

Abstract
We found that TGF-β1, a cytokine that previously has been reported to have neuroprotective effects, was able to prevent the toxicity induced by the HIV-1 coat protein gp120 in hippocampal pyramidal neuron cultures. In the presence of glia, gp120 induced time- and dose-dependent cell death, which was more pronounced in mature (7–19 d in culture) than in young neurons (2–7 d in culture). Staining with nuclear dyes (propidium iodide and Hoechst 33342),in situ detection of DNA fragments, and DNA analysis on agarose gels indicated that apoptosis was mainly responsible for the death caused by the viral protein. However, after several days of treatment, death-displaying necrotic features also occurred. Neurotoxicity induced by gp120 was dependent on the activation of NMDA receptors and required the presence of glia as well as new protein synthesis. Thus, the effect of gp120 was abolished by the NMDA receptor antagonist APV and partially reduced by cycloheximide. Only modest neurotoxicity was observed in pure neuronal cultures deprived of the glia feeder layer. Fura-2-based videoimaging showed that treatment with gp120 enhanced the ability of NMDA to increase neuronal [Ca2+]i. The impairment of neuronal Ca2+ homeostasis was prevented completely by TGF-β1. Therefore, it is likely that the neuroprotective action of the cytokine is attributable to its ability to stabilize neuronal [Ca2+]i.
Keywords: AIDS, HIV-1, NMDA receptors, intracellular calcium, cell death, neurotoxicity
In a recent study on lymph nodes from HIV-infected children, Finkel et al. (1995) observed that DNA fragmentation was rare in productively infected cells and that the virus was detected rarely in apoptotic cells. This suggests that apoptosis occurs predominantly in uninfected cells. Similar conclusions come from studies on local cell death in lymph nodes from mice infected by different HIV-1 isolates (Mosier et al., 1993; Reinhardt et al., 1994). Only the death of uninfected “bystander” cells was noted. It was observed that the differences between HIV-1 isolates that determine the rate of CD4+ T-cell loss mapped to the gene encoding the HIV-1 envelope protein gp120.
An analogous situation occurs in the central nervous system of AIDS patients. Despite the inability of HIV-1 to infect neurons directly, severe neuronal loss occurs in different brain areas, resulting in multiple types of neurological disorders (Navia et al., 1986; Price et al., 1988; Wiley et al., 1991). In vitro studies on central neurons have found that gp120, probably shed by the virus and/or by infected microglia in vivo, produces neurotoxicity in several types of rodent and human neurons (e.g., hippocampal, retinal ganglion, cortical, and cerebellar granule neurons) (Brenneman et al., 1988; Dreyer et al., 1990; Lipton et al., 1991; Muller et al., 1992;Savio and Levi, 1993). Also, the pathogenic potential of gp120 has been demonstrated in the intact brain by inducing the expression of the viral coat protein in GFAP–gp120 transgenic mice (Toggas et al., 1994). However, it should be noted that, in this study, only gp120 mRNA, rather than protein, was detected and only in the nucleus and not in the cytoplasm. Thus, it is quite possible that the gliosis and subsequent neuronal damage found in this study resulted only from the expression of viral RNA.
The cellular mechanisms responsible for gp120-induced neurotoxicity are understood only partially. It is still unclear, for example, whether the envelope protein has a direct effect on neurons, as most of the studies have been performed on mixed cultures of neurons and glia. However, there is evidence showing that the presence of non-neuronal cells (macro and microglia) is necessary for HIV-related neurotoxicity to occur (for review, see Lipton, 1994a,b). Now, it generally is believed that the gp120 shed by the virus may interact with macrophages/microglia and astrocytes and stimulate the release of neurotoxins, “potentiating” the activation of NMDA receptors by endogenous glutamate. Ca2+ influx through voltage-sensitive Ca2+ channels as well as Ca2+-permeable glutamate receptors and the subsequent production of nitric oxide (NO) (Dreyer et al., 1990; Lipton et al., 1991; Dawson et al., 1993) seem to be involved in the toxic effects of gp120 on neurons. As a result, studies on the development of therapeutic agents that potentially could prevent HIV-associated neuronal damage have focused on glutamate receptor antagonists (Lipton, 1994b).
TGF-β1 is a cytokine with neuroprotective effects that is able to protect neurons from different kinds of insult both in vivoand in culture (Prehn et al., 1993, 1994, 1996). The protective action of this cytokine has been correlated with its ability to stabilize Ca2+ homeostasis and increase the expression of the anti-apoptotic proteins bcl-2 and bcl-xl. It was, therefore, of interest to examine the effect of TGF-β1 on gp120-induced neurotoxicity in rat hippocampal cultures. Picomolar concentrations of gp120 were found to cause significant neuronal injury in the presence of glia. A- poptotic as well as necrotic cell death occurred at the same time. A gp120-induced impairment in neuronal Ca2+ homeostasis was observed also. We found that TGF-β1 prevented the gp120-induced death as well as the neuronal Ca2+ overloading, consistent with the effects of this cytokine in other models of neurodegeneration (Prehn et al., 1994,1996).
MATERIALS AND METHODS
Hippocampal neuronal cultures
Treatments in the presence of glia. Neuronal cultures were prepared from the hippocampi of 17- to 18-d-old rat embryos. Dissection and dissociation were performed in a Ca2+/Mg2+-free solution (D1) containing glucose (3.25 gm/l), sucrose (7.5 gm/l), HEPES (10 mm), NaCl (8 gm/l), KCl (400 mg/l), Na2HPO4 (180 mg/l), KH2PO4 (30 mg/l), pH 7.4 (320–330 mOsm). Tissues were incubated for 20 min in 0.25% (w/v) trypsin, washed twice in D1, and dissociated by trituration in the presence of DNase (150 μg/ml). Cells were plated in DMEM containing 10% horse serum at a density of 5 × 104cells/cm2 on 15 mm glass coverslips coated with polylisine (1 mg/ml). After 2–4 hr, coverslips were transferred to plates containing N2.1-defined media (2 ml) and a confluent bed of astroglia (Abele et al., 1990). AraC (10 μm) was added to the cultures within 48 hr from plating. Treatments were started after 2 or 7 d in culture by replacing 500 μl of culture medium with fresh medium plus gp120 and/or the other drugs.
Treatments in the absence of glia. Neurons treated in the absence of glia were dispersed and plated as above. After 2–4 hr, they were moved to 35 mm culture dishes containing defined neurobasal medium (Life Technologies, Gaithersburg, MD) supplemented with 0.5 mml-glutamine, 25 μm glutamate, and 5 μg/l human fibronectin. In these cultures, gp120 was added at 7 d in culture (DIC).
