

Abstract
There was remarkable progress in the understanding of the role genetic risk factors in chronic pancreatitis. These factors seem to be much more important than thought in the past. The rare autosomal-dominant mutations N29I and R122H of PRSS1 (cationic trypsinogen) as well as the variant N34S of SPINK1 (pancreatic secretory trypsin inhibitor) are associated to a disease onset in childhood or youth. Compared to chronic alcoholic pancreatitis the progression is slow so that for a long time only signs of acute-recurrent pancreatitis are found. Only at later time points (more than 10-15 years) there is evidence for chronic pancreatitis in the majority of patients. Acute recurrent pancreatitis may therefore be regarded as a transition state until definite signs of chronic pancreatitis are detectable.
Keywords: Genetic disorders, Acute recurrent pancreatitis
INTRODUCTION: FROM ACUTE TO ACUTE-RECURRENT AND TO CHRONIC PANCREATITIS
Pancreatitis is general is divided into acute and chronic forms. Acute pancreatitis is defined as a disease in which both exocrine and endocrine function of the gland may recover to normal (restitution ad integrum). In some patients with severe acute pancreatitis, a residual functional defect may persistent but there is no recurrent episode (Figure 1). Whereas acute pancreatitis may be related to either bile duct stones or alcohol, the main cause of chronic pancreatitis is heavy alcohol consumption[1]. Patients presenting with an acute flare of alcoholic pancreatitis often show evidence for chronic alteration of the duct, therefore already having definite signs of chronic pancreatitis. The pathogenesis of alcoholic chronic pancreatitis is under debate. Whereas one group of investigators believe that acute alcoholic pancreatitis proceeds to chronic pancreatitis, others think that this is primarily a chronic disease. It is furthermore under discussion whether alcoholic pancreatitis, when initiated, may, even at complete cessation of alcohol consumption, ever recover to normal or not.
Figure 1.
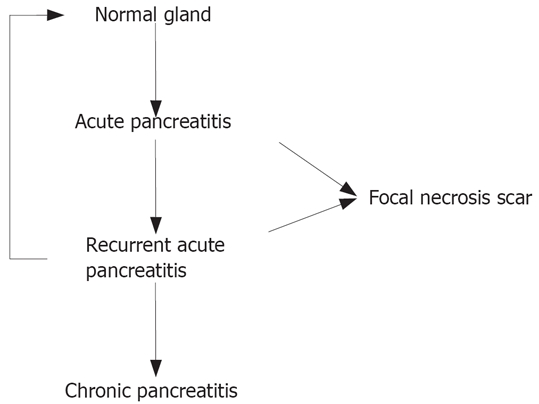
Relationship between acute, acute-recurrent and chronic pancreatitis.
Acute-recurrent pancreatitis may be regarded as a transition state between acute and chronic pancreatitis that is characterized by repetitive acute episodes without evidence for chronic inflammation (Figure 1). This patient group is difficult to define as we only have a description of the disease within a limited time interval. It is unclear at the moment of the investigation whether the pancreatitis resumes completely or progresses to chronic disease. Only the investigation of the long-term course of the disease will show us to what group the respective patient belongs to.
In the recent years several genetic risk factors were identified in pancreatitis[2]. In particular, mutations of the cationic trypsinogen (PRSS1), secretory pancreatic trypsin inhibitor 1 (SPINK1), and the cystic fibrosis transmembrane conductance regulator (CFTR) were described. The mutation G191R of the anionic trypsinogen (PRSS2) was shown to act as a factor protecting from pancreatitis[3]. In patients with autosomal- dominant traits the mutations N29I and R122H of PRSS1 as well as a triplication of the trypsinogen was found[4]. The other mutations were mainly found in sporadic, but also in alcohol-induced disease[5]. In this short review I will discuss the classification of hereditary or idiopathic pancreatitis, but mainly focus on the description of the course of disease in patients with genetic risk factor in order to rule out whether an acute-recurrent form exists at all. These data are mainly based on an investigation of patients and their families[6,7].
HEREDITARY OR IDIOPATHIC PANCREATITIS
The term “hereditary pancreatitis” was created in the fifties of the last century and described an accumulation of patients with pancreatitis in one family[8]. Its use is not uniform as several different definitions can be found in the literature. Most authors request 2 patients within one generation or more than 2 patients in more than one generation. Due to its origin in the pre-genetic area the term is purely phenomenological. After identification of mutations of the cationic trypsinogen (PRSS1), of the pancreatic secretory trypsin inhibitor (SPINK1) and of the cystic fibrosis transmembrane conductance regulator (CFTR) in patients with pancreatitis[2] it became evident that only less than 5% of the patients with genetic risk factors fulfil any of these definitions of “hereditary pancreatitis”. In our group of patients, for example, 35% of families with autosomal-dominant trypsinogen mutations (N29I and R122H) cannot be classified as suffering from “hereditary” pancreatitis. In consequence, the definition of “hereditary pancreatitis” will have to be changed.
