

Abstract
The ligase IV/XRCC4 complex plays a central role in DNA double-strand break repair by non-homologous end joining (NHEJ). During adenovirus infection, NHEJ is inhibited by viral proteins E4 34k and E1B 55k, which redirect the Cul5/Rbx1/Elongin BC ubiquitin E3 ligase to polyubiquitinate and promote degradation of ligase IV. In cells infected with E1B 55k-deficient adenovirus, ligase IV could not be found in XRCC4-containing complexes and was observed in a novel ligase IV/E4 34k/Cul5/Elongin BC complex. These observations suggest that dissociation of the ligase IV/XRCC4 complex occurs at an early stage in E4 34k-mediated degradation of ligase IV and indicate a role for E4 34k in dissociation of the ligase IV/XRCCC4 complex. Expression of E4 34k alone was not sufficient to dissociate the ligase IV/XRCC4 complex, which indicates a requirement for an additional, as yet unidentified, factor in E1B 55k-independent dissociation of the ligase IV/XRCC4 complex.
Keywords: Adenovirus, Non-homologous end joining, XRCC4, Ligase IV, E4 34k, E4orf6, E1B 55k
Introduction
DNA damage occurs in all living cells either as a result of environmental insults or normal metabolic processes and presents a major threat to the integrity of the genome. In particular, DNA doublestrand breaks (DSBs) can lead to genetic rearrangements that promote tumor formation. Eukaryotic cells repair DSBs by two mechanisms: homologous recombination, which is used for repair of DSBs that occur during late-S or G2 phases of the cell cycle, and nonhomologous end joining (NHEJ), which is used during G0 and G1 phases of the cell cycle (Lieber et al. 2004, Sonoda et al. 2006, Weterings and Chen 2008). In addition to spontaneous DSBs, somatic cell recombination processes required for immunoglobulin (Ig) production and diversification are initiated by DSBs that are repaired by NHEJ (Lieber et al. 2004, Weterings and Chen 2008). Because of the large proportion of non-cycling cells in multicellular organisms and the added role of DSBs in Ig production, the majority of DSBs in humans are repaired by NHEJ (Lieber et al. 2004, Sonoda et al. 2006).
The repair of the DSBs by NHEJ involves recognition of DSBs, localization of repair enzymes to the DSB, processing of the DNA ends to yield ligatable termini, and ligation to reseal the break. In mammalian cells, DSB recognition requires phosphorylation of histone H2AX and processing of DNA ends may involve one of several factors, which include aprataxin and polynucleotide kinase (Chappell et al. 2002, Koch et al. 2004, Ahel et al. 2006). Mammalian NHEJ also requires the heterotrimeric DNA-dependent protein kinase (DNA-PK), a nuclear serine/threonine protein kinase belonging to the phosphatidylinositol 3-related protein kinase (PI3K) family (Smith and Jackson 1999, Weterings and Chen 2008). This heterotrimeric kinase consists of a 460 kDa catalytic subunit (DNA-PKcs) and a heterodimeric DNA binding subunit (Ku) (Smith and Jackson 1999). Ku is thought to be involved in the recognition of DSBs based on its abundance in nuclei and its affinity for DNA ends (Smith and Jackson 1999, Weterings and Chen 2008). In mice, disruption of genes encoding DNA-PKcs or either of the Ku subunits results in a spectrum of DSB repair and Igproduction defects (Smith and Jackson 1999, Weterings and Chen 2008).
In mammalian NHEJ, the ligation event is catalyzed by DNA ligase IV, which forms a complex with the DNA-binding proteins XRCC4 and XLF (Bryans et al. 1999, Ahnesorg et al. 2006, Buck et al. 2006, Sonoda et al. 2006, Weterings and Chen 2008). XRCC4 makes direct contact with ligase IV and XLF, stimulates ligase IV activity and is essential for ligase IV stability (Critchlow et al. 1997, Grawunder et al. 1998, Ahnesorg et al. 2006, Buck et al. 2006, Sonoda et al. 2006, Weterings and Chen 2008). Structural studies have shown that both XRCC4 and XLF form homodimers with remarkable similarity in overall fold (Junop et al. 2000, Andres et al. 2007, Li et al. 2007). Direct-binding and structural studies paint a picture of a complex composed of 1 ligase IV: 2 XRCC4: 2 XLF that is held together through strong, direct ligase IV/XRCC4 and XRCC4/XLF interactions with weaker, possibly indirect, ligase IV/XLF contacts (Deshpande and Wilson 2007).
Cells lacking ligase IV, XRCC4 or XLF cannot carryout NHEJ and are therefore radiation-sensitive and unable to carry out Ig-gene rearrangement by V(D)J recombination (Barnes et al. 1998, Frank et al. 1998, Lieber et al. 2004, Buck et al. 2006, Weterings and Chen 2008). In mice, disruption of ligase IV or XRCC4 results in embryonic lethality (Barnes et al. 1998, Frank et al. 1998, Weterings and Chen 2008). The severe phenotype associated with ligase IV or XRCC4 gene ablation likely reflects the central role of the ligase IV complex in mammalian NHEJ. Interaction between the DNA-PK and ligase IV/XRCC4/XLF complexes has been observed (McElhinny et al. 2000, Hsu et al. 2002, Costantini et al. 2007, Yano et al. 2008). Previous reports have described ligase IV/Ku and XRCC4/DNA-PKcs interactions, which are believed to be important in coordinating the efforts of these two large multi-protein complexes (Hsu et al. 2002, Costantini et al. 2007).
During infection of human cells with adenovirus, replication of the linear dsDNA adenovirus genome and the presence of adenovirusgenome termini can activate the host cell DSB repair response (Weitzman and Ornelles 2005). Unperturbed, this response can result in adenovirus genome concatamer formation and inhibits productive lytic infection. Adenovirus genome concatenation is an NHEJ-dependent process that requires DNA-PK, ligase IV and the Mre11 protein (Boyer et al. 1999, Stracker et al. 2002, Weitzman and Ornelles 2005). To prevent genome concatenation, human adenovirus type 5 (Ad5) encodes early proteins that specifically target ligase IV for polyubiquitination and proteasome-mediated degradation (Baker et al. 2007). This process requires viral proteins encoded by early region 1B (E1B 55k) and early region 4 (E4) open reading frame 6 gene product (E4orf6, E4 34k), which also target Mre11 and p53 to effectively disable DNA damage sensing, DSB repair and apoptosis (Carson et al. 2003, Berk 2005). To further prevent genome concatenation, the E4 open reading frame 3 gene product (E4orf3, E4 11k) causes mis-localization of the nuclear Mre11 protein to cytoplasmic aggresomes, which prevents Mre11 from acting on the newly replicated Ad5 genomes (Araujo et al. 2005).