When gp120 was added at 2 DIC, neurons were cultured in the presence of glia for the first 48 hr. Before gp120 was added, coverslips with neurons were moved to a new culture dish containing their old, conditioned medium (DMEM + N2.1).
Cell survival and apoptosis detection
Fluorescein diacetate (10 μm) and propidium iodide (5 μm) were used to stain viable and dead cells, respectively. Hoechst 33342 (5 μm) was used to evaluate differences between normal and apoptotic nuclei. Cells were counted by using a Leitz microscope (Nuhsbaum, McHenry, IL) with optics for fluorescence and Nomarski. Five to ten microscopic fields were counted for each coverslip, and two to three coverslips per treatment were used for each experiment. Because the cell death induced by gp120 occurred progressively and was caused mainly by apoptosis (see below), only neurons positive to fluorescein were counted for the final analysis of cell survival. Indeed, long treatments were necessary to see a significant effect of gp120, and dead cells could have been removed by changing the culture medium and/or by the action of microglia. Furthermore, the maintenance of the membrane integrity in the early stage of apoptosis did not allow the entrance of propidium iodide into the nucleus of apoptotic cells. Also, the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) technique (Gavrieli et al., 1992) for in situ apoptosis detection was used to determine the number of apoptotic cells. Briefly, cells were fixed with Bouin’s solution (Sigma, St. Louis, MO) for 5 min at room temperature, permeabilized with 1% Tween (15 min), incubated with terminal deoxytransferase (TdT) enzyme (1 hr at 37°C) and with anti-digoxygenin–peroxidase (30 min at room temperature), and finally stained using 0.05% DAB (5 min).
Analysis of DNA on agarose gel
Neurons were grown on 35 mm culture dishes previously coated with polylisine (0.5 mg/ml), and glia was cultured on perforated plastic coverslips, as described previously (Prehn et al., 1994). Treatments started after 7 d from plating by the direct addition of gp120 (100×) to the culture media. After 72 hr, coverslips with glia were removed, and neurons were scraped and collected in centrifuge tubes. Neurons (8 × 106 cells per treatment) were lysed at 37°C in a buffer containing 0.5% Triton X-100 and 20 mm EDTA, pH 7.4, and then centrifuged at 4°C for 10 min in a microfuge (14,000 rpm). Then the supernatant was incubated for 4 hr at 50°C in the presence of proteinase K (200 μg/ml); subsequently, DNA was extracted by adding an equal volume of phenol and precipitated with 10% ammonium acetate (3 m) and 2.5 vol of absolute ethanol (−70°C for at least 15 min). Samples were loaded in a 1.5% agarose gel after a 30 min incubation at 37°C and a 10 min incubation at 65°C in the presence of RNase (10 mg/ml).
Fura-2 videoimaging
Cells were loaded with 2 μm fura-2 acetoxymethylester using a balanced salt solution (standard buffer) of the following composition (in mm): NaCl 159, KCl 5, MgSO4 0.4, MgCl2 0.5, KH2PO4 0.64, NaHCO3 3, HEPES 20, glucose 5, Na2HPO4 0.33, CaCl2 2, and BSA 0.2% (330 mOsm/kg), pH-adjusted to 7.35. The cells were incubated with fura-2 for 30 min at room temperature to avoid probe compartmentalization and then incubated for a further 30 min at room temperature to allow deesterification of the fura-2 dye. Coverslips were mounted on a coverslip chamber for fluorescence measurements. All measurements were made at room temperature, as previously described (Meucci et al., 1995), using standard buffer supplemented with 10 μmglycine. Each cell in the image was analyzed independently for each time point in the captured sequence. All individual cell [Ca2+]i traces shown are representative responses for a given field of cells. For the calibration of fluorescent signals, we used cells loaded with fura-2;Rmax and Rmin, ratios at saturating and zero Ca2+, respectively, were obtained by perfusing cells with standard buffer containing 10 mm CaCl2 and 4 μm ionomycin and, subsequently, with a Ca2+-free solution containing 10 mm EGTA. The values of the obtainedRmax and Rmin, expressed as gray-level mean, were used to calculate the calibration curve by TARDIS software. The [Ca2+]i was determined according to the equation of Grynkiewicz et al. (1985).
Materials
Recombinant HIV-1 gp120 from a baculovirus expression system was purchased from Intracel Corporation (Cambridge, MA) as lyophilized powder. Aliquots of reconstituted protein (100 ng/μl) were stored at −70°C. Working solutions (100× in PBS) were prepared just before starting treatments and added to the neurons as described in Materials and Methods. TGF-β1 was purchased from R & D Systems (Minneapolis, MN). TdT enzyme and anti-digoxygenin–peroxidase were purchased from Oncor (Gaithersburg, MD). DMEM, neurobasal medium, and sera were obtained from Life Technologies. Propidium iodide and Hoechst 33342 were obtained from Molecular Probes (Eugene, OR). All the other reagents, unless otherwise specified, were purchased from Sigma.
Statistical analysis
One way ANOVA and the Student–Newman–Keuls method have been used for the analysis of survival experiments, and the Student’st test has been performed for the analysis of calcium experiments.
RESULTS
The effect of recombinant gp120 on the survival of hippocampal pyramidal neurons depends on the age of the culture and the presence of glia
Purified gp120 has been shown to cause significant neuronal cell loss in rodent hippocampal cultures (Brenneman et al., 1988; Dreyer et al., 1990). To see whether the neurotoxic effect of gp120 was related to the age of the cultures and/or to the presence of non-neuronal cells, we treated rat hippocampal pyramidal neurons with recombinant gp120 under different experimental conditions. Treatments were started after either 2 DIC or 7 DIC, both in the presence and in the absence of a glia feeder layer (see Materials and Methods). Cells were counted from 4 to 12 d after the addition of gp120. Two concentrations of gp120, 20 and 200 pm (2.4 and 24 ng/ml, respectively), were used.
In the presence of glia, the addition of gp120 to differentiated neurons (7 DIC) that showed a complex network of neurites clearly affected neuronal survival and caused a time- and dose-dependent reduction in the number of surviving cells (Fig.1A). Neuronal death developed slowly, and at least 4–5 d of treatment were required to see a significant reduction in the number of viable cells.
Fig. 1.

Effect of recombinant gp120 on the survival of differentiated hippocampal neurons. Treatment started at 7 DIC in the presence (A) or in the absence (B) of glia (see Materials and Methods). Survival was evaluated 4, 5, 6, and 12 d after the addition of gp120 in A and after 4, 6, and 9 d in B. Data shown in the figure are mean ± SEM and come from three different experiments for each treatment. (*p < 0.05 vs control).