Another problem is the use of “diopathic” pancreatitis in patients with genetic risk factor. Recently, even the term “amilial-idiopathic pancreatitis” s created for those families having 2 members with pancreatitis[9]. Both concepts are contradictions in term as “diopathic”means absence of any risk factor and familial accumulation or mutations always point to a risk factor. It is my suggestion to substitute the term “diopathic” by “poradic” that is also used in other diseases. “Autosomal-dominant” and “familial pancreatitis” (in case of absence of an autosomal-dominant trait) could be used to formally describe the familial association of the disease.
HOW FREQUENT ARE MUTATION IN PANCREATITIS?
After the first description of trypsinogen mutations several reports were published which showed up to 10% of autosomal-dominant mutations N29I or R122H in patients with “idiopathic” pancreatitis[10,11]. This is typical for an initial enthusiasm that overestimates the significance of a new finding. Later, the investigation of larger groups of patients revealed that trypsinogen mutations were present much less frequently. Interestingly, the smaller the study was, the larger were the percentages of mutations and the higher were the impact factors of the journals in which the data were published. As noted already[12] this was nice example for a publication bias favouring “positive” findings that are preferably published in journals with high impact factor.
Table 1 shows an overview on the frequency of the various mutations in the different groups of patients. The data largely based on our own genetic findings in more than 1000 patients with non-alcoholic chronic pancreatitis[5,12] and on several other studies[13–17]. The different groups are defined by family history (autosomal-dominant, familiar, sporadic) and the alcohol consumption. In approximately 75% of the patients with autosomal-dominant disease mutations of the cationic trypsinogen (N29I or R122H) were found. Recently a triplication of the trypsinogen was identified[4], but its frequency is unclear at the moment. A positive family history without clear autosomal-dominant trait (familial pancreatitis) predisposes to the SPINK1 mutation N34S, but also PRSS1 variants (A16V, N29I, R122H) can be seen. In patients with sporadic disease all three genes (PRSS1, SPINK1, CFTR) contribute to the risk of the disease, but, as already mentioned above, autosomal-dominant mutations are extremely rare.
Table 1.
Mutations in different groups of patients suffering from pancreatitis
| Clinical characteristic of chronic pancreatitis | % of all patients |
With mutation |
Without mutation | |
| Type of mutation | % | % | ||
| Autosomal-dominant | 1 | PRSS1:R122H, N29I | 70-80 | 10-20 |
| PRSS1:triplication | 10-20 | |||
| Familial | 3 | PRSS1:R122H, N29I | 15 | 50 |
| PRSS1:A16V | < 5 | |||
| SPINK1:N34S | 30 | |||
| Other SPINK1 mutations | 5 | |||
| Sporadic | 20 | PRSS1:R122H, N29I | << 1 | 15-50 |
| PRSS1:A16V | < 5 | |||
| SPINK1:N34S | 20-40 | |||
| CFTR:various | 20-40 | |||
| Alcoholic | 75-80 | SPINK1:N34S | 5 | 85 |
| CFTR:various | 10 | |||
| Pancreas divisum | 3 | CFTR:various | 20 | 80 |
| Tropical pancreatitis | ? | SPINK1:N34S | 50 | 50 |
| All groups: protective mutation | 1 | PRSS1:G191R | In controls: 3 | |
In alcoholic chronic pancreatitis 15% of the patient exhibit SPINK1 or CFTR mutations[13,14]. In 22% of patients with pancreas divisium, mutations of the CFTR molecule can be identified[16]. In tropical pancreatitis half of the patients were affected from the variant N34S of SPINK1[17]. Recently Witt et al[3] reported the identification of a PRSS1 mutation (G191R) protecting from pancreatitis. This supports the central role of trypsin in the pathogenesis of pancreatitis. Taken together, in approximately 25% of the patients with chronic pancreatitis a genetic risk factor can be identified. A regularly updated list of PRSS1 and SPINK1 mutations can be found in the internet (http://www.uni-leipzig.de/pancreasmutation).
