should result in the localized destruction of malignant cells even in the presence of normal neighboring cells.
The successful application of BNCT depends on the identification and production of boron-containing compounds, which are accumulated in significant amounts (>15 μg of B per g of tumor; ref. 2) by the tumor through natural mechanisms, or, alternatively, on the identification of a tumor-specific delivery modality, which would enable the delivery of boron-containing compounds that have no inherent tumor specificity. Although the utility of tumor-targeted monoclonal antibody conjugates has been investigated, limited success has been achieved because of the highly competitive loss of conjugate to the liver once sufficient amounts of boron have been conjugated to the antibody (3).
The ideal delivery modality should be able to incorporate large quantities of boron without affecting the selective delivery of the boron to the tumor. Small unilamellar liposomes encapsulating concentrated aqueous solutions of polyhedral borane salts and injected intravenously have been shown to deliver therapeutic quantities of boron selectively to tumors in vivo (4, 5). Although essentially any water-soluble boron-containing compound can be encapsulated in the aqueous core of the liposomes and delivered to the tumor, only polyhedral borane salts that possess the potential to form covalent bonds with intracellular protein moieties have been retained in significant quantity by the tumor. Compounds that lack this chemical reactivity were cleared from all tissues, including tumor (4, 5). The necessity of liposomal incorporation for tumor accretion has been substantiated by the clearance of unincorporated (“free”) boron-containing compounds from all tissues, including tumor, in murine biodistribution experiments (4, 5).
The polyhedral borane anions investigated for liposomal encapsulation have been based on the normal isomer of [B20H18]2−. Initial interest in this compound was derived from the large boron content per unit charge and the known susceptibility of the anion to nucleophilic attack. The anion is also synthesized in two high yield reactions, amenable to boron-10 enrichment, from decaborane, B10H14 (6, 7).
The substitution chemistry of the normal isomer of [B20H18]2− was first investigated by Hawthorne and coworkers (8, 9). Nucleophilic attack on the electron-deficient three-center two-electron bonds of [B20H18]2− by hydroxide ion produced an apical–equatorial isomer of [B20H17OH]4−, designated [ae-B20H17OH]4−, with the hydroxide substituent located on the equatorial belt adjacent to the intercage linkage, as shown in Scheme 1, 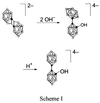 where ○ = BH and ● = B. Acid-catalyzed rearrangement of the apical–equatorial isomer yielded the thermodynamically stable apical–apical (a2) isomer. Analogous reactions also have been reported for the synthesis of alkoxy and amine derivatives (5, 9, 10).
where ○ = BH and ● = B. Acid-catalyzed rearrangement of the apical–equatorial isomer yielded the thermodynamically stable apical–apical (a2) isomer. Analogous reactions also have been reported for the synthesis of alkoxy and amine derivatives (5, 9, 10).
In addition, the normal isomer of [B20H18]2− can be photoisomerized to a different isomer, designated [i-B20H18]2−, which is also susceptible to nucleophilic attack (4). Nucleophilic attack on the photoisomer by the hydroxide ion produced an equatorial–equatorial isomer of [B20H17OH]4−, designated [e2-B20H17OH]4−, as shown in Scheme 2 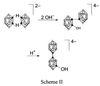 , where ○ = BH and ● = B. Acid-catalyzed rearrangement of the equatorial–equatorial isomer yielded the thermodynamically stable apical–apical isomer with the hydroxide substituent located on the equatorial belt adjacent to the terminal boron apex.
, where ○ = BH and ● = B. Acid-catalyzed rearrangement of the equatorial–equatorial isomer yielded the thermodynamically stable apical–apical isomer with the hydroxide substituent located on the equatorial belt adjacent to the terminal boron apex.