Experiments on younger cultures in the presence of glia (treatments started at 2 DIC) showed that immature neurons were less sensitive to gp120 toxicity. For treatments shorter than 7 d, only the highest concentration of gp120 induced significant cell death. After longer treatments, both 20 and 200 pm gp120 effectively reduced the percentage of live cells (Table1).
Table 1.
Effect of gp120 on neuronal survival in young cultures
| Cell survival (% of control) | |||
|---|---|---|---|
| After 4 d | After 6 d | After 9 d | |
| gp120 (20 pm) | 84 ± 9 | 88 ± 12 | 64 ± 12* |
| gp120 (200 pm) | 76 ± 15 | 62 ± 16* | 33 ± 15* |
Treatment of neurons with gp120 started at 2DIC in the presence of glia. The number of surviving cells (stained by fluorescein diacetate) was assessed at the indicated times. Data are mean ± SEM (n = 3, *p < 0.05).
To test the effect of gp120 in the absence of glia, two different experimental protocols were used for immature (2 DIC) and mature (7 DIC) neurons, because the older cultures could not be deprived of glia without affecting their survival. Thus, when gp120 was added at 2 DIC, neurons were grown for the first 48 hr in the presence of the glia feeder layer. At the time of treatment, neurons were moved to a culture plate without glia while still in the presence of their conditioned medium. In these experiments, a modest effect of the gp120 was observed (80 ± 2.9 and 74 ± 4.4% survival for 20 and 200 pm, respectively, vs control after 8 d of treatment; n = 3). Similar results were obtained with mature neurons grown in the absence of glia by the use of a chemically defined medium (see Materials and Methods) and treated with gp120 at 7 DIC (Fig. 1B). In these experiments, cell survival after a 4 d treatment with gp120 was ∼20% less than for untreated neurons (control = 100%, 20 pm gp120 = 72 ± 3.7%, 200 pm gp120 = 85 ± 2.5%;n = 4). Longer treatments with gp120 did not produce a further decrease in cell survival. (Maximum length of treatment was 9 d.)
Both apoptosis and necrosis are responsible for gp120-induced neuronal death
Although necrosis and apoptosis generally have been considered to be two distinct types of cell death exhibiting different biochemical and morphological characteristics, sometimes it can be difficult to evaluate which cell-death type is responsible for cell loss. Thus, both necrotic and apoptotic cell death may coexist in the same cell population (Bonfoco et al., 1995; van Lookeren Campagne et al., 1995). This seems to be the case for the neuronal damage induced by gp120 under our experimental conditions. Neuronal injury was characterized by a progressive decrease in the number of viable neurons, a reduction in the neuritic network, the appearance of cell shrinkage, and an increase in the number of dead cells displaying a condensed or fragmented nucleus (Figs. 2, 3). Typically, these are features that are associated with apoptosis. Less frequently, morphological alterations characteristic of necrosis were observed, including groups of cells that had lost their nuclear membrane integrity and displayed an enlarged nucleus that stained with propidium iodide (Fig. 2E). A large number of dead cells with enlarged nuclei occurred, particularly at the higher concentration of gp120 (200 pm). During the first 3 d of treatment, the number of dead cells was quite low. This may be attributable to the fact that, at early stages of apoptosis, the integrity of the cell membrane is maintained and cells are not stained by propidium iodide. We therefore performed experiments using the dye Hoechst 33342. This compound is able to enter the nuclei of both living and dead cells. We found that, after a 24–48 hr treatment with gp120, most of the cells stained with Hoechst 33342 but not with propidium iodide, and their nuclei seemed normal. However, after 72 hr, alterations in the nuclear density of the cells stained exclusively by Hoechst 33342 became evident (Fig. 2A–D, arrows), and cells with enlarged nuclei also were present (Fig. 2E,F,arrows). At this stage, sometimes “blebs” on the neurites were visible (Fig. 2C). Staining of neurons after prolonged treatments (4–5 d or more) showed that all of the dead cells were stained with both dyes and had condensed and/or fragmented nuclei (data not shown).
Fig. 2.

Alterations in the nuclear density of neurons during treatment with gp120. Neurons are stained with fluorescein diacetate (yellow/green), propidium iodide (red), and Hoechst 33342 (white/blue). After 72 hr of treatment, the first changes in the nuclei, which are still negative to propidium iodide, are visible (A–D, large arrows). At this stage, blebs on the neurites are also present (C, small arrow). Dead neurons (not stained with fluorescein diacetate; A, B, small arrows) are stained with both propidium iodide and Hoechst 33342 and display either an enlarged (E, F, smallarrows) or a fragmented (E, F,large arrows) nucleus. Note that nuclei stained with both dyes are brighter (white) than normal nuclei, which are stained only with Hoechst 33342 (blue) (magnification, 500×).
Fig. 3.
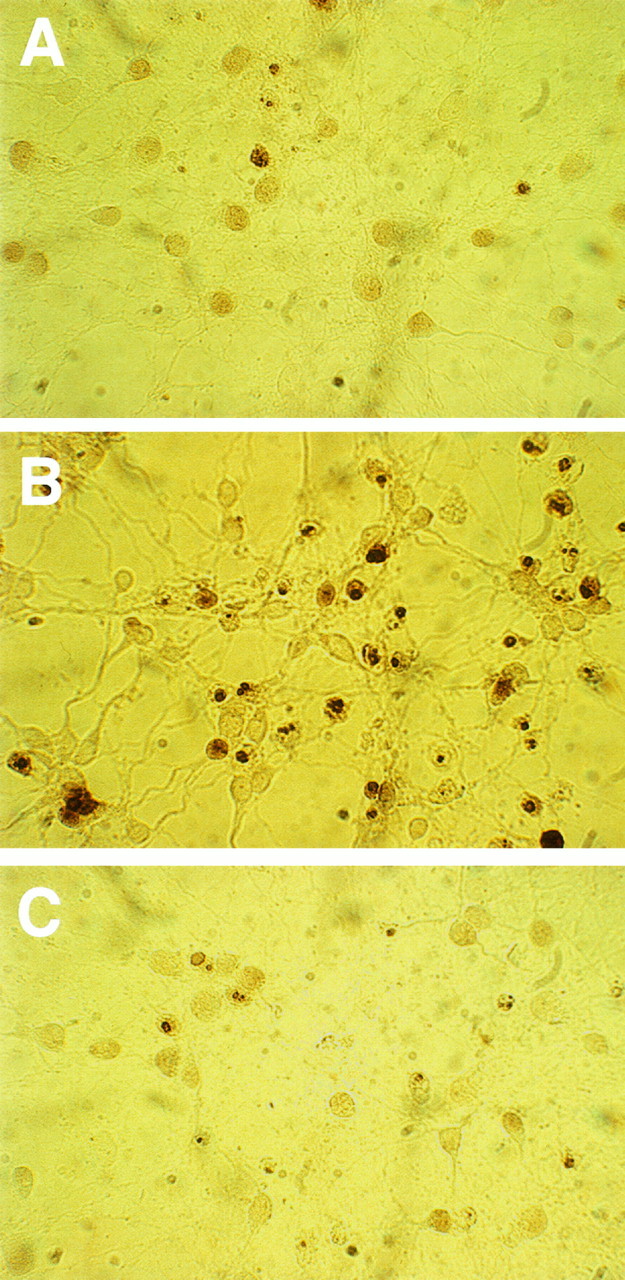
In situ DNA fragmentation induced by gp120 in hippocampal neurons assessed by the TUNEL technique.A, Control neurons at 11 DIC; B, gp120-treated neurons (20 pm); C, gp120/TGF-treated (5 ng/ml) neurons. Treatments were started at 7 DIC.