Study of the molecular mechanism that underlies adenovirusdirected protein degradation has shown that the Ad5 E1B 55k and E4 34k proteins form a complex that re-directs a host ubiquitin E3 ligase to polyubiquitinate host factors (Querido et al. 2001, Blanchette et al. 2004, Cheng et al. 2007). This system has been most thoroughly studied in the context of p53 degradation during Ad5 infection. The Cul5/Rbx1/Elongin BC ubiquitin E3 ligase is bound by E4 34k through direct contact with Cul5 and the Elongin BC complex to form an Ad5-specific E4 34k ubiquitin E3 ligase (Blanchette et al. 2004, Cheng et al. 2007). It is thought that efficient selection and delivery of p53 to the E4 34k ubiquitin E3 ligase is mediated by E1B 55k, which binds to E4 34k and delivers p53 only after assembly of the Ad5-specific E4 34k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase (Blanchette et al. 2004). Cul5-dependent polyubiquitination of p53 has been observed, and because polyubiquitination acts as a general signal for proteasome-mediated degradation, viral redirection of the Cul5/Rbx1/Elongin BC ubiquitin E3 ligase results in effective elimination of targeted host proteins (Querido et al. 2001, Blanchette et al. 2004, Cheng et al. 2007).
Ad5-directed degradation of ligase IV requires the same factors necessary for degradation of p53 and is thought to proceed by the same mechanism. Interestingly, while ligase IV is known to form a stable complex with the XRCC4 and XLF proteins, Ad5-directed degradation of ligase IV does not appear to affect XRCC4 or XLF protein levels (Baker et al. 2007). This observation suggests that the ligase IV/XRCC4/XLF complex is dissociated at some stage during the virus-specific pathway that targets ligase IV for degradation. Previous observation of an E1B 55k/ligase IV complex suggests that ligase IV, like p53, is specifically recognized by E1B 55k and then delivered to the E4 34k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase.
In this communication, we show that in the absence of E1B 55k, ligase IV is bound by E4 34k to form a complex that contains Elongins B and C, but does not contain XRCC4. Our findings show that ligase IV, like p53, is recognized by both E1B 55k and E4 34k and indicate an important role for E4 34k in dissociation of the ligase IV/XRCC4 complex. Because expression of E4 34k alone was not sufficient to cause dissociation of the ligase IV/XRCC4 complex, our observations suggest the possibility that an additional factor is required for E4 34k-dependent, E1B 55k-independent dissociation of the ligase IV/XRCC4 complex.
Results
Infection with Ad5 deletion mutants reveals that viral expression of E4 34k compromises the DNA-PKcs/ligase IV interaction in an E1B 55k-independent fashion
While DNA-PK has been shown to functionally and physically interact with the ligase IV/XRCC4 complex (Hsu et al. 2002), during Ad5 infection, DNA-PK and XRCC4 appear to be unaffected, while ligase IV is degraded (Baker et al. 2007). Because E4 34k participates in ligase IV degradation and co-immunoprecipitates (co-IP) with DNAPKcs (Boyer et al. 1999, Baker et al. 2007), we wanted to examine the effect of Ad5 infection on the interactions between the DNA-PK and ligase IV/XRCC4 complexes. To address this question we prepared lysates from cells infected with wild type or deletion mutants of Ad5, used immunoprecipitation (IP) to isolate DNA-PKcs-containing protein complexes and then used Western blot analysis to determine if XRCC4 and ligase IV were associated with DNA-PKcs. In extracts prepared from mock-infected cells, we were able to detect both ligase IV and XRCC4 in the input lysate and in the DNA-PKcs co-IP (Fig. 1, top). We interpret these data as the result of direct protein-protein interactions between XRCC4 and DNA-PKcs (Hsu et al. 2002) and between XRCC4 and ligase IV (Critchlow et al. 1997, Grawunder et al. 1998), which result in an indirect (i.e. through XRCC4) interaction between ligase IV and DNA-PKcs (Fig. 1). In extracts prepared from cells infected with wild type Ad5, it is worth noting that ligase IV was absent and the co-IP between XRCC4 and DNA-PKcs was still observable (Fig. 1, top). Similar results were obtained with extracts prepared from cells infected with H5dl1013 and H5pm1020, which express both E1B 55k and E4 34k (Table 1). As anticipated, degradation of ligase IV was not observed in lysates prepared from cells infected with viruses that did not express E4 34k, and we were able to observe the interaction between the DNA-PK and ligase IV/XRCC4 complexes (Fig. 1 H5dl1004, H5dl1014 with H5dl1015).
Fig. 1.
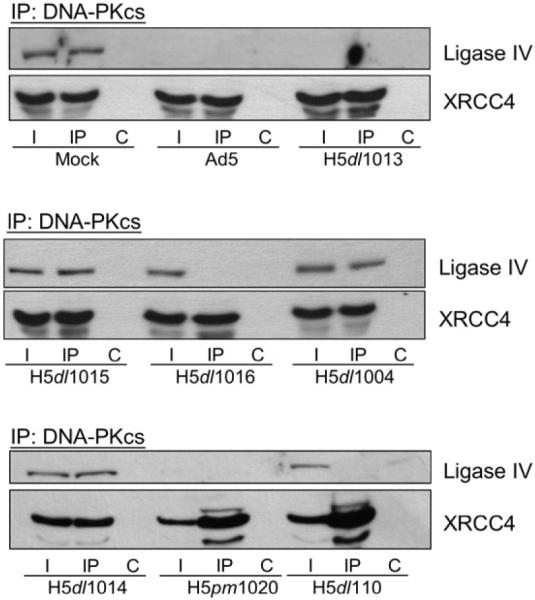
E1B 55k-independent loss of ligase IV interaction with DNA-PKcs. (A) HeLa cells were Mock, Ad5 or Ad5-mutant (H5dl1013, H5dl1015, H5dl1016, H5dl1004, H5dl1014, H5pm1020 or H5dl110) infected and lysed in NP40 lysis buffer 20 h post-infection. 200 μg of the total extracts were subjected to immunoprecipitation (IP) with anti-DNAPKcs antibodies. DNA-PKcs-containing complexes were recovered, resolved by SDS-PAGE, Western transferred and probed for Ligase IV and XRCC4. I, 15% of input lysate; IP, immunoprecipitation using anti-DNA-PKcs antibodies; C, control IP.
Table 1.
Genotypes of E4 and E1B mutants used in this study. +, intact open reading frame; -, disrupted open reading frame
| Mutant | E4orf1 | E4orf2 | E4orf3 (E4 11k) | E4orf4 | E4orf6 (E4 34k) | E4orf3/4 | E4orf6/7 | E1B 55k |
|---|---|---|---|---|---|---|---|---|
| Ad5 | + | + | + | + | + | + | + | + |
| H5dl1004 | + | - | - | - | - | - | - | + |
| H5dl1011 | - | - | - | - | - | - | - | + |
| H5dl1013 | - | - | - | + | + | - | - | + |
| H5dl1014 | - | - | - | + | - | - | - | + |
| H5dl1015 | - | - | + | + | - | + | - | + |
| H5dl1016 | - | - | - | + | + | - | - | - |
| H5pm1020 | + | + | + | - | + | + | + | + |
| H5dl110 | + | + | + | + | + | + | + | - |
All mutations are deletions except for the ATG mutation that inactivates ORF 4 in H5pm1020 and an 8 bp insertion that interrupts ORF 7 in H5dl1013.