MUTATIONS AND RISK OF PANCREATITIS
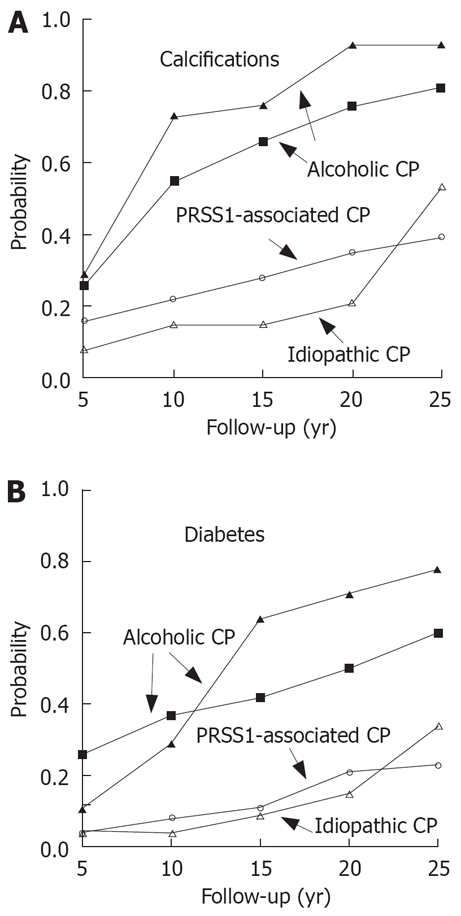
The different mutations in pancreatitis confer a different risk of pancreatitis. This may be calculated from the frequencies of mutations in the healthy general publication and in patients. In trypsinogen mutations N29I and R122H approximately 80% of the subjects suffer from disease[7,16]. Apart from families with autosomal-dominant disease the mutations are obviously absent in healthy subjects. At a prevalence of chronic pancreatitis of 50-100/100.000 it may be estimated that PRSS1 mutation increase the risk for pancreatitis by a factor of at least 1000-2000. The homozygous SPINK1 N34S is detected in healthy subjects at a rate of 1/10.000, and in patients with pancreatitis it is found in approximately 5%-10% of those with N34S-associated disease[19]. Therefore it may be assumed that the homozygous N34S increased the risk by a factor of about 500. The heterozygous N34S of SPINK1 as well as CFTR mutations are associated with a 20-40-fold risk of pancreatitis[18]. This is in difference to the role of alcohol that increased the risk of disease only by a factor of 3, 5-11[20]. A conclusion from these data would be that genetic risk factors confer a much higher risk than the consumption of alcohol. It may also be concluded that alcohol is a very frequent but weak risk factor of chronic pancreatitis.
Figure 2 shows an attempt for a graphical representation of these data. Autosomal-dominant trypsinogen mutations or homozygous N34S may be regarded as strong risk factors (× 100-1000). Variants of SPINK1 or CFTR confer an intermediate risk (× 10-100). Weak factors (increase up to 10 fold) like alcohol consumption are supplementary and may modify the course of chronic pancreatitis. The graph shows that interaction of environmental and genetic factors (i.e. N34S + alcohol or PRSS1 + smoking) further increased the probability of a disease. It is also shown that SPINK1 mutations are stronger risk factors than CFTR variants and in their homozygous form they were similar to the autosomal-dominant trypsinogen mutations. Therefore, it is no longer justified to degrade them to “disease modifier” as done recently[21].
Figure 2.
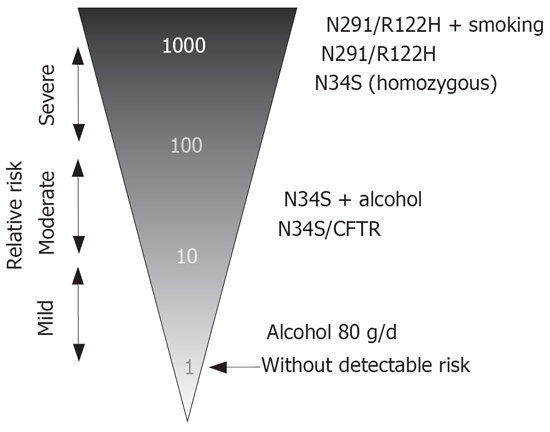
Strength of genetic and environmental risk factors of chronic pancreatitis.