In situ labeling for DNA fragmentation, assessed by using the TUNEL technique, also showed that gp120 greatly increased the number of apoptotic cells, as shown in Figure 3. (Also note that most of the cells stained show condensed nuclei.) Using this method, we estimated that apoptotic cell death increased from 10 ± 3.6% in control to 47 ± 5.5 and 50 ± 2.6% in neurons treated for 4 d with 20 and 200 pm gp120, respectively (n = 3; 307 cells for control, 142 cells for 20 pm, and 148 cells for 200 pm gp120). Similar results have been described very recently in hippocampal organotypic explants after a 48 hr treatment with gp120 (Charriaut-Marlangue et al., 1996).
Analysis of DNA on agarose gels also showed that soluble DNA obtained from neurons treated with gp120 for 3 d displayed a fragmentation pattern, although the amount of fragmented DNA was very low (data not shown). In summary, these data show that both apoptosis and necrosis may occur during gp120-induced neurotoxicity in rat hippocampal cultures. In particular, although apoptosis seems to represent the main pathway of death during the first 4 d, the situation becomes more complex after longer treatment and/or at high concentrations of gp120.
NMDA-receptor activation and protein synthesis are necessary for gp120-induced neuronal death
There is evidence suggesting that the neuronal damage caused by gp120 is related to its ability to potentiate glutamatergic neurotransmission, Ca2+ influx, and NO production (Dreyer et al., 1990; Lipton et al., 1991; Dawson et al., 1993). Ca2+ and NO seem to play major roles in several types of neurotoxicity associated with both apoptotic and necrotic forms of cell death (Choi, 1988; Randall and Thayer, 1992; Bonfoco et al., 1995; Scorziello et al., 1996) .
Consistent with these findings, the NMDA receptor antagonist 2-amino-5-phosphonovalerate (APV; 25 μm) was able to inhibit NMDA-stimulated [Ca2+]i increases by ∼70% under our experimental conditions (data not shown). As shown in Figure 4, APV also was able to almost completely prevent neuronal death caused by gp120 (95 ± 4% viability in the presence of 20 pm gp120 and 91 ± 6.7% in the presence of 200 pm gp120), thus confirming previous observations suggesting a role of NMDA receptors in gp120-mediated neurotoxicity (Lipton, 1994b).
Fig. 4.

Effect of the NMDA receptor antagonist APV (25 μm) and the protein synthesis inhibitor CHX (0.5 μg/ml) on gp120-induced neurotoxicity of mature hippocampal cultures. Treatments started at 7 DIC, and survival was assessed after 5 d. Data are expressed as mean ± SEM from three experiments (*p < 0.05 vs control).
The protein synthesis inhibitor cycloheximide (CHX; 0.5 μg/ml) also was able to reduce gp120-induced neurotoxicity, although it caused a 10–15% decrease in neuronal survival by itself (Fig. 4). The protective action of CHX could be caused by interference with some active processes in the cell-death program triggered by gp120, as reported for other cases of apoptosis (Martin et al., 1988; Galli et al., 1995) or by prevention of oxidative stress-induced apoptosis (Ratan et al., 1994). Alternatively, CHX merely might decrease the release of gp120-induced neurotoxins produced by non-neuronal cells (Giulian et al., 1993).
Impairment of intracellular calcium responses caused by long-term treatment with gp120
Because NMDA receptor activation is an important event in gp120 neurotoxicity, we studied the effect of the long-term treatment (3–4 d) with gp120 on NMDA-induced [Ca2+]i responses in hippocampal neurons. Figure 5A shows the typical [Ca2+]i response we observed on the addition of NMDA (50 μm, 4 min) in most control neurons (82%)—a rapid increase of [Ca2+]i (1672 ± 120 nm; mean ± SEM; n = 59) and a plateau (849 ± 396 nm), which returned to basal levels after agonist removal (see also Prehn et al., 1994).
Fig. 5.