Ad5-deletion mutants H5dl1016 and H5dl110 do not express E1B 55k (Table 1) and are, therefore, impaired for targeted degradation of ligase IV. In lysates prepared from H5dl1016- or H5dl110-infected cells, we detected ligase IV and XRCC4 in the input lysate, although the amount of ligase IV appeared to be slightly reduced when compared with extracts prepared from mock-infected cells. Interestingly, although the ligase IV polypeptide was present in lysates prepared from H5dl1016- or H5dl110-infected cells and we were able to observe the direct interaction between XRCC4 and DNA-PKcs, in neither lysate were we able to observe the co-IP between ligase IV and DNA-PKcs (Fig. 1, center). These data suggest that while the ligase IV and XRCC4 polypeptides remain intact in H5dl1016- or H5dl110-infected cells, the direct interaction between these NHEJ factors has been compromised.
Although E4 11k is known to act on host DSB repair factors, expression of E4 11k had no effect on ligase IV, or on its indirect interaction with DNA-PKcs (Fig. 1, compare H5dl1014 with H5dl1015). Similarly, disruption of the indirect DNA-PKcs/ligase IV interaction was independent of E4orf1-3, E4orf3/4 and E4orf6/7 expression (Fig. 1A, compare H5dl110 with H5dl1016). Western blots of lysates prepared from cells infected with the adenoviruses used did not show high molecular weight forms of ligase IV, which would be consistent with ubiquitination. Taken together, the data presented in Fig. 1 shows that viral expression of E4 34k in the absence of E1B 55k can abrogate the co-IP between DNA-PKcs and ligase IV. These data suggest that important functional interaction between XRCC4 and ligase IV had been compromised by an E1B 55k-independent mechanism.
E1B 55k-independent disruption of the ligase IV/XRCC4 direct interaction
To confirm that loss of an observable DNA-PKcs/ligase IV co-IP reflected loss of the stable ligase IV/XRCC4 direct interaction, we compared XRCC4 IPs using extracts prepared from mock- and Ad5-infected cells with extracts prepared from cells infected with the E1B 55k-, E4 11k-deficient adenovirus H5dl1016. As shown in Fig. 2A, in extracts prepared from mock-infected cells, both the XRCC4 and ligase IV polypeptides were detected in the input lysate and in the XRCC4 IP. In Ad5-infected cell lysates, degradation of ligase IV precluded observation of ligase IV in the lysate or the XRCC4 IP. In extracts prepared from H5dl1016-infected cells we again found that the amount of ligase IV detected in the input lysate was slightly reduced relative to the amount observed in extracts prepared from mock-infected cells. Significantly, in extracts prepared from H5dl101- infected cells, ligase IV did not co-IP with XRCC4. Western blot analysis confirmed the presence of E1B 55k in extracts prepared from Ad5-infected cells, but not in lysates of mock- or H5dl1016-infected cells (data not shown). These data show that, in the absence of E1B 55k, the strong, functionally important direct interaction between ligase IV and XRCC4 had, in fact, been compromised.
Fig. 2.
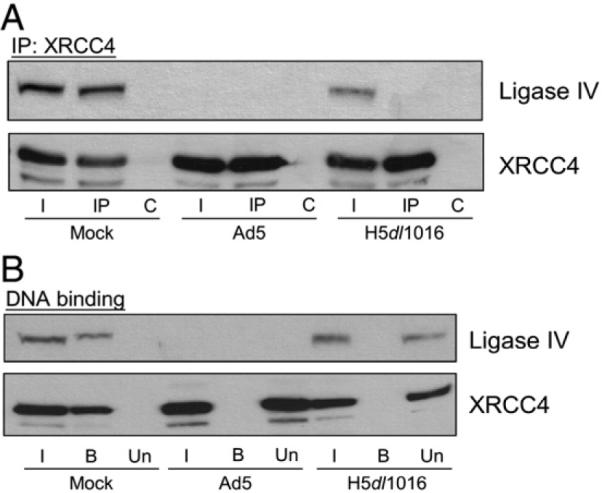
Loss of ligase IV/XRCC4 interaction in lysates from H5dl1016-infected cells. HeLa cells were Mock-, Ad5- or H5dl1016-infected and lysates were prepared 20 h post-infection. (A) Ligase IV and XRCC4 failed to co-IP from H5dl1016-infected cell lysate. Cells were lysed in NP40 lysis buffer and 200 μg of the total extracts were subject to IP with anti-XRCC4 antibodies. XRCC4-containing complexes were recovered, resolved by SDS-PAGE, Western transferred and probed for Ligase IV and XRCC4. I, 15% of input lysate; IP, immunoprecipitation using anti-XRCC4 antibodies, C, control IP. (B) DNA binding by XRCC4 and ligase IV is inhibited in extracts from H5dl1016-infected cells. Mini-WCE was prepared and 50 μg of mWCE was used for DNA-cellulose collection of DNA-binding proteins. I, 15% of the input extract; B, collected DNA-binding proteins; U, the extract following removal of DNA-binding proteins (unbound). Fractions were resolved by SDS-PAGE, Western transferred and probed for Ligase IV or XRCC4.
DNA binding by the ligase IV/XRCC4 complex has previously been described (Marchetti et al. 2006) and this biochemical activity is thought to be important to the biology of this complex. To determine whether the loss of ligase IV/XRCC4 direct interaction affected the dsDNA binding by XRCC4 or ligase IV we prepared mini whole-cell extracts (mWCE) from mock-, Ad5- or H5dl1016-infected cells and used dsDNA cellulose to capture DNA-binding proteins; the bound fraction. This procedure left proteins that lack affinity for dsDNA in the unbound fraction. Input, bound and unbound fractions were subject to Western blot analysis to determine if ligase IV and XRCC4 were capable of binding DNA. In Fig. 2B we show that in control mWCE prepared from mock-infected cells, XRCC4 and ligase IV were present in the input and bound fractions. In Ad5-infected cell mWCE, targeted degradation of ligase IV resulted in the loss of observable DNA binding by XRCC4, which was found in the unbound fraction. In mWCE prepared from H5dl1016-infected cells, the XRCC4 polypeptide was observed in the input lysate and in the unbound fraction. Similar results were obtained for ligase IV. In control experiments, we found that in mWCE prepared from mock-, Ad5- and H5dl1016-infected cells the Ku heterodimer and DNA-PKcs bound dsDNA equally well (data not shown), indicating that the dsDNA used in these experiments was accessible for recognition by DNA-binding proteins. The presence of DNA-PKcs and Ku in the DNA-bound fraction also indicated that NHEJ factors with DNA-binding properties similar to those of the ligase IV/XRCC4 complex are present in the mWCE and retain DNA-binding activity through the extraction procedure. Taken together, data presented in Fig. 2 shows E1B 55k-independent disruption of the ligase IV/XRCC4 protein-protein interaction that was coincident with loss of DNA binding by ligase IV and XRCC4.