The utility of this class of compounds for BNCT has been investigated (4, 5). Tumor-selective unilamellar liposomes, capable of delivering their contents intracellularly (11), were used as delivery vehicles for the compounds, which have little inherent tumor specificity. Although the liposomes successfully delivered the species to the tumor, the utility of the compounds for BNCT depends on the retention of the drug by the tumor. In biodistribution experiments, compounds unsuitable for application in BNCT were cleared from the tumor, as they were cleared from blood and normal tissue, resulting in negligible tumor to normal tissue concentration differentials. Previous research has shown that retention of the boron-containing species depends on the substituents present in the compound as well as the reactivity of the compound (4, 5). The tumor retention of [B12H11SH]2− (BSH), a compound currently used in Japan and in clinical trials in Europe for BNCT, is enhanced compared with the unsubstituted [B12H12]2− anion because of the presence of the thiol substituent (4). Compounds known to be susceptible to nucleophilic attack, such as the isomers of the [B20H18]2− ion, are better retained by tumors (4). Additionally, compounds that have the potential to be converted into a more reactive species in vivo, such as the isomers of Na3[B20H17NH3], accreted in and were retained by tumors in small-animal experiments (5). The combination of these attributes that are known to enhance tumor retention into a single compound should result in substantially increased retention. Therefore, synthesis of the thiol derivative of [B20H18]4−, [B20H17SH]4−, would provide a compound that possesses both the reactive thiol substituent and the potential to be oxidized into the more reactive [B20H17SH]2− ion. The thiol derivative would have two possible mechanisms for tumor retention (as shown in Scheme 3 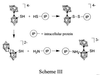 where ○ = BH and ● = B): (i) direct reaction of the thiol substituent with intracellular protein moieties and (ii) oxidation to the more reactive [B20H17SH]2− ion, which should be susceptible to nucleophilic attack by intracellular protein substituents. To test this hypothesis, the sodium salt of [B20H17SH]4− was synthesized and encapsulated in unilamellar liposomes, and its fate was investigated in murine biodistribution experiments.
where ○ = BH and ● = B): (i) direct reaction of the thiol substituent with intracellular protein moieties and (ii) oxidation to the more reactive [B20H17SH]2− ion, which should be susceptible to nucleophilic attack by intracellular protein substituents. To test this hypothesis, the sodium salt of [B20H17SH]4− was synthesized and encapsulated in unilamellar liposomes, and its fate was investigated in murine biodistribution experiments.
MATERIALS AND METHODS
Materials.
Synthetic reactions were performed under an argon atmosphere by using Schlenk techniques where necessary. The polyhedral borane starting materials were prepared by published methods (6, 7). The Bender’s salt, K[SC(O)OC(CH3)3], was prepared by using the methods reported by Daly and Lee (12). Tetrahydrofuran was distilled from sodium metal before use. Distearoylphosphatidylcholine was obtained from Avanti Polar Lipids, and the cholesterol was supplied by Sigma.
Physical Measurements.
The 1H and 11B Fourier transform (FT) NMR spectra were obtained with a Varian INOVA instrument operating at 400 MHz and 128 MHz, respectively. Proton chemical shifts were referenced to residual solvent protons. Boron chemical shifts were externally referenced to BF3⋅Et2O in C62H6; peaks up-field of the reference are designated as negative. The FT IR spectra were obtained as Nujol mulls by using a Perkin–Elmer 1300 instrument.
K4[1-(1′-B10H9)-6-SC(O)O(CH3)3-B10H8], Designated K4[a2-B20H17SC(O)O(CH3)3].
The Bender’s salt, K[SC(O)O(CH3)3] (8.23 g; 47.8 mmol), [Et3NH]2[B20H18] (4.12 g; 9.6 mmol), and ≈500 ml of freshly distilled tetrahydrofuran were transferred to a reaction vessel and refluxed overnight. The product, K4[a2-B20H17SC(O)O(CH3)3], is insoluble in tetrahydrofuran. Therefore, the reaction was monitored by the disappearance of the B–H absorption in the FT IR spectrum of reaction aliquots, which were reduced in volume. After reaction completion, the product was filtered under an argon atmosphere, dried, and recrystallized from water/ethanol to yield a white solid in 68% yield. {1H}11B NMR (ppm; H2O; multiplicity in parenthesis): 11.6 (s), 7.9 (s), −0.5 (s), −5.2 (d), −8.9 (d), −22.3 (d), −23.5 (d), −28.2 (d), −33.8 (d). 1H NMR (ppm, 2H2O): 1.3 [s, —C(CH3)3]. FT IR (cm−1, Nujol): 1590 (C⩵O), 2457 (B-H).
K4[1-(1′-B10H9)-6-SH-B10H8], Designated K4[a2-B20H17SH].
The protected starting material, K4[a2-B20H17SC(O)O(CH3)3] (0.50 g, 1.0 mmol), was transferred to a vessel and dissolved in a minimum amount of water (≈1 ml). Aqueous HCl (3 ml; 0.09 M) was added to the solution, and the mixture was stirred at room temperature for ≈45 minutes. The pH of the solution was increased to ≈11 by the addition of aqueous potassium hydroxide. Isolation of the product in 71% yield was achieved by the addition of absolute ethanol. The 11B NMR of the product is the same as that obtained for K4[a2-B20H17SC(O)O(CH3)3]. The product showed no carbonyl absorption in the IR spectrum and no t-butyl peak in the 1H NMR spectrum. The compound was ion-exchanged to the sodium form in aqueous solution by using Bio-Rad AG50W-X8, 50- to 100-mesh cation exchange resin.