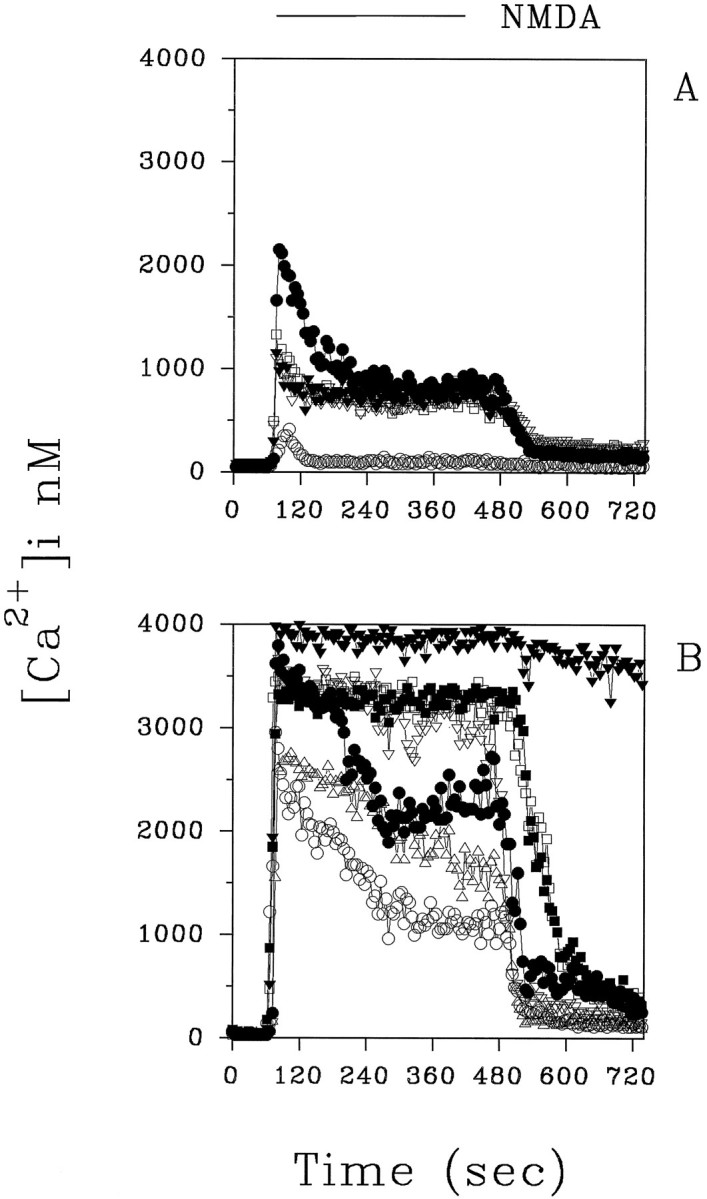
Typical NMDA-evoked (50 μm) [Ca2+]i increases in control neurons (A) and in gp120-treated neurons (B). Experiments were performed at 10 DIC in the presence of glycine (10 μm) and Mg2+(see Materials and Methods). Treatment with gp120 (20 pm) started at 7 DIC. Representative traces from control and treated neurons are shown in the figure.
In neurons treated previously with gp120 (3 d, 20 pm), the increases in [Ca2+]i evoked by NMDA were more pronounced [2722 ± 186 nm (mean ± SEM; n = 47; p < 0.05) vs 1672 ± 120 nm] and frequently were continuous with a high plateau (2724 ± 833 nm). Indeed, only 36 ± 9% of the neurons examined exhibited a “peak and plateau” [Ca2+]i response to NMDA as in untreated neurons. In the majority of neurons (64%), a monophasic response of high amplitude occurred (Fig. 5B). Only 5 of 52 cells examined showed a [Ca2+]i response within the range of those observed in control neurons (peak [Ca2+]i value, 1476 ± 226 nm). In control neurons, a second stimulus with NMDA, 15 min after washout of the first challenge, also induced a peak and plateau slightly smaller than the initial response (first NMDA peak, 1864 ± 138 nm; second NMDA peak, 1194 ± 120 nm; mean ± SEM; n = 41;p < 0.05) (Fig. 6A,B). We rarely observed a reduction in the [Ca2+]i response to a second NMDA challenge in most of the gp120-treated neurons (Fig.6C,D). Indeed, many cells did not recover at all after the second NMDA stimulus (data not shown). The [Ca2+]i rise induced by the first NMDA application in these cells was 2763 ± 132 nm; mean ± SEM; n = 38. The [Ca2+]i response to the second NMDA stimulus was just as large in 79% of neurons (2437 ± 160 nm; mean ± SEM; n = 30), and it was reduced in the remaining cells (1166 ± 133 nm; mean ± SEM; n = 8;p < 0.05).
Fig. 6.

Representative traces of the effect of two consecutive NMDA stimuli on [Ca2+]i in control (A, B) and gp120-treated (C,D) neurons. The second NMDA application (B,D) occurs 15 min after washout of the first challenge.
Despite the marked effect of long-term treatment with gp120 on the NMDA-induced [Ca2+]irise, we never observed potentiation of NMDA responses after short incubations (30 sec to 30 min) with the protein under the same experimental conditions. As reported previously by Lo et al. (1992), we found that the addition of gp120 to neurons induced [Ca2+]i spikes or oscillations by itself in a small percentage of cells (<5%; data not shown). Previously, we found that analogous [Ca2+]i transients also were evoked by β-amyloid peptide 25-35 (Brorson et al., 1994).
Also, we tested the effect of gp120 on [Ca2+]i increases induced by KCl (50 mm). In neurons treated for 3 d with 20 pm gp120, we found that exposure to the glycoprotein did not affect the [Ca2+]i responses evoked by KCl in the presence of 25 μm APV (data not shown). However, in the absence of APV, the [Ca2+]i increase evoked by KCl was enhanced slightly in gp120-treated neurons. In these experiments, the peak [Ca2+]i value was 729 ± 58 nm in control neurons (n = 12) and 1021 ± 120 nm in treated neurons (n = 5). Therefore, it is likely that an NMDA component (attributable to glutamate release induced by KCl depolarization) is involved in the [Ca2+]iresponse evoked by KCl. This suggests that the action of gp120 is specific for NMDA-induced [Ca2+]i signals.
TGF-β1 prevents gp120-induced neuronal death and Ca2+ overload
Previous studies from this laboratory have shown that the cytokine TGF-β1 protects primary hippocampal neurons from a number of insults (Prehn et al., 1994, 1996). This is associated with increased expression of bcl-2 and bcl-xl as well as other effects (Prehn et al., 1994, 1996). Therefore, we tested the effect of this cytokine on gp120-induced neuronal damage. As illustrated in Figure 7, we found that TGF-β1 (5 ng/ml) was able to block the neurotoxicity produced by both 20 and 200 pm gp120. The viability of TGF-β1-treated neurons in the presence of 20 and 200 pm gp120 was 101 ± 8% and 88 ± 6%, respectively (44 microscopic field observed in three different neuronal preparations). In situdetection of fragmented nuclei also showed that TGF-β1 was able to counteract apoptosis induced by gp120 (Fig. 3C). Only 14 ± 4% of the cells treated with gp120 (20 pm) in the presence of TGF-β1 were positive by TUNEL staining (n = 57; Fig. 8).
Fig. 7.

Effect of TGF-β1 (5 ng/ml) on gp120-induced neurotoxicity. Data are from three different experiments and show the percentage of survived cells after 5 d of treatment (mean ± SEM; *p < 0.05 vs control). Treatments with vehicle, gp120, and TGF-β1 started at 7 DIC.
Fig. 8.

Inhibition of gp120-induced apoptosis (20 pm for 5 d) by TGF-β1 (5 ng/ml). The number of apoptotic cells was assessed by in situ detection of fragmented nuclei. Data are mean ± SEM (*p < 0.05 vs gp120 alone) from three different neuronal preparations.