PROGRESSION OF GENETIC PANCREATITIS
The majority of subjects with autosomal-dominant trypsinogen mutations N29I and R122H get ill in childhood[6], but this is also true for those with the variant N34S of SPINK1[7] and a considerable number of patients without any detectable risk factor (“arly onset idiopathic”). The data from our patient group were shown in Figure 3[7]. The progression of disease was analysed in both patient groups. As shown in Figure 4, in the majority of the patients with the PRSS1 mutations N29I and R122H the initial event was a severe acute episode leading to treatment in a hospital. At the same time point there was no evidence for definite signs of chronic pancreatitis in the majority of these patients. Only several years later there is a slowly increasing rate of duct dilatation or calcification that may be regarded as definite prove of chronic pancreatitis. However, even after a follow-up time of 20 years after the first symptoms, only 30% of the patients show evidence for calcifications or duct dilatation. The onset of diabetes seems to be even later as after 10 years less than 10% and after 20 years less than 20% were diabetic (Figure 4). This shows that the signs of chronic pancreatitis may be absent when patients were not observed long enough.
Figure 3.
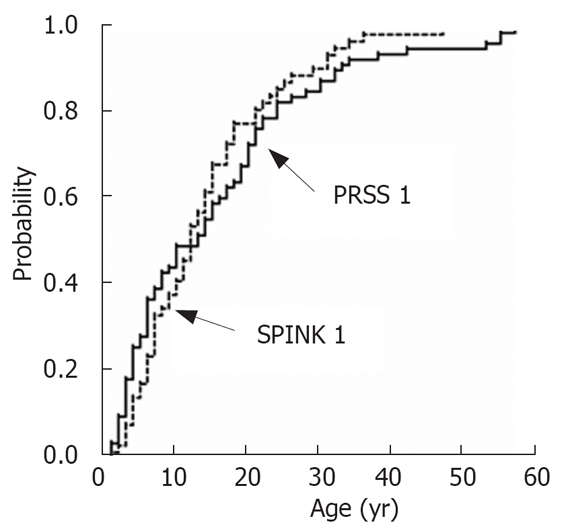
Age at onset of pancreatitis in patients with mutations N29I and R122H of the cationic trypsinogen and mutation N34S of SPINK1.
Figure 4.
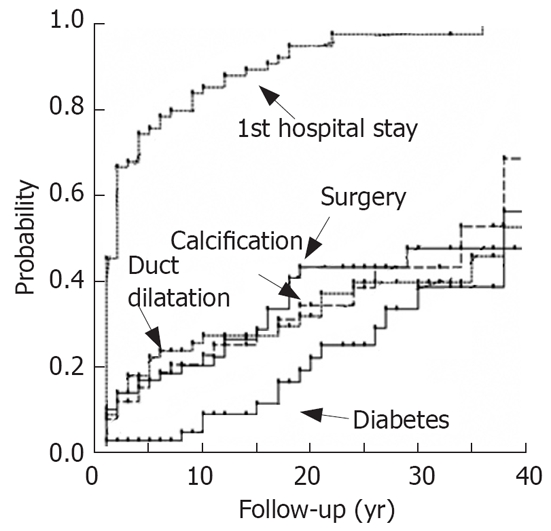
Progression of pancreatitis in patients with PRSS1 mutations N29I and R122H.
The progression of disease in patients with N34S of SPINK1 was rather similar. There was a hospital stay early in the course of the disease, a slow appearance of duct dilatation or calcifications and a late onset of diabetes (Figure 5). Compared to patients with alcoholic chronic pancreatitis (Figure 6) the progression of disease is much slower, as in more than 50% of the alcoholics diabetes and calcification were observed already after a follow-up time of less than 15 years.
Figure 5.
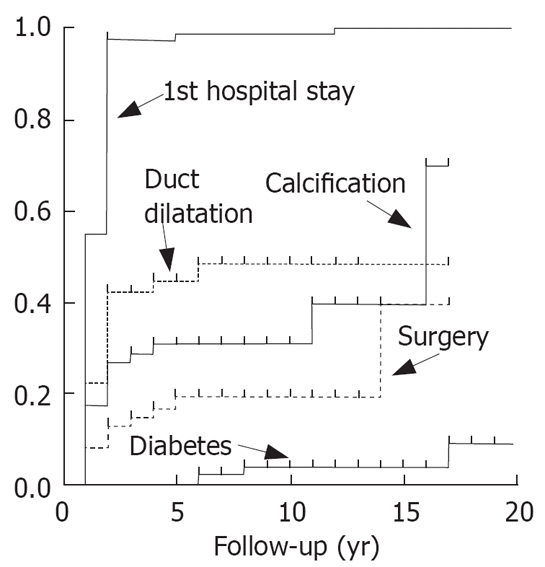
Progression of pancreatitis in patients with mutation N34S of SPINK1.