E4 34k forms a complex with DNA ligase IV
Ligase IV is unstable and subject to degradation in the absence of XRCC4 (Bryans et al. 1999). In extracts prepared from H5dl1016-infected cells ligase IV is no longer in contact with XRCC4, yet is not quantitatively degraded. These observations suggest that in extracts prepared from H5dl1016-infected cells, ligase IV may be stabilized through interaction with other factors of host or viral origin. Previous observations have shown that targets for the adenoviral E4 34k/E1B 55k ubiquitin E3 ligase can be bound by E4 34k and E1B 55k. Both p53 and Mre11 are targets for the adenoviral ubiquitin E3 ligase and while p53 can be bound by E4 34k and E1B 55k independently, Mre11 is not recognized by E4 34k and only bound by E1B 55k. In the absence of E4 34k, ligase IV can form a complex with E1B 55k (Baker et al. 2007). To determine if ligase IV, like p53, can also form a complex with E4 34k in the absence of E1B 55k we prepared extracts from mock-, Ad5-, or H5dl1016-infected cells, immunoprecipitated E4 34k from these lysates and used Western blot analysis to determine if ligase IV and E4 34k are part of a new complex.
As shown in Fig. 3, ligase IV and XRCC4 were detected in lysates from mock-infected cells, which contain no E4 34k. A control E4 34k IP using mock-infected cell lysates shows no retention of ligase IV or XRCC4 in the absence of E4 34k. In extracts prepared from Ad5-infected cells, while degradation precluded detection of ligase IV, XRCC4 and E4 34k were observed, but did not co-IP. In H5dl1016-infected cell lysates ligase IV, XRCC4 and E4 34k were all detected and we observed E4 34k co-IP of ligase IV, but not of XRCC4. Taken together, these data show that in lysates prepared from cells infected with H5dl1016, the ligase IV/XRCC4 complex was disrupted and a new complex E4 34k/ligase IV, which did not contain XRCC4, was observed. These findings suggest the intriguing possibility that E4 34k may be responsible for dissociation of the ligase IV/XRCC4 complex prior to polyubiquitination of ligase IV.
Fig. 3.
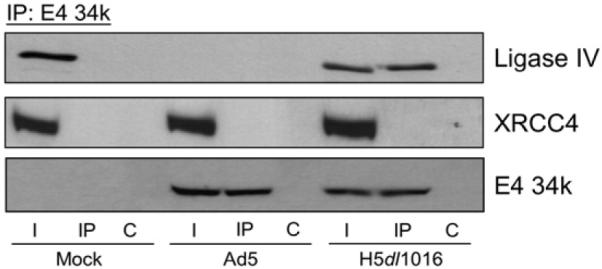
An E4 34k/ligase IV complex in extracts from H5dl1016-infected cells. HeLa cells were Mock-, Ad5- or H5dl1016-infected and lysed in NP40 lysis buffer 20 h post-infection. 200 μg of extract was subject to IP with anti-E4 34k antibodies. E4 34k-containing complexes were resolved on SDS-PAGE, Western transferred and probed for Ligase IV, XRCC4 and E4 34k. I, 15% of input lysate; IP, immunoprecipitation using anti-E4 34k antibodies; C, control IP.
The E4 34k/ligase IV complex contains the Elongin BC complex and Cullin5
E4 34k contains three BC box motifs that mediate direct binding of the Elongin BC complex as part of the E4 34k ubiquitin E3 ligase (E4 34k/Cul5/Rbx1/Elongin BC) (Blanchette et al. 2004). It is thought that assembly of the E4 34k ubiquitin E3 ligase is a prerequisite to binding by E1B 55k, which does so through direct contact with E4 34k (Cheng et al. 2007). Because we have observed E4 34k and ligase IV in the same complex, we wanted to determine if this complex represented binding by E4 34k alone, or assembly of the E4 34k ubiquitin E3 ligase. To do this, we prepared lysates from HeLa cells that had been mock-, Ad5- or H5dl1016-infected, immunoprecipitated either Elongin B or Elongin C then used Western blot analysis to determine if E4 34k and ligase IV co-IP with the Elongins. In extracts prepared from mock-infected cells, ligase IV was detected in the input lysate, but was not found to co-IP with the Elongin B or C (Fig. 4). In Ad5-infected cell extracts, we observed E4 34k in the input lysate and in co-IPs with both Elongin B and C (Fig. 4). Degradation of ligase IV in Ad5-infected cells, however, precluded detection of ligase IV. In extracts prepared from cells infected with E1B 55k-deficient H5dl1016, we found that both E4 34k and ligase IV could be detected in the input lysate and that both proteins co-IP with Elongins B and C (Fig. 4).
Fig. 4.
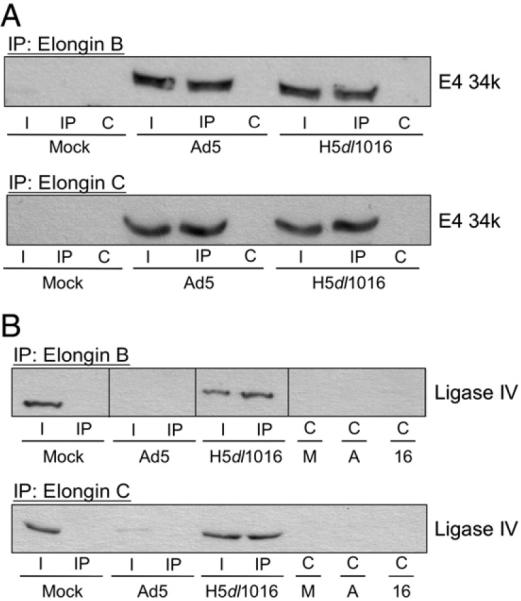
The E4 34k/ligase IV complex observed in extracts from H5dl1016-infected cells contained Elongins C and B. HeLa cells were Mock-, Ad5- or H5dl1016-infected and lysed in NP40 lysis buffer 20 h post-infection. (A) 200 μg of extract was subject to IP with antibodies directed against Elongin B (top) or Elongin C (bottom). Elongin-containing complexes were resolved by SDS-PAGE, Western transferred and probed for E4 34k. (B) Complexes containing Elongin B (top) and C (bottom) were isolated as described in (A), resolved by SDS-PAGE, Western transferred and probed for Ligase IV. I, 15% of input lysate; IP, immunoprecipitation using anti-Elongin antibodies; C, control IP. M, A and 16; control IP carried out with lysates from Mock-, Ad5- or H5dl1016-infected cells, respectively. IP: Elongin B panel was assembled from lanes from a single Western blot.