As TGF-β1 can enhance acutely the [Ca2+]i buffering in hippocampal neurons exposed to NMDA (Prehn et al., 1994), we tested whether it stabilized Ca2+ homeostasis in gp120-treated cultures. Neurons cultured in the presence of TGF-β1 (5 ng/ml) and gp120 (20 pm) showed [Ca2+]i responses to NMDA similar to those observed in control cells. In fact, in 78% of these neurons, the [Ca2+]ipeaks evoked by the first and the second NMDA challenge were 1872 ± 129 nm and 1399 ± 162 nm, respectively (n = 30; p < 0.05). In all cases the average of [Ca2+]i peaks induced by NMDA was significantly lower than in gp120-treated cells [2165 ± 139 (n = 37; p < 0.05) vs 2633 ± 135] (Fig. 9). Although many cells (43%) still showed relatively high [Ca2+]iduring the plateau phase, they never approached the values observed in neurons treated with gp120 alone (1431 ± 862 vs 2724 ± 833 nm). All of the cells examined in these experiments fully recovered after the NMDA washout. These results suggest that the neuroprotective action of TGF-β1 may be associated, at least in part, with its ability to stabilize Ca2+ homeostasis in these cells.
Fig. 9.
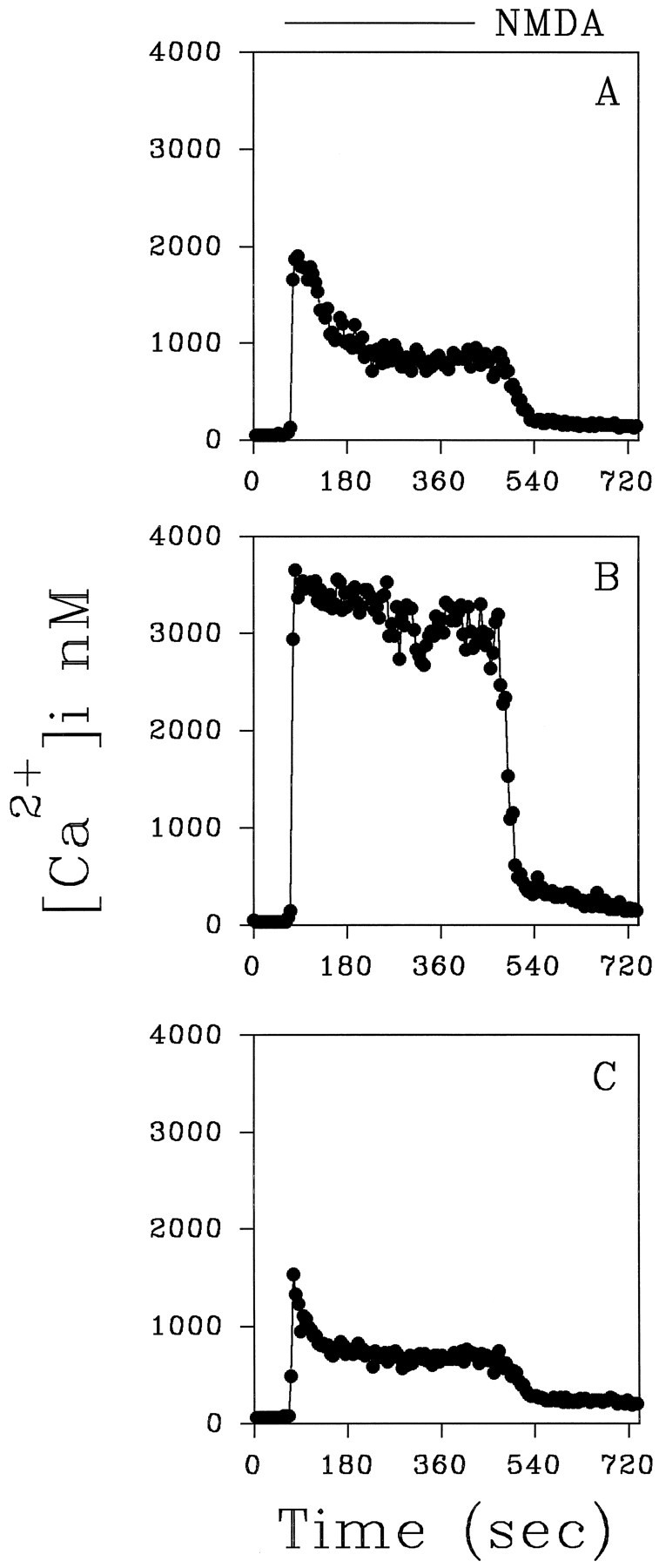
Representative traces showing the increase in [Ca2+]i induced by NMDA (50 μm) in neurons treated for 3 d with 20 pm gp120 in the absence (B) or in the presence (C) of TGF-β1 (5 ng/ml). A “peak and plateau” response similar to the one observed in control neurons (A) was found in the presence of TGF-β1.
DISCUSSION
The data presented in this study show that the neuronal injury caused by the HIV-1 coat protein gp120 on hippocampal neuronsin vitro is associated with features of both apoptosis and necrosis. Although the apoptotic pathway seems dominant, the intra/intercellular mechanisms leading to neuronal damage all result from the activation of NMDA receptors. Thus, the NMDA antagonist APV completely prevented gp120-induced neurotoxicity, consistent with previous data in the literature (Lipton et al., 1991; Lipton, 1994b). gp120 did not affect the survival of immature neurons (2–7 DIC), i.e., before the time they express NMDA receptors. NMDA-induced neurotoxicity usually has been associated with necrosis (Meldrum and Garthwaite, 1990). However, a growing corpus of data has shown that both apoptosis and necrosis can be involved in excitotoxicity (Bonfoco et al., 1995; van Lookeren et al., 1995). In particular, a study on cortical neurons showed that mild or intense NMDA stimuli can result in predominantly apoptotic or necrotic neuronal damage, respectively (Bonfoco et al., 1995). Therefore, if the enhancing effects of gp120 on the NMDA response in different neurons vary somewhat, this variation may result in neuronal death ultimately via one process or the other.