Figure 6.
Progression of pancreatitis in patients with alcoholic, idopathic or PRSS1-induced disease.
DOES ACUTE RECURRENT PANCREATITIS EXIST IN PATIENTS WITH GENETIC RISK FACTORS?
The data shown above suggest that in a significant number of patients definite signs of chronic pancreatitis may be found only after a long follow-up time. From this finding it may be suspected that acute-recurrent pancreatitis is only a transition state that finally ends in chronic pancreatitis. To analyse this presence of either calcifications or duct dilatations in patients with PRSS1 (N29I and R122H) or SPINK1 (N34S) mutations were analysed by Kaplan-Meier. As shown in Figure 7 in approximately 80% of the patients with SPINK1 show at least one of the definite signs of chronic pancreatitis after a follow-up time of 15-20 years. In 50% of the patients with a PRSS1 mutation a chronic pancreatitis can be diagnosed after 25 years of follow-up. This may, however, be an underestimation of chronic pancreatitis as only two signs were evaluated. Histological specimens were available in only a few patients and the retrospective study design tends to overlook patients with chronic disease. The data suggests that recurrent acute pancreatitis is present in patients with genetic risk factors, but with time there is a slow progression to chronic pancreatitis. In the majority of patients, however, one has to wait a long time and in some others recurrent pancreatitis may persist.
Figure 7.
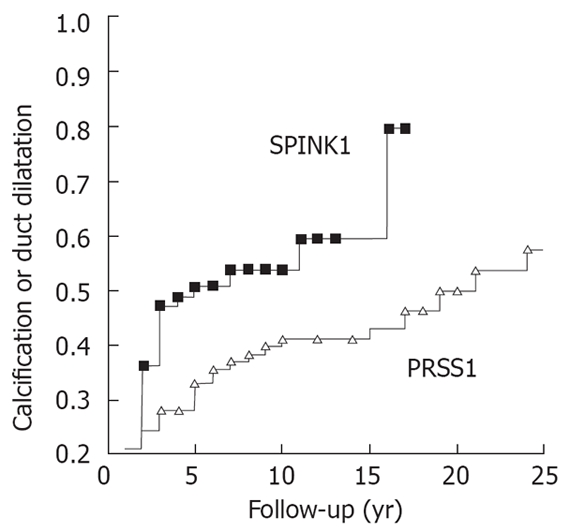
Probability of either calcification or duct dilatation in patients with N34S mutation of SPINK1.
CONCLUSION
The autosomal-dominant mutations N29I and R122H of PRSS1 (cationic trypsinogen), N34S of SPINK1 (pancreatic secretory trypsin inhibitor) lead to a disease onset in childhood or youth. Compared to chronic alcoholic pancreatitis the progression is low so that for a long time only signs of acute-recurrent pancreatitis are found. At later time points (more than 15-20 years) there is evidence for chronic pancreatitis (duct dilatation, calcification, histological prove of chronic pancreatitis). In patients with genetic risk factors acute recurrent pancreatitis may therefore be regarded as a transition state until definite signs of chronic pancreatitis are detectable. The progression of disease, however, is slow so that the clinical picture is compatible with an acute-recurrent pancreatitis for a very long time.