Vesicle Preparation.
The liposome suspension was prepared by probe sonication of a 300-mg dried film composed of equimolar amounts of distearoylphosphatidylcholine and cholesterol with 6.0 ml of the hydrating solution (160 mM in Na4B20H17SH) at 65°C for 20 min. Vesicles were separated from the remaining free polyhedral borane salt by elution through a column of Sephadex G-25 (medium) with isotonic Hepes-buffered saline. The liposomal formulation was diluted with Hepes-buffered saline to a lipid concentration of 23–24 mg/ml and sterilized by filtration through a 0.22-μm Millipore membrane. The integrity of the encapsulated borane salt was confirmed by 11B NMR at 128 MHz. The volume-weighted mean vesicle diameter of the liposomes, obtained by dynamic light scattering, was 40 nm. The unencapsulated borane salt was recovered from the column by aqueous elution and isolated as the potassium salt by precipitation with a saturated solution of potassium acetate in absolute ethanol.
Murine Studies.
Murine biodistribution studies used female BALB/c mice (16–20 g), with EMT6 tumors implanted in the right flank 7–10 days before the experiment. Tumor mass was 125–350 mg at the time the mice were killed. Injections of the liposome suspension (200 μl; 210 mg of boron) were made in the tail vein. Each data point represents the average value obtained from seven mice. Details of the murine experiments have been reported (4). Boron analyses of tissues and of the liposome suspension were performed by inductively coupled plasma-atomic emission spectroscopy (13) at the Idaho National Engineering and Environmental Laboratory (Idaho Falls, ID).
RESULTS
The reaction of the protected thiol anion, [SC(O)OC(CH3)3]− with [B20H18]2− in tetrahydrofuran at room temperature produces an unexpected apical–apical isomer of [1-(1′n-B10H9)-6-SC(O)OC(CH3)3B10H8]4−, designated [a2-B20H17SC(O)OC(CH3)3]4−, as shown in Scheme 4 ![]() , where ○ = BH and ● = B. The 1H and 11B NMR spectra of this species are consistent with this structural assignment. In the 1H NMR spectrum, one signal, at 1.3 ppm, corresponding to the protons on the three methyl groups of the tertiary butyl substituent, is present. In the 11B NMR (note axis in Fig. 1), two terminal apical boron signals, at −5.2 and −8.9 ppm, appear as singlets in the proton-decoupled spectrum and as doublets in the proton-coupled spectrum. Two broad singlets, at 11.6 and 7.9 ppm, are assigned to the two boron atoms involved in the intercage connection. The remaining broad singlet at −0.5 ppm is assigned to the boron atom attached to the sulfur atom of the substituent. The remaining up-field signals are assigned to the remaining equatorial boron atoms. The two-dimensional 11B NMR spectrum (Fig. 1) shows a cross-coupling peak between the two boron atoms of the intercage connection (11.6 and 7.9 ppm), as well as a cross-coupling peak between the signal corresponding to the boron atom bearing the sulfur substituent (−0.5 ppm) and one of the signals corresponding to an apical boron atom (−8.9 ppm).
, where ○ = BH and ● = B. The 1H and 11B NMR spectra of this species are consistent with this structural assignment. In the 1H NMR spectrum, one signal, at 1.3 ppm, corresponding to the protons on the three methyl groups of the tertiary butyl substituent, is present. In the 11B NMR (note axis in Fig. 1), two terminal apical boron signals, at −5.2 and −8.9 ppm, appear as singlets in the proton-decoupled spectrum and as doublets in the proton-coupled spectrum. Two broad singlets, at 11.6 and 7.9 ppm, are assigned to the two boron atoms involved in the intercage connection. The remaining broad singlet at −0.5 ppm is assigned to the boron atom attached to the sulfur atom of the substituent. The remaining up-field signals are assigned to the remaining equatorial boron atoms. The two-dimensional 11B NMR spectrum (Fig. 1) shows a cross-coupling peak between the two boron atoms of the intercage connection (11.6 and 7.9 ppm), as well as a cross-coupling peak between the signal corresponding to the boron atom bearing the sulfur substituent (−0.5 ppm) and one of the signals corresponding to an apical boron atom (−8.9 ppm).