E4 34k is thought to mediate selection of Cullin5 by the viral ubiquitin E3 ligase, possibly through direct protein-protein interactions. While attempts to identify a specific Cullin5-binding motif in E4 34k were not successful, it is clear that Cullin5 is the predominant Cullin present in E4 34k-containing complexes (Querido et al. 2001, Cheng et al. 2007). To determine if the E4 34k-ligase IV complex contained Cullin5, in addition to the Elongin BC complex, antibodies against Cullin5 were used to immunoprecipitate Cullin5 from lysates prepared from mock-, Ad5- or H5dl1016-infected HeLa cells and the isolated Cullin5-containing complexes were probed for the presence of E4 34k and ligase IV. As shown in Fig. 5, lysates prepared from mock-infected cells showed that host factors Cullin5 and ligase IV did not interact in the absence of E4 viral proteins. In lysates prepared from Ad5-infected cells, degradation of ligase IV did not affect formation or stability of the E4 34k/Cullin5 complex, which was visualized by co-IP. As previously observed with the Elongin co-IPs, both ligase IV and E4 34k were detected in the Cullin5 co-IP from extracts prepared from H5dl1016-infected HeLa cells.
Fig. 5.
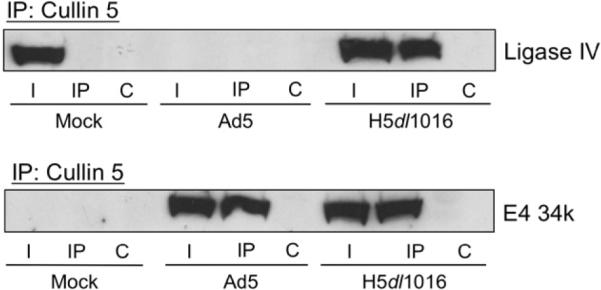
The E4 34k/ligase IV complex observed in extracts from H5dl1016-infected cells contains Cullin5. HeLa cells were Mock-, Ad5- or H5dl1016-infected and lysed in NP40 lysis buffer 20 h post-infection. 200 μg of extract was subject to IP with antibodies directed against Cullin5. Cul5-containing complexes were resolved by SDS-PAGE, Western transferred and probed for Ligase IV or E4 34k. I, 15% of input lysate; IP, immunoprecipitation using anti-Cul5 antibodies; C, control IP.
The observation of a ligase IV/E4 34k/Cul5/Elongin BC complex in the absence of a detectable ligase IV/XRCC4 complex suggests that assembly of the E4 34k ubiquitin E3 ligase and binding of the ubiquitination substrate, ligase IV, can take place in the absence of E1B 55k and is coincident with disruption of the ligase IV/XRCC4 complex. Moreover, these findings indicate an important role for E4 34k in dissociation of the ligase IV/XRCC4 complex.
Expression of E4 34k is not sufficient to disrupt the ligase IV/XRCC4 complex
Thus far, our data are consistent with the hypothesis that E4 34k can bind ligase IV, independently of E1B 55k, and that binding of E4 34k to ligase IV results in disruption of the ligase IV/XRCC4 complex. Because the E4 34k/ligase IV complex observed in H5dl1016-infected HeLa cells also contained Cullin5 and the Elongin BC complex, we reasoned that these small proteins may be required for stabilization of E4 34k/ligase IV interactions and therefore necessary for detection of the E4 34k/ligase IV complex by co-IP. Additionally, Cullin5, Elongins B and C, and possibly other human host-cell proteins, might be required for ligase IV/XRCC4 complex dissociation. To address this possibility we transiently expressed E4 34k in HeLa cells and found that, despite detectable levels of E4 34k, the ligase IV/XRCC4 co-IP remained intact (data not shown). While these results were consistent with the hypothesis that E4 34k alone is not sufficient to promote dissociation of the ligase IV/XRCC4 complex, our observations could also be explained by insufficient transfection efficiency.
Radiosensitive RKO-E4 cells were derived from RKO cells and stably express E4 34k (Hart et al. 2005). Western blot analysis confirmed expression of E4 34k in RKO-E4 cells and showed that the amount of E4 34k in mock-infected RKO-E4 cell lysates actually exceed that observed in lysates prepared from Ad5-infected RKO cells (Fig. 6A). Control experiments showed that when RKO cells were infected with E4-deletion mutant H5dl1011 (Table 1), the lack of E4 34k prevented expression of late viral proteins (Fig. 6A). In contrast, the E4 34k expressed in RKO-E4 cells complemented the E4 34k-deficiency of H5dl1011 and late viral proteins were observed 48 h post-infection (Fig. 6A). We also observed equal expression of XRCC4 by RKO and RKO-E4 cells, which did not change with Ad5 or H5dl1011 infection. These data demonstrate expression of functional E4 34k in RKO-E4 cells.
Fig. 6.

Expression of E4 34k alone is not sufficient to disrupt the ligase IV/XRCC4 complex in human cells. (A) Expression of Ad5 late proteins in H5dl1011-infected RKO-E4 cells. RKO and RKO-E4 cells were mock-(M), Ad5-, or H5dl1011-infected (1011). Extracts were prepared 24 or 48 h post-infection (HPI), resolved by SDS-PAGE, Western transferred and probed for adenoviral late proteins, E4 34k and XRCC4. Top panel showing late protein expression was assembled from lanes from a single Western blot. (B) Control RKO and E4 34k-expressing RKO-E4 cells were lysed in NP40 lysis buffer and 200 μgof extract was subject to IP with antibodies directed against XRCC4. Immune complexes were resolved by SDS-PAGE, Western transferred and probed for Ligase IV, XRCC4 and Actin. I, 7.5% of input lysate; IP, immunoprecipitation using anti-XRCC4 antibodies; C, control IPs. (C) RKO and RKO-E4 cells were lysed in NP40 lysis buffer and 200 μgof extract was subject to IP with antibodies directed against E4 34k. Immune complexes were resolved by SDS-PAGE, Western transferred and probed for E4 34k, Ligase IV, Elongin B, Elongin C and Actin. I, 10% of input lysate; IP, immunoprecipitation using anti-E4 34k antibodies; C, control IPs.