Peer reviewer: Giuseppe Brisinda, MD, Department of Surgery, Catholic School of Medicine “Agostino Gemelli”, Largo Agostino Gemelli 8-00168 Rome, Italy
S- Editor Liu Y E- Editor Yin DH
References
- 1.Gupta V, Toskes PP. Diagnosis and management of chronic pancreatitis. Postgrad Med J. 2005;81:491–497. doi: 10.1136/pgmj.2003.009761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teich N, Rosendahl J, Toth M, Mossner J, Sahin-Toth M. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat. 2006;27:721–730. doi: 10.1002/humu.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witt H, Sahin-Toth M, Landt O, Chen JM, Kähne T, Drenth JP, Kukor Z, Szepessy E, Halangk W, Dahm S, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet. 2006;38:668–673. doi: 10.1038/ng1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Marechal C, Masson E, Chen JM, Morel F, Ruszniewski P, Levy P, Ferec C. Hereditary pancreatitis caused by triplication of the trypsinogen locus. Nat Genet. 2006;38:1372–1374. doi: 10.1038/ng1904. [DOI] [PubMed] [Google Scholar]
- 5.Teich N, Bauer N, Mossner J, Keim V. Mutational screening of patients with nonalcoholic chronic pancreatitis: identification of further trypsinogen variants. Am J Gastroenterol. 2002;97:341–346. doi: 10.1111/j.1572-0241.2002.05467.x. [DOI] [PubMed] [Google Scholar]
- 6.Keim V, Bauer N, Teich N, Simon P, Lerch MM, Mossner J. Clinical characterization of patients with hereditary pancreatitis and mutations in the cationic trypsinogen gene. Am J Med. 2001;111:622–626. doi: 10.1016/s0002-9343(01)00958-5. [DOI] [PubMed] [Google Scholar]
- 7.Keim V, Witt H, Bauer N, Bodeker H, Rosendahl J, Teich N, Mossner J. The course of genetically determined chronic pancreatitis. JOP. 2003;4:146–154. [PubMed] [Google Scholar]
- 8.Comfort MW, Steinberg AG. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology. 1952;21:54–63. [PubMed] [Google Scholar]
- 9.Threadgold J, Greenhalf W, Ellis I, Howes N, Lerch MM, Simon P, Jansen J, Charnley R, Laugier R, Frulloni L, et al. The N34S mutation of SPINK1 (PSTI) is associated with a familial pattern of idiopathic chronic pancreatitis but does not cause the disease. Gut. 2002;50:675–681. doi: 10.1136/gut.50.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creighton J, Lyall R, Wilson DI, Curtis A, Charnley R. Mutations of the cationic trypsinogen gene in patients with chronic pancreatitis. Lancet. 1999;354:42–43. doi: 10.1016/S0140-6736(99)01814-0. [DOI] [PubMed] [Google Scholar]
- 11.Simon P, Weiss FU, Zimmer KP, Rand S, Brinkmann B, Domschke W, Lerch MM. Spontaneous and sporadic trypsinogen mutations in idiopathic pancreatitis. JAMA. 2002;288:2122. doi: 10.1001/jama.288.17.2122. [DOI] [PubMed] [Google Scholar]
- 12.Keim V, Teich N. Idiopathic vs hereditary pancreatitis. JAMA. 2003;289:983–984; author reply 985. [PubMed] [Google Scholar]
- 13.Truninger K, Malik N, Ammann RW, Muellhaupt B, Seifert B, Muller HJ, Blum HE. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. Am J Gastroenterol. 2001;96:2657–2661. doi: 10.1111/j.1572-0241.2001.04047.x. [DOI] [PubMed] [Google Scholar]
- 14.Witt H, Luck W, Becker M, Bohmig M, Kage A, Truninger K, Ammann RW, O'Reilly D, Kingsnorth A, Schulz HU, et al. Mutation in the SPINK1 trypsin inhibitor gene, alcohol use, and chronic pancreatitis. JAMA. 2001;285:2716–2717. doi: 10.1001/jama.285.21.2716-a. [DOI] [PubMed] [Google Scholar]
- 15.Choudari CP, Imperiale TF, Sherman S, Fogel E, Lehman GA. Risk of pancreatitis with mutation of the cystic fibrosis gene. Am J Gastroenterol. 2004;99:1358–1363. doi: 10.1111/j.1572-0241.2004.30655.x. [DOI] [PubMed] [Google Scholar]
- 16.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, Truninger K, Ammann R, Cavallini G, Charnley RM, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 17.Rossi L, Pfützer RH, Parvin S, Ali L, Sattar S, Kahn AK, Gyr N, Whitcomb DC. SPINK1/PSTI mutations are associated with tropical pancreatitis in Bangladesh. A preliminary report. Pancreatology. 2001;1(3):242–245. doi: 10.1159/000055818. [DOI] [PubMed] [Google Scholar]
- 18.Noone PG, Zhou Z, Silverman LM, Jowell PS, Knowles MR, Cohn JA. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology. 2001;121:1310–1319. doi: 10.1053/gast.2001.29673. [DOI] [PubMed] [Google Scholar]
- 19.Witt H, Luck W, Hennies HC, Classen M, Kage A, Lass U, Landt O, Becker M. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 20.White IR, Altmann DR, Nanchahal K. Alcohol consumption and mortality: modelling risks for men and women at different ages. BMJ. 2002;325:191. doi: 10.1136/bmj.325.7357.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfutzer RH, Barmada MM, Brunskill AP, Finch R, Hart PS, Neoptolemos J, Furey WF, Whitcomb DC. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119:615–623. doi: 10.1053/gast.2000.18017. [DOI] [PubMed] [Google Scholar]