Figure 1.

Two-dimensional proton-decoupled 11B NMR spectrum in 2H2O.
Aqueous acidification of K4[a2-B20H17SC(O)OC(CH3)3] results in the elimination of carbon dioxide and isobutylene (12) to yield the unprotected, bridge-protonated K3[B20H18SH] as shown in Scheme 5, 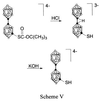 where ○ = BH and ● = B. Deprotection of the sulfur moiety was indicated by the loss of the infrared carbonyl absorption at 1,590 cm−1 and the disappearance of the t-butyl substituent peak in the 1H NMR (1.3 ppm). The bridge proton is removed by the addition of an aqueous solution of potassium hydroxide. The pKa of this acidic bridge proton of K4[a2-B20H18SH] has been estimated to be 6.8 by using the half-equivalence point of the titration.
where ○ = BH and ● = B. Deprotection of the sulfur moiety was indicated by the loss of the infrared carbonyl absorption at 1,590 cm−1 and the disappearance of the t-butyl substituent peak in the 1H NMR (1.3 ppm). The bridge proton is removed by the addition of an aqueous solution of potassium hydroxide. The pKa of this acidic bridge proton of K4[a2-B20H18SH] has been estimated to be 6.8 by using the half-equivalence point of the titration.
The biodistribution of liposomes loaded with Na4[a2-B20H17SH] is shown in Fig. 2 (i.d. = 210 μg of B; ≈10.5 mg of B per kg of body weight). The following six tissues were analyzed: tumor, skin, liver, kidney, brain, and blood. The initial measured tumor boron concentration (23.7 μg of B per g of tumor) represented 11.3% of the i.d. per g of tissue. Over a period of 48 h, the tumor boron concentration increased continuously to the observed maximum (46.7 μg of B per g of tumor) at 48 h, which was 22.2% of the i.d. per g of tissue. The final measured tumor to blood boron ratio was 7.7. At the 48-h time point, all tissues examined had lower boron concentrations than the tumor. Kidney, liver, and blood boron concentrations were initially high relative to other tissues but generally decreased over the 48-h time period, resulting in final measured boron concentrations of 17.8 μg of B per g of tissue, 36.4 μg of B per g of tissue, and 6.0 μg of B per g of tissue, respectively. Brain and skin boron concentrations were relatively low, with maximum measured boron concentrations of 1.3 μg of B per g of tissue at 6 h and 9.3 μg of B per g of tissue at 30 h, respectively.
Figure 2.

Murine tissue boron concentrations from the liposomal delivery of Na4[a2-B20H17SH] [210 mg of B (10.5 mg of B per kg of body weight)]: ▫, tumor; ▪, skin; ▿, liver; ▾, kidney; ○, brain; and ●, blood. For clarity, error bars are not shown in the graphical data; SDs were typically <10% of the average values.
DISCUSSION
Nucleophilic attack of the protected thiol anion, [SC(O)OC(CH3)3]−, on the electron-deficient bonding region in the normal isomer of [B20H18]2− forms, directly and in good yield, an unexpected isomer of [B20H17SC(O)OC(CH3)3]4− under mild conditions. The isomer produced in the reaction is characterized by an apical–apical boron atom intercage connection as well as the thiol substituent location on the equatorial belt adjacent to the terminal boron apex. Although this isomeric assignment has been reported as a product of the nucleophilic attack of hydroxide on the photoisomer of [B20H18]2−, to our knowledge, no reports of the isomer being formed by the nucleophilic attack on the normal isomer of [B20H18]2− have been made.