To determine if expression of E4 34k alone is sufficient to disrupt the ligase IV/XRCC4 complex we used anti-XRCC4 antibodies to IP XRCC4-containing complexes from lysates prepared from RKO and RKO-E4 cells. As shown in Fig. 6B, ligase IV was observed in the input lysate from RKO and RKO-E4 cells and in the XRCC4 co-IP from both cell lines. Actin, used as a loading control, demonstrated the specific retention of XRCC4 and ligase IV by anti-XRCC4 antibodies (Fig. 6B). Immunoprecipitaiton of E4 34k-containing complexes from lysates prepared from RKO and RKO-E4 cells show that while E4 34k is associated with Elongins B and C, ligase IV is not part of that complex (Fig. 6C). Again, detection of actin serves as both a loading control and demonstrates the specificity of the E4 34k IP. Taken together, Figs. 6B and C show that ligase IV and E4 34k are part of separate multiprotein complexes.
We conclude from these data that while E4 34k is required for E1B 55k-independent dissociation of the ligase IV/XRCC4 complex, expression of E4 34k alone in human cells is not sufficient to separate ligase IV from XRCC4. These findings suggest the intriguing possibility that one or more additional factors are required for E4 34k-mediated dissociation of the ligase IV/XRCC4 complex.
Discussion
During adenoviral infection, host proteins are degraded through the action of viral proteins E4 34k and E1B 55k. We previously reported that ligase IV is targeted for degradation during Ad5 infection, and while ligase IV forms a functional complex with XRCC4 and XLF, protein levels of these ligase IV-binding factors were unchanged during infection (Baker et al. 2007). Because nuclear export and translation of host-cell mRNAs are inhibited during adenovirus infection (Schneider and Mohr 2003, Weitzman and Ornelles 2005), synthesis of host-cell factors XRCC4 and XLF would be greatly reduced. The observation of XRCC4 and XLF in the absence of ligase IV in adenovirus-infected cells therefore suggests that the ligase IV complex is disassembled during virus-mediated ligase IV degradation. Adenovirus-mediated degradation of host-cell factors is a multi-step process in which host-cell target proteins are: i) bound by viral factors, ii) delivered to the virus-specific E4 34k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase, iii) polyubiquitinated then iv) degraded by the proteasome. It is plausible that dissociation of the ligase IV complex might occur at any stage of this complex process. Formation of new protein-protein contacts might disrupt the ligase IV complex during recognition of ligase IV by viral target-selection factors or by binding to the viral ubiquitin E3 ligase. Alteration of ligase IV during polyubiquitination or degradation could also result in disassembly of the ligase IV complex.
Here, we provide evidence that dissociation of the ligase IV complex occurs prior to virus-directed degradation of ligase IV. Using Ad5 deletion mutant H5dl1016, which does not express E1B 55k but retains E4 34k expression, we have observed that the functional, direct interaction between ligase IV and XRCC4 is lost, as is the DNA-binding activity of this complex. Under these conditions, a ligase IV/E4 34k/Cul5/Elongin BC complex was evident. We did not observe the high molecular weight forms of ligase IV that would indicate polyubiquitination. Nor did we observe significant degradation of ligase IV. Our findings, summarized in Fig. 7A, predict that Rbx1 will be part of the ligase IV/E4 34k/Cul5/Elongin BC complex and suggest that ligase IV polyubiquitination and proteasome-mediated degradation follow ligase IV/XRCC4 complex dissociation. Our data implicate recognition of ligase IV by viral factors and binding of ligase IV by the viral ubiquitin E3 ligase as the processes most likely to separate ligase IV from XRCC4. Our model predicts that in the presence of E1B 55k, assembly of the E1B 55k/E4 34k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase causes ligase IV to dissociate from XRCC4 before polyubiquitination and ligase IV degradation (Fig. 7B).
Fig. 7.

Schematic representation of E4 34k-mediated ligase IV recognition in adenovirus-infected cells. (A) In cells infected with adenovirus expressing E4 34k, but not E1B 55k, observation of an E4 34k/Cul5/Elongin BC/ligase IV complex and coincident observation of XRCC4/DNA-PKcs complexes that lack ligase IV indicate that assembly of the E4 34k/Cul5/Elongin BC ubiquitin E3 ligase and binding to ligase IV results in dissociation of the strong, functional XRCC4-ligase IV direct interaction. Rbx1 (white) is predicted to assemble with the E4 34k/Cul5/Elongin BC ubiquitin E3 ligase. (B) Model predicts that in the cells infected with adenovirus expressing both E4 34k and E1B 55k, assembly of the E4 34k/E1B 55k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase and binding to ligase IV results in ligase IV/XRCC4 dissociation. Polyubiquitination of ligase IV and proteasome-mediated degradation of ligase IV follows.
Host cell target proteins fall into two groups: those recognized only by E1B 55k (e.g. Mre11) and those that can be independently bound by E1B 55k and E4 34k (e.g. p53). While p53 can be bound by E4 34k in the absence of E1B 55k, p53 is not efficiently degraded without the participation of E1B 55k. It is generally thought that E1B 55k mediates selection of host factors and delivery to the E4 34k ubiquitin E3 ligase. We have previously shown that E1B 55k can bind ligase IV in the absence of E4 34k (Baker et al. 2007). Here, we show that, like p53, ligase IV can also be recognized by E4 34k and is not efficiently degraded in the absence of E1B 55k. Our findings suggest an unappreciated E1B-independent role for E4 34k in selection of some substrates of the viral ubiquitin E3 ligase prior to ubiquitination and degradation of ligase IV. Our observations also indicate that structural similarities may exist between p53 and ligase IV that facilitate recognition by E1B 55k and E4 34k. Investigation of interactions between host and viral factors often illuminates biologically relevant aspects of host-factor biochemistry. To gain further insight into the stability of the ligase IV complex, we are presently working to separate the effects of ligase IV selection from those of ligase IV binding by the viral ubiquitin E3 ligase to determine which of these two steps results in dissociation of ligase IV from XRCC4. We are also working to determine the minimal requirements for recognition of ligase IV by E4 34k.
Assembly of the E4 34k/Cul5/Rbx1/Elongin BC ubiquitin E3 ligase has been studied in great detail, and it appears that only E4 34k and E1B 55k are necessary for degradation of p53 and ligase IV (Querido et al. 2001, Blanchette et al. 2004, Baker et al. 2007, Cheng et al. 2007). In attempting to elucidate the minimal requirements for E1B 55k-independent E4 34k-mediated dissociation of the ligase IV/XRCC4 complex, we have determined that while expression of E4 34k in human cells resulted in radiosensitivity (Hart et al. 2005), the ligase IV/XRCC4 complex was intact. These findings suggest that one or more additional factors, of host or virus origin, are required for dissociation of the ligase IV/XRCC4 complex. It has been shown that during H5dl1016 infection, adenoviral late proteins were not produced at detectable levels and viral DNA and late mRNA accumulation was severely impaired (Bridge and Ketner 1990). The observation that ligase IV and XRCC4 were separated in lysates prepared from H5dl1016-infected cells allows us to rule out the participation of adenovirus late proteins in ligase IV/XRCC4 complex dissociation. Taken together, our data indicate that one, or more, early viral proteins or virus-induced host proteins may collaborate with E4 34k to dissociate the ligase IV/XRCC4 complex in the absence of E1B 55k.
