

Abstract
In the recent study by Preissig and Rigby in Critical Care, the authors argue that critical illness hyperglycemia in children with both respiratory failure and cardiovascular failure is due to a primary failure of the beta-cell. However, alternative explanations that the failure is secondary to an increase in insulin resistance leading to beta-cell exhaustion, or a negative impact of exogenous glucocorticoid therapy, may be equally likely.
In their study on hyperglycemia in critically ill children Preissig and Rigby [1] observed that children in the pediatric intensive care unit are unlikely to have critical illness hyperglycemia (CIH) in the absence of respiratory failure (RF) or cardiovascular failure (CVF) (0 of 12 patients studied), whereas those with RF but without CVF may (9 of 18) or may not (9 of 18), and virtually all patients with both RF and CVF do (10 of 11). The key observation was that the C-peptide level in children without CIH was similar for those with RF versus those without RF or CVF (2.3 versus 5.3 ng/ml), whereas in children with CIH, C-peptide was significantly higher with RF alone than with RF and CVF (11.5 versus 4.4 ng/ml; data reproduced in Figure 1). Importantly, the RF and CVF cohort uniformly received exogenous glucocorticoid therapy, unlike the other cohorts (100% versus 44% to 50%). The authors concluded from their data that elevated insulin resistance (high C-peptide) was the prominent cause of CIH in children with RF only, whereas beta-cell dysfunction (low C-peptide) was the primary cause in children with RF and CVF.
Figure 1.
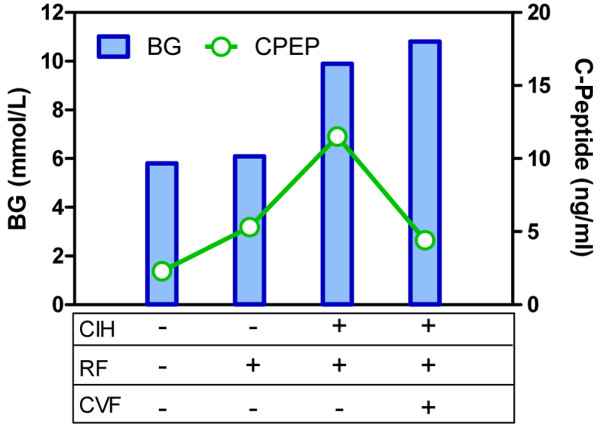
Blood glucose (BG) and C-peptide (CPEP) levels in children with (+) or without (-) critical illness hyperglycemia (CIH), with or without respiratory failure (RF) or cardiovascular failure (CVF). Data taken from [1].
We find the results very interesting but would take care in concluding that CIH in children with both RF and CVF is due primarily to a failure of the beta-cell. The beta-cell has a very complex response to hyperglycemia both acutely (first and second phase responses over just minutes [2,3]) and over the course of several days (increased mass [4,5]). Also, beta-cell exhaustion is a well known phenomenon characterized by an ability to increase secretion up to a certain level and thereafter fail in response to further demand [6,7]. Finally, exogenous glucocorticoid therapy, given to all patients with RF and CVF together, can also have complex interactions with insulin secretion, suppressing it in some in vitro studies [8,9] and enhancing it in other in vivo studies [10,11]. Support for these mechanisms playing a major role in CIH is variable, but there is insufficient evidence to dismiss the mechanisms.
Arguments that the beta-cell can rapidly adapt, or become exhausted, when faced with increased demand provide an alternative hypothesis to that put forth by Preissig and Rigby. With adaptation, the difference in C-peptide levels observed in the RF group without CIH compared to the group that had neither RF nor CVF (5.3 versus 2.3 ng/ml) could indicate beta-cell adaptation, with the twofold increase in secretion having failed to achieve statistical significance due to the small number of subjects in each group (9 and 12 subjects, respectively). That the two groups had similar glucose levels (5.8 versus 6.1 mmol/l) with different secretion (C-peptide) could occur if the beta-cells' second phase response is interpreted as increasing insulin secretion when glucose is above a threshold [12]. The remaining 9 out of 18 patients with RF and no CVF may have had CIH only because the beta-cell did not have sufficient time to normalize the level. That is, in the study CIH was defined as two blood glucose values taken 1 to 2 hours apart both above 7.7 mmol/l. This definition does not preclude the possibility that glucose was decreasing at the time CIH was assessed. The unarguable failure of the beta-cell to normalize glucose in children with RF+CVF could be attributed to beta-cell exhaustion (low C-peptide levels) secondary to further increases in insulin resistance in this group. By the authors' own estimates, the RF+CVF subjects required longer, and at times up to 50% more, exogenous insulin to normalize glucose. Using the values reported, one could argue that the beta-cell had sufficient capacity to meet a peak demand of 0.13 U/kg/h for 5.8 days (RF group) but an insufficient capacity to meet a peak demand of 0.19 U/kg/h for 8.7 days (RF+CVF group).