Although a single crystal x-ray diffraction study of the product has not been possible because of the high disorder present in the obtained crystals, the one-dimensional and two-dimensional 11B NMR spectra of the compound unambiguously confirm the isomeric assignment. The apical–apical assignment is validated by the presence of two apical boron signals, which are singlets in the proton-decoupled 11B NMR spectrum and doublets in the proton-coupled 11B NMR spectrum. An apical–equatorial isomer would have three apical boron signals with the same splitting properties. The two-dimensional 11B NMR spectrum establishes the location of the sulfur substituent on the equatorial belt adjacent to the terminal boron apex as well as the presence of the boron–boron intercage connection. The existence of cross-coupling peaks between one of the terminal apical boron signals and the substituted boron atom, as well as the cross-coupling peaks between the two boron atoms in the intercage connection, is unequivocal. Additionally, comparison of the obtained 11B NMR spectrum with the 11B NMR spectrum of the known apical–apical hydroxy derivative that results from the nucleophilic attack of hydroxide on the photoisomer of [B20H18]2− (4, 8, 9) provides further support of the current isomeric assignment. Although the location of the thiol substituent in the thiol product obtained from the normal isomer of [B20H18]2− would require cage rearrangement, as determined by the study on the mechanisms reported in ref. 9, to our knowledge, no synthetic investigations have been performed to date with such a sterically demanding nucleophile. To investigate the potential steric effects that may control the reaction, direct reaction of [B20H18]2− with Na[SH] was attempted; however, complicated reaction mixtures resulted regardless of reaction solvent and experimental conditions, possibly because of the potential oxidative reactivity of the thiol anion. A variety of protected thiol anions can be readily synthesized (12) to investigate the potential steric factors present in this reaction sequence (D.A.F., unpublished results).
Acidification of [a2-B20H17SC(O)OC(CH3)3]4− results in the deprotection of the thiol substituent by the elimination of carbon dioxide and isobutylene (12) and the formation of a bridge-protonated species, [B20H18SH]3−. Removal of the bridge proton by using aqueous potassium hydroxide results in the formation of [a2-B20H17SH]4−. The pKa of the bridge proton (≈6.8) is consistent with analogous polyhedral borane anions, such as [B20H19]3− (8). The acid–base process can be monitored by using 11B NMR spectroscopy. The 11B NMR spectrum of the unprotected thiol product is not altered by the loss of the protecting group. The removal of the protecting group is confirmed by the absence of the carbonyl absorption in the FT IR spectrum and the absence of the tertiary butyl signal in the 1H NMR spectrum. Although BSH is known to undergo facile dimerization through disulfide bond formation (14) to produce [B12H11S-SB12H11]4−, [a2-B20H17SH]4− seems to be immune to an analogous oxidation process in air.
The murine biodistribution of Na4[a2-B20H17SH], encapsulated in unilamellar liposomes, has an initial measured tumor boron concentration that is approximately twice that observed with BSH (4), as would be anticipated by changing from a 12-boron atom to a 20-boron atom species. The value is comparable to values observed for the [B20H17NH3]3− series investigated in previous experiments (5). However, in the case of [B20H17SH]4−, the boron concentration in the tumor increases over the entire time-course experiment, and even higher tumor boron concentrations would be anticipated if the time period was extended, a property not observed in the analogous biodistributions of BSH or the [B20H17NH3]3− series. Rapid clearance of the boron concentration in the tumor was observed in the biodistribution of liposomally encapsulated BSH (4), indicating a lack of binding ability by the polyhedral borane dianion. In the case of the [B20H17NH3]3− series (5), tumor accretion was observed over a ≈30-h time period and was followed by slow clearance from the tumor, possibly indicating a decrease in the binding ability of these species over time. The thiol product, [B20H17SH]4−, combines the favorable properties of both BSH and the [B20H17NH3]3− series, resulting in an accretion of tumor boron concentration over the entire time-course experiment. The final measured tumor boron concentration is comparable to the highest percentage of the i.d. per gram of tissue obtained by liposomal delivery to date and approaches results anticipated from antibody delivery. Therapeutic levels of boron (>15 μg of B per g of tumor) are present in the tumor at all points in the time-course experiment. The extended lifetime of the compound in the tumor enables the clearance of the boron from all tissues, resulting in a therapeutically favorable final tumor to blood boron ratio of 7.7 and tissue boron concentrations that are all lower than the tumor boron concentration. Based on previous results, the necessity of liposomal delivery has been established (4, 5).
CONCLUSIONS
The encapsulation of water-soluble polyhedral borane anions in unilamellar liposomes has proven to be the most generally attractive delivery modality for application in BNCT. Previous research has provided a basis to design boron-containing compounds that can be incorporated into the liposomes and that should be retained to a high degree in tumors. Based on the enhanced retention provided by the thiol substituent in BSH and the oxidative propensity of the isomers of Na3[B20H17NH3], the proposal of the synthesis of the thiol derivative of [B20H18]4−, [B20H17SH]4−, was made. Reaction of the nucleophilic Bender’s salt, K[SC(O)OC(CH3)3], and the normal isomer of [B20H18]2− yielded an unexpected isomer of the desired product, K4[B20H17SC(O)OC(CH3)3]. Although this isomer has not been obtained by the nucleophilic attack of small alkoxide or amine nucleophiles on the normal isomer of [B20H18]2−, spectroscopic evidence has unambiguously established the isomeric assignment. Deprotection of the thiol substituent under acidic conditions, followed by aqueous neutralization, yields the desired K4[a2-B20H17SH] in moderate yield.