At least two or three different cell types, including diverse neuronal subtypes, microglia, and astocytes, are supposed to contribute to the neurotoxicity caused by gp120 in vitro (Merril and Chen, 1991; Dawson et al., 1993; Giulian et al., 1993). Evidence for a direct toxic effect of gp120 on neurons has never been clearly provided because of the presence of glia in neuronal cultures, although recent findings on human embryonic neurons support this possibility (Lannuzel et al., 1995). We found that the effect of gp120 was reduced in purified neuronal cultures devoid of glia, thus supporting the idea that glial cells play a major role in gp120-related neurotoxicity. However, even in these cultures, we observed a small reduction in neuronal survival. This suggests that gp120-induced neurotoxicity may be caused also by a direct action of the viral protein on neurons. As the neuronal damage observed under these conditions is modest and does not increase with the length of time, it may be that only a subpopulation of neurons is the target for a direct effect of gp120. Another explanation for the lower sensitivity of purified neuronal cultures to gp120 may be that, in cultures deprived of glia, a particular neuronal subtype that normally counteracts the effects of gp120 (i.e., GABAergic neurons) may occur. Finally, the possible contribution of a small amount of glial contamination (<5%) to the observed toxicity cannot be ruled out.
There have been some reports suggesting the presence of binding sites for gp120 on neurons (Bhat et al., 1991; Schneider-Schaulies et al., 1992; Apostolski et al., 1993), but the precise nature of this “neuronal receptor” is unclear. On the other hand, it has been found that gp120 exerts diverse direct effects on human and rat astroglia and microglia (Levi et al., 1993; Benos et al., 1994; Bubien et al., 1995). This might lead to glutamate release, overactivation of NMDA receptors, and neuronal death. Hence, it is not surprising that Ca2+, NO, and reactive oxygen species (ROS) have been identified as mediators of gp120 neurotoxicity (Dreyer et al., 1990; Lipton et al., 1991, Dawson et al., 1993). Consistent with these results, we observed that treatment with gp120 altered the [Ca2+]i response of neurons produced by NMDA, resulting in a substantial potentiation of the [Ca2+]i rise. This might be attributable to increased Ca2+ influx or reduced Ca2+ buffering or both. These effects of gp120 were observed in neurons treated ∼12–24 hr before the appearance of significant cell death and did not require the presence of the viral protein during recordings. Thus, it is likely that a complex series of events is responsible for the alteration of Ca2+ homeostasis in gp120-treated cultures. The absence of any rapid effects of gp120 on NMDA-elicited [Ca2+]i increases in our experiments, as well as data reporting no effect of the glycoprotein on NMDA currents (Lipton et al., 1991; Lannuzel et al., 1995), suggests that Ca2+ may not be the initial mediator of gp120 neurotoxicity, even though Ca2+ influx seems to be necessary, ultimately, for the neuronal damage to occur (Dreyer et al., 1990; Lipton et al., 1991). Other possible mediators of the neuronal injury produced by gp120 include arachidonic acid (Ushijima et al., 1995), superoxide, and NO (Dawson et al., 1993; Hewett et al, 1994). These factors can be released in the extracellular medium either by astrocytes and microglia (Hewett et al., 1994; Mallat and Chamak, 1994) or by NO synthose-containing neurons (Dawson et al., 1993). Furthermore, augmented production of ROS in neurons, caused by glutamate accumulation as a consequence of gp120-induced glutamate efflux from astroglia (Benos et al., 1994), also could contribute to the neurotoxicity.
The ability of TGF-β1 to counteract the toxic effect of gp120 may be attributable to several different effects. Recently, this cytokine was found to protect hippocampal neurons exposed to NMDA from Ca2+ overload and to prevent ROS-mediated neuronal injury. TGF-β1 also was shown to increase the concentrations of bcl-2 and bcl-xl in hippocampal pyramidal neurons, both of which have anti-apoptotic effects (Prehn et al., 1994,1996). TGF-β1 also has been reported to inhibit the proliferation and activation of microglia, which produce NO, and to reduce their release of O−2 (for review, see Mallat and Chamak, 1994). Finally, this cytokine is able to reduce astroglial proliferation, making glia more supportive for neuronal survival (Wang et al., 1994). All of these features make TGF-β1 an extremely interesting tool for the study of AIDS-associated neuronal injury and other neurodegenerative diseases.
Footnotes
This research was supported by National Institutes of Health Grants DAO2121, DAO25475, and MH40165. O.M. was supported by Ministero della Sanitá, Italy (borsa AIDS, 1994–1995 and VIII AIDS Grant 9306-21, 1995).
Correspondence should be addressed to Richard J. Miller, Department of Pharmacological and Physiological Sciences, The University of Chicago, 947 East 58th Street, Chicago, IL 60637.
Dr. Meucci is on leave from Unitá di Neuroscienze, Centro di Biotecnologie Avanzate, Largo, Rosanna Benzi 10, 16132 Genova, Italy.
REFERENCES
- 1.Abele AE, Scholz KP, Scholz WK, Miller RJ. Excitotoxicity induced by enhanced excitatory neurotransmission in cultured hippocampal pyramidal neurons. Neuron. 1990;2:413–419. doi: 10.1016/0896-6273(90)90053-i. [DOI] [PubMed] [Google Scholar]
- 2.Apostolski S, McAlarney T, Quattrini A. The gp120 glycoprotein of human immunodeficiency virus type 1 binds to sensory ganglion neurons. Ann Neurol. 1993;34:855–863. doi: 10.1002/ana.410340616. [DOI] [PubMed] [Google Scholar]
- 3.Bhat S, Spitalnik SL, Gonzalez-Scarano F, Silberberg DH. Galactosyl ceramide or a derivative is an essential component of the neural receptor for human immunodeficiency virus type 1 envelope glycoprotein gp120. Proc Natl Acad Sci USA. 1991;88:7131–7134. doi: 10.1073/pnas.88.16.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benos DJ, Hahn BH, Bubien JK, Ghosh SK, Mashburn NA, Chaikin MA, Shaw GM, Benveniste E. Envelope glycoprotein gp120 of human immunodeficiency virus type 1 alters ion transport in astrocytes: implication for AIDS dementia complex. Proc Natl Acad Sci USA. 1994;91:494–498. doi: 10.1073/pnas.91.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton S. A- poptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, Pert CB. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. 1988;335:639–642. doi: 10.1038/335639a0. [DOI] [PubMed] [Google Scholar]