Materials and methods
Cell culture and viral infections
HeLa monolayer cell cultures were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 10 μg/mL penicillin, and 10 μg/mL Streptomycin. Wild type Ad5 and E4 mutants used in these experiments (H5dl1004, H5dl1013, H5dl1014, H5dl1015 and H5pm1020; and the E1B 55k deletion mutants H5dl1016 and H5dl110) have been described previously (Babiss and Ginsberg 1984, Bridge and Ketner 1989, Bridge and Ketner 1990, Medghalchi et al. 1997). The viruses were propagated on W162 cells, and titered as plaque forming units (PFU)/mL. For viral infection, culture media was aspirated and replaced with fresh FBS-free media and the virus was added at 20 PFU/mL. Virus was allowed to adsorb for 2 h after which time the media was aspirated and replaced with DMEM containing 10% FBS. The infections were carried out for 20 h post-infection (HPI). RKO (a generous gift from Steve Balin, JHMI) and RKO-E4 cells (a generous gift from Constantinos Koumenis, U. Penn.) were cultured in McCoy's 5A media with 10% FBS. RKO-E4 cultures also contained 100 μg/mL Neomycin Sulfate.
Lysate (extract) preparation and immunoprecipitation
HeLa cells (7 × 105) were seeded in 35 mm dishes and infected as described above. Following infection, cells were harvested by scraping, washed twice with ice cold phosphate buffer saline (PBS) and lysed in 200 μL of ice cold NP40 lysis buffer (50 mM Tris HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 10% glycerol, 1 mM DTT and 1% NP40) for 20 min at 4 °C by end-over-end mixing. Cellular debris was removed by centrifugation (16,500 ×g, 10 min, 4 °C). For immunoprecipitation (IP), 150-200 μg of the extract was treated with the respective antibody for 2 h at 4 °C with gentle end-over-end mixing. XRCC4 IPs: rabbit polyclonal XRCC4 (Novus), 1:200 dilution. DNAPKcs IPs: rabbit polyclonal anti-DNA-PKcs (Serotec), 1:200 dilution. Elongin C IPs: anti-Elongin C mAb (BD transduction laboratories), 1:10 dilution. Elongin B IPs: rabbit polyclonal anti-Elongin B (Santa Cruz), 1:10 dilution. Cullin5 (Cul5) IPs: anti-Cul5 mAb (described below), 1:100 dilution. For control IP's, FBS was added in place of antibody. 15% of the treated extract was saved as the input and 20- 30 μl of washed protein-A sepharose beads (GE) was added after being washed 3 times in lysis buffer. The mixture was then incubated for 1 h at 4 °C with gentle end-over-end mixing. The samples were washed 3-8 times in lysis buffer, and resuspended in 2× protein sample buffer.
Preparation of mini whole-cell extracts (mWCE) and DNA binding assay
Mini whole-cell extracts (mWCE) were prepared as previously described (Smeaton et al. 2007). Briefly, HeLa cells, cultured as monolayers, were harvested using a cell scraper, washed twice in twice in PBS, snap frozen on dry ice and stored at -80 °C. Frozen cell pellets were resuspended in 70-90 μL of hypotonic lysis buffer (10 mM Tris pH 8.0, 1 mM EDTA), incubated on ice for 20 min then subject to vigorous vortexing for 30 sec. Nuclei were collected by gentle centrifugation (1300 ×g, 2 min, room temp) and the supernatant was reserved. Nuclei were resuspended in 35-45 μL of nuclear extract buffer (25 mM Tris pH 8.0, 0.33 M KCl, 1.5 mM EDTA) and incubated on ice for 20 min. The reserved cytoplasmic extract was added back to the nuclei, cell debris was removed by centrifugation (16,500 ×g, 10 min, 4 °C) and the resulting supernatant was collected as mWCE. For DNA-binding assays, 20 μL of a 50% native-DNA cellulose (SIGMA) slurry was added directly to 50 μg of mWCE, incubated for 3 h at room temperature with turning. DNA cellulose and DNA-bound species were collected by centrifugation, washed 3 times in 1 mL HEK buffer (20 mM HEPES, pH 7.6, 0.1 M KOAc, 0.5 mM EDTA) and DNA-bound proteins were eluted with 2× protein sample buffer.
Western blotting and antibodies
For detection of adenoviral late proteins, ligase IV or XRCC4, samples were heated to 100 °C for 5 min, resolved by SDS-PAGE (8%) and transferred to 0.45 μM PVDF membrane in the absence of SDS. Adenoviral late proteins were detected using rabbit polyclonal antibody (1:2000). XRCC4 was detected with rabbit polyclonal anti-XRCC4 (Serotec, 1:3000). Ligase IV was detected with rabbit polyclonal anti-ligase IV (Serotec, 1:1000). For detection of Elongins B and C, samples were resolved by 15% SDS-PAGE and transferred to 0.2 μM PVDF membrane in the absence of SDS. Elongin C was detected with anti-Elongin C mAb (BD transduction laboratories, 1:500). Elongin B was detected with rabbit polyclonal anti-Elongin B (Santa Cruz, 1:1000). For detection of E4 34k, samples were not heated prior to fractionation by SDS-PAGE (15%), after which they were transferred to PVDF membrane in the presence of 0.037% SDS and E4 34k was detected using anti-E4 34k rabbit polyclonal antibody raised against C-terminal octapeptide; 1:1000 dilution. Generation of monoclonal antibody against human Cullin5 (anti-Cul5 mAb) (Yu et al. 2003): cDNA encoding 138 amino acids of the N-terminus of Cul5 was amplified by PCR with the forward primer 5′-ggatcccatggcgacgtctaatctg-3′ and reverse primer 5′-gaattccctaaagctttcgaacaatactg-3′. The cDNA was inserted into the expression vector pRSETB, and the sequences were confirmed by direct DNA sequence analysis. E. coli strain BL21(DE3) (Invitrogen) was used as host to express the Cul5-His fusion protein. The soluble fusion protein was obtained by IPTG (0.5 mM) induction at 37 °C for 3 h and purified using the ProBondTM purification system (Invitrogen) according to manufacturers instructions. BALB/c mice were immunized with purified protein to produce polyclonal antibodies according to conventional procedures: 100 μgof protein was injected into multiple subcutaneous sites, and mice were boosted twice at 2-week intervals. Seven days after the last injection, anti-Cul5 serum was obtained, and the titer and specificity were determined by immunoblotting using purified Cul5-His protein. Splenic lymphocytes from the mouse with highest anti-Cul5 antibody titer were fused with murine myeloma cells using polyethylene glycol 1000, hybridomas were selected in medium containing hypoxanthine, thymidine, and aminopterin, and the media were screened by ELISA using purified Cul5-His. One of monoclonal antibody against human Cul5 was confirmed by immunoblotting using purified Cul5-His fusion protein and was used in this study.