The clinical entity of RF+CVF represents a temporal and clinical progression from the state where only one is present – the key question during this progression being what changes are occurring in peripheral insulin sensitivity and beta-cell function. In the authors' data, the issue becomes whether the low C-peptide levels in the RF+CVF group are indicative of a lower level of insulin resistance (Preissig and Rigby's argument), or whether the higher exogenous insulin required to bring these patients to target is indicative of higher resistance. Discerning whether beta-cell exhaustion, or events secondary to the glucocorticoid therapy, contributed to the low insulin secretion will be essential in guiding future treatment strategies.
Abbreviations
CIH: critical illness hyperglycemia; CVF: cardiovascular failure; RF: respiratory failure.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
MSDA is presently conducting an on-going trial of tight glycemic control following cardiac surgery in a cohort of children less than 3 years old (US National Institutes of Health R01HL088448).
See related research by Preissig and Rigby, http://ccforum.com/content/13/1/R27
Contributor Information
Garry M Steil, Email: garry.steil@childrens.harvard.edu.
Michael SD Agus, Email: michael.agus@childrens.harvard.edu.
References
- Preissig CM, Rigby MR. Hyperglycaemia results from beta-cell dysfunction in critically ill children with respiratory and cardiovascular failure: a prospective observational study. Crit Care. 2009;13:R27. doi: 10.1186/cc7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahi D. In praise of the hyperglycemic clamp. A method for assessment of B-cell sensitivity and insulin resistance. Diabetes Care. 1996;19:278–286. doi: 10.2337/diacare.19.3.278. [DOI] [PubMed] [Google Scholar]
- Toschi E, Camastra S, Sironi AM, Masoni A, Gastaldelli A, Mari A, Ferrannini E, Natali A. Effect of acute hyperglycemia on insulin secretion in humans. Diabetes. 2002;51(Suppl 1):S130–S133. doi: 10.2337/diabetes.51.2007.S130. [DOI] [PubMed] [Google Scholar]
- Jonas JC, Laybutt DR, Steil GM, Trivedi N, Pertusa JG, Casteele M Van de, Weir GC, Henquin JC. High glucose stimulates early response gene c-Myc expression in rat pancreatic beta cells. J Biol Chem. 2001;276:35375–35381. doi: 10.1074/jbc.M105020200. [DOI] [PubMed] [Google Scholar]
- Steil GM, Trivedi N, Jonas JC, Hasenkamp WM, Sharma A, Bonner-Weir S, Weir GC. Adaptation of beta-cell mass to substrate oversupply: enhanced function with normal gene expression. Am J Physiol Endocrinol Metab. 2001;280:E788–E796. doi: 10.1152/ajpendo.2001.280.5.E788. [DOI] [PubMed] [Google Scholar]
- Grill V, Bjorklund A. Impact of metabolic abnormalities for beta cell function: clinical significance and underlying mechanisms. Mol Cell Endocrinol. 2009;297:86–92. doi: 10.1016/j.mce.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Steil GM, Doria A, Soeldner JS, Warram JH. Heterogeneity of B-cell compensation during progression to NIDDM. Diabetes. 1997;46(Suppl 1):0944. [Google Scholar]
- Barseghian G, Levine R, Epps P. Direct effect of cortisol and cortisone on insulin and glucagon secretion. Endocrinology. 1982;111:1648–1651. doi: 10.1210/endo-111-5-1648. [DOI] [PubMed] [Google Scholar]
- Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest. 1997;99:414–423. doi: 10.1172/JCI119175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvik B, Clodi M, Kautzky-Willer A, Capek M, Hartter E, Pacini G, Prager R. Effect of dexamethasone on insulin sensitivity, islet amyloid polypeptide and insulin secretion in humans. Diabetologia. 1993;36:84–87. doi: 10.1007/BF00399099. [DOI] [PubMed] [Google Scholar]
- Hollingdal M, Juhl CB, Dall R, Sturis J, Veldhuis JD, Schmitz O, Pørksen N. Glucocorticoid induced insulin resistance impairs basal but not glucose entrained high-frequency insulin pulsatility in humans. Diabetologia. 2002;45:49–55. doi: 10.1007/s125-002-8244-y. [DOI] [PubMed] [Google Scholar]
- Steil GM, Clark B, Kanderian S, Rebrin K. Modeling insulin action for development of a closed-loop artificial pancreas. Diabetes Technol Ther. 2005;7:94–108. doi: 10.1089/dia.2005.7.94. [DOI] [PubMed] [Google Scholar]