The sodium salt of the thiol derivative, [a2-B20H17SH]4−, encapsulated in unilamellar liposomes, represents one of the most promising results obtained for water-soluble polyhedral boranes to date. The biodistribution of the liposomal formulation is characterized by tumor boron concentrations that increase throughout the time-course experiment and continuously exceed therapeutically desired values. The retention of the thiol derivative by the tumor enables the boron concentration present in the other tissues to decrease sufficiently, resulting in final measured boron concentrations that are lower than the tumor boron concentration.
Acknowledgments
The authors wish to thank Dr. Patrick Gavin (Washington State University, Pullman, WA) for assistance with the murine biodistribution studies, Dr. William Bauer (Idaho National Engineering and Environmental Laboratory, Idaho Falls, ID) for the total boron analysis, and Dr. Kenneth Shelly (University of California, Los Angeles) for the dynamic light scattering measurements. This research was supported by the Robert A. Welch Foundation Grant AI1292, Research Corporation Grant CC3913, and a grant from Southwest Texas State University.
ABBREVIATIONS
- BNCT
boron neutron capture therapy
- BSH
[B12H11SH]2−
- i.d.
injected dose
- FT
Fourier transform
- [i-B20H18]2−
photoisomer of [B20H18]2−
- [ae-B20H17OH]4−
[1-(2′-B10H9)-2-OHB10H8]4−
- [e2-B20H17OH]4−
[2-(2′-B10H9)-6-OHB10H8]4−
- [a2-B20H17SC(O)OC(CH3)3]4−
[1-(1′-B10H9)-6-SC(O)OC(CH3)3B10H8]4−
- [a2-B20H17SH]4−
[1-(1′-B10H9)-6-SHB10H8]4−
References
- 1.Locher G L. Am J Roentgenol Radium Ther Nucl Med. 1936;36:1–13. [Google Scholar]
- 2.Fairchild R G, Bond V P. J Radiat Oncol Biol Phys. 1985;11:831–840. doi: 10.1016/0360-3016(85)90318-9. [DOI] [PubMed] [Google Scholar]
- 3.Hawthorne M F. Pure Appl Chem. 1991;63:327–334. [Google Scholar]
- 4.Shelly K, Feakes D A, Hawthorne M F, Schmidt P G, Krisch T A, Bauer W F. Proc Natl Acad Sci USA. 1992;89:9039–9043. doi: 10.1073/pnas.89.19.9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feakes D A, Shelly K, Knobler C B, Hawthorne M F. Proc Natl Acad Sci USA. 1994;91:3029–3033. doi: 10.1073/pnas.91.8.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawthorne M F, Pilling R L. Inorg Synth. 1967;9:16–19. [Google Scholar]
- 7.Chamberland B L, Muetterties E L. Inorg Chem. 1964;3:1450–1456. [Google Scholar]
- 8.Hawthorne M F, Pilling R L, Stokely P F, Garrett P M. J Am Chem Soc. 1963;85:3704. [Google Scholar]
- 9.Hawthorne M F, Pilling R L, Garrett P M. J Am Chem Soc. 1965;87:4740–4746. doi: 10.1021/ja00949a014. [DOI] [PubMed] [Google Scholar]
- 10.Georgiev E M, Shelly K, Feakes D A, Kuniyoshi J, Romano S, Hawthorne M F. Inorg Chem. 1996;35:5412–5416. doi: 10.1021/ic960171y. [DOI] [PubMed] [Google Scholar]
- 11.Straubinger R M, Papahadjopoulos D, Hong K. Biochemistry. 1990;29:4929–4939. doi: 10.1021/bi00472a025. [DOI] [PubMed] [Google Scholar]
- 12.Daly W H, Lee C S. Polym Pepr, Am Chem Soc Div Polym Chem. 1973;14:1238–1243. [Google Scholar]
- 13.Tamat S R, Moore D E, Allen B J. Anal Chem. 1987;59:2161–2164. doi: 10.1021/ac00144a033. [DOI] [PubMed] [Google Scholar]
- 14.Wellum G R, Tolpin E I, Soloway A H, Kaczmarczyk A. Inorg Chem. 1977;16:2120–2122. [Google Scholar]