Acknowledgments
We thank Amy Baker, Joyce Cheung, B.T. Rantipole and Brenda Salerno for many thoughtful discussions. We would also like to thank Steve Balin (JHMI) and Constantinos Koumenis (U. Penn.) for their generous gifts of RKO and RKO-E4 cells. This work was supported by the National Institutes of Health (GM070639-1 to LAH), (5R01CA082127 to GK), and by the Johns Hopkins University Bloomberg School of Public Health Faculty Research Initiatives Fund (to LAH and GK).
References
- Ahel I, Rass U, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006;443(7112):713–716. doi: 10.1038/nature05164. [DOI] [PubMed] [Google Scholar]
- Ahnesorg P, Smith P, et al. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Andres SN, Modesti M, et al. Crystal structure of human XLF: a twist in nonhomologous DNA end-joining. Mol. Cell. 2007;28(6):1093–1101. doi: 10.1016/j.molcel.2007.10.024. [DOI] [PubMed] [Google Scholar]
- Araujo FD, Stracker TH, et al. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 2005;79(17):11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiss LE, Ginsberg HS. Adenovirus type 5 early region 1b gene product is required for efficient shutoff of host protein synthesis. J. Virol. 1984;50(1):202–212. doi: 10.1128/jvi.50.1.202-212.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker A, Rohleder KJ, et al. Adenovirus E4 34k and E1b 55k Oncoproteins Target Host DNA Ligase IV for Proteasomal Degradation. J. Virol. 2007;81(13):7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, et al. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- Berk AJ. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene. 2005;24(52):7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- Blanchette P, Cheng CY, et al. Both BC-box motifs of adenovirus protein E4orf6 are required to efficiently assemble an E3 ligase complex that degrades p53. Mol. Cell. Biol. 2004;24(21):9619–9629. doi: 10.1128/MCB.24.21.9619-9629.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J, Rohleder K, et al. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology. 1999;263(2):307–312. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- Bridge E, Ketner G. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 1989;63(2):631–638. doi: 10.1128/jvi.63.2.631-638.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge E, Ketner G. Interaction of adenoviral E4 and E1b products in late gene expression. Virology. 1990;174(2):345–353. doi: 10.1016/0042-6822(90)90088-9. [DOI] [PubMed] [Google Scholar]
- Bryans M, Valenzano MC, et al. Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by XRCC4. Mutat. Res. DNA Repair. 1999;433(1):53–58. doi: 10.1016/s0921-8777(98)00063-9. [DOI] [PubMed] [Google Scholar]
- Buck D, Malivert L, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, et al. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22(24):6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, et al. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002;21:2827–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, Blanchette P, et al. The adenovirus E4orf6 E3 ubiquitin ligase complex assembles in a novel fashion. Virology. 2007;364(1):36–44. doi: 10.1016/j.virol.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Costantini S, Woodbine L, et al. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) 2007;6(6):712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Critchlow SE, Bowater RP, et al. Mammalian DNA double-strand break repair protein Xrcc4 interacts with DNA ligase IV. Curr. Biol. 1997;7(8):588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- Deshpande RA, Wilson TE. Modes of interaction among yeast Nej1, Lif1 and Dnl4 proteins and comparison to human XLF, XRCC4 and Lig4. DNA Repair (Amst) 2007;6(10):1507–1516. doi: 10.1016/j.dnarep.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sekiguchi JM, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396(6707):173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, et al. Requirement for an interaction of Xrcc4 with DNA ligase IV for wild-type V(D)J recombination and DNA double-strand break repair in vivo. J. Biol. Chem. 1998;273(38):24708–24714. doi: 10.1074/jbc.273.38.24708. [DOI] [PubMed] [Google Scholar]
- Hart LS, Yannone SM, et al. The adenovirus E4orf6 protein inhibits DNA double strand break repair and radiosensitizes human tumor cells in an E1B-55K-independent manner. J. Biol. Chem. 2005;280(2):1474–1481. doi: 10.1074/jbc.M409934200. [DOI] [PubMed] [Google Scholar]
- Hsu HL, Yannone SM, et al. Defining interactions between DNA-PK and ligase IV/XRCC4. DNA Repair (Amst) 2002;1(3):225–235. doi: 10.1016/s1568-7864(01)00018-0. [DOI] [PubMed] [Google Scholar]
- Junop MS, Modesti M, et al. Crystal structure of the XRCC4 DNA repair protein and implications for end joining. EMBO J. 2000;19:5962–5970. doi: 10.1093/emboj/19.22.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CA, Agyei R, et al. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J. 2004;23(19):3874–3885. doi: 10.1038/sj.emboj.7600375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chirgadze DY, et al. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. EMBO J. 2007;27:290–300. doi: 10.1038/sj.emboj.7601942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Ma Y, et al. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst) 2004;3(89):817–826. doi: 10.1016/j.dnarep.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Walker SA, et al. Identification of a novel motif in DNA ligases exemplified by DNA ligase IV. DNA Repair (Amst) 2006;5(7):788–798. doi: 10.1016/j.dnarep.2006.03.011. [DOI] [PubMed] [Google Scholar]
- McElhinny SAN, Snowden CM, et al. Ku recruits the XRCC4-ligase IV complex to DNA ends. Mol. Cell. Biol. 2000;20(9):2996–3003. doi: 10.1128/mcb.20.9.2996-3003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medghalchi S, Padmanabhan R, et al. Early region 4 modulates adenovirus DNA replication by two genetically separable mechanisms. Virology. 1997;236(1):8–17. doi: 10.1006/viro.1997.8737. [DOI] [PubMed] [Google Scholar]
- Querido E, Blanchette P, et al. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15(23):3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RJ, Mohr I. Translation initiation and viral tricks. Trends Biochem. Sci. 2003;28(3):130–136. doi: 10.1016/S0968-0004(03)00029-X. [DOI] [PubMed] [Google Scholar]
- Smeaton MB, Miller PS, et al. Small-scale extracts for the study of nucleotide excision repair and non-homologous end joining. Nucleic Acids Res. 2007;35(22):e152. doi: 10.1093/nar/gkm974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev. 1999;13(8):916–934. doi: 10.1101/gad.13.8.916. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Hochegger H, et al. Differential usage of non-homologous endjoining and homologous recombination in double strand break repair. DNA Repair (Amst) 2006;5(910):1021–1029. doi: 10.1016/j.dnarep.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, et al. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418(6895):348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Weitzman MD, Ornelles DA. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene. 2005;24(52):7686–7696. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18(1):114–124. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- Yano K, Morotomi-Yano K, et al. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9(1):91–96. doi: 10.1038/sj.embor.7401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, et al. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302(5647):1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]