- 7.Brorson JR, Bindokas VP, Iwama T, Marcuccilli CJ, Chisholm JC, Miller RJ. The Ca2+ influx induced by β-amyloid 25-25 in cultured hippocampal neurons results from network excitation. J Neurobiol. 1994;26:325–338. doi: 10.1002/neu.480260305. [DOI] [PubMed] [Google Scholar]
- 8.Bubien JK, Benveniste E, Benos DJ (1995) HIV–gp120 activates large-conductance apamin-sensitive potassium channels in rat astrocytes. Am J Physiol C1440–C1449. [DOI] [PubMed]
- 9.Charriaut-Marlangue C, Aggoun-Zouaoui D, Represa A, Ben-Ari Y. Apoptotic features of selective neuronal death in ischemia, epilepsy, and gp120 toxicity. Trends Neurosci. 1996;19:109–114. doi: 10.1016/s0166-2236(96)80039-7. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW. Calcium-mediated neurotoxicity: relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988;11:465–469. doi: 10.1016/0166-2236(88)90200-7. [DOI] [PubMed] [Google Scholar]
- 11.Dawson VL, Dawson TM, Uhl GR, Snyder SH. Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cultures. Proc Natl Acad Sci USA. 1993;90:3256–3259. doi: 10.1073/pnas.90.8.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. [DOI] [PubMed] [Google Scholar]
- 13.Finkel TH, Tudorwilliams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba TW, Rubrecht RM, Kubfer A. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat Med. 1995;1:129–134. doi: 10.1038/nm0295-129. [DOI] [PubMed] [Google Scholar]
- 14.Galli C, Meucci O, Scorziello A, Werge TM, Calissano P, Schettini G. Apoptosis in cerebellar granule cells is blocked by high KCl, forskolin, and IGF-1 through distinct mechanisms of action: the involvement of intracellular calcium and RNA synthesis. J Neurosci. 1995;15:1172–1179. doi: 10.1523/JNEUROSCI.15-02-01172.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci USA. 1993;90:2769–2773. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 18.Hewett SJ, Csernansky CA, Choi DW. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron. 1994;13:487–494. doi: 10.1016/0896-6273(94)90362-x. [DOI] [PubMed] [Google Scholar]
- 19.Lannuzel A, Lledo P-M, Lamghitnia HO, Vincent J-D, Tardieu M. HIV-1 envelope proteins gp120 and gp160 potentiate NMDA-induced [Ca2+]iincrease, alter [Ca2+]ihomeostasis, and induce neurotoxicity in human embryonic neurons. Eur J Neurosci. 1995;7:2285–2293. doi: 10.1111/j.1460-9568.1995.tb00649.x. [DOI] [PubMed] [Google Scholar]
- 20.Levi G, Patrizio M, Bernardo A, Petrucci T, Agresti C. Human immunodeficiency virus coat protein gp120 inhibits the β-adrenergic regulation of astroglial and microglia functions. Proc Natl Acad Sci USA. 1993;90:1541–1545. doi: 10.1073/pnas.90.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lipton SA. HIV coat protein induces soluble neurotoxins in culture medium. Neurosci Res Commun. 1994a;15:31–37. [Google Scholar]
- 22.Lipton SA. Ca2+, N -methyl-d-aspartate receptors, and AIDS-related neuronal injury. Int Rev Neurobiol. 1994b;36:1–27. doi: 10.1016/s0074-7742(08)60301-3. [DOI] [PubMed] [Google Scholar]
- 23.Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB. Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron. 1991;7:111–118. doi: 10.1016/0896-6273(91)90079-f. [DOI] [PubMed] [Google Scholar]
- 24.Lo T-M, Fallert CJ, Piser TM, Thayer SA. HIV-1 envelope protein evokes intracellular calcium oscillations in rat hippocampal neurons. Brain Res. 1992;594:189–196. doi: 10.1016/0006-8993(92)91125-x. [DOI] [PubMed] [Google Scholar]
- 25.Mallat M, Chamak B. Brain macrophages: neurotoxicity or neurotrophic effector cells? J Leukoc Biol. 1994;56:416–422. doi: 10.1002/jlb.56.3.416. [DOI] [PubMed] [Google Scholar]
- 26.Martin DP, Schmidt RE, Di Stefano P, Lowry S, Carter OH, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factors deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meldrum B, Garthwaite J. Excitatory amino acid and neurodegenerative disease. Trends Neurosci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 28.Merril JE, Chen ISY. HIV-1, macrophages, glial cells, and cytokines in AIDS nervous disease. FASEB J. 1991;5:2391–2397. doi: 10.1096/fasebj.5.10.2065887. [DOI] [PubMed] [Google Scholar]
- 29.Meucci O, Fatatis A, Holzwarth JA, Miller RJ. Developmental regulation of the toxin sensitivity of Ca2+ permeable AMPA receptors in cortical glia. J Neurosci. 1996;16:519–530. doi: 10.1523/JNEUROSCI.16-02-00519.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosier DE, Gulizia RJ, MacIsaac PD, Torbett BE, Levi JA. Rapid loss of CD+ T cells in human-PBL-SCID mice by noncytopathic HIV isolates. Science. 1993;260:689–692. doi: 10.1126/science.8097595. [DOI] [PubMed] [Google Scholar]
- 31.Muller WEG, Schoder HC, Ushijima H, Dapper J, Bormann J. gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: prevention by nemantine. Eur J Pharmacol. 1992;226:209–214. doi: 10.1016/0922-4106(92)90063-2. [DOI] [PubMed] [Google Scholar]
- 32.Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex. II. Neuropathology. Ann Neurol. 1986;19:525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 33.Prehn JH, Backhauß C, Krieglstein J. Transforming growth factor-β1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemia injury in vivo. J Cereb Blood Flow Metab. 1993;13:521–525. doi: 10.1038/jcbfm.1993.67. [DOI] [PubMed] [Google Scholar]
- 34.Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal bcl-2 protein expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prehn JH, Bindokas VP, Jordan J, Galindo M, Ghadge GD, Roos RP, Boise LH, Thompson CB, Krajewski S, Reed JC, Miller RJ. Protective effect of transforming growth factor-β1 on β-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–327. [PubMed] [Google Scholar]
- 36.Price RW, Brew B, Sidtis J, Resenblum M, Scheck AC, Clearly P. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science. 1988;239:586–592. doi: 10.1126/science.3277272. [DOI] [PubMed] [Google Scholar]
- 37.Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cystein from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reinhardt B, Torbett BE, Gulizia RJ, Reinhart PP, Spector SA, Mosier DE. Human immunodeficiency virus type 1 infection of neonatal severe combined immunodeficiency mice xenografted with human cord blood cells. AIDS Res Hum Retroviruses. 1994;10:131–141. doi: 10.1089/aid.1994.10.131. [DOI] [PubMed] [Google Scholar]
- 40.Savio T, Levi G. Neurotoxicity of HIV coat protein gp120, NMDA receptors, and protein kinase C: a study with rat cerebellar granule cell cultures. J Neurosci Res. 1993;34:265–272. doi: 10.1002/jnr.490340303. [DOI] [PubMed] [Google Scholar]
- 41.Schneider-Schaulies J, Schneider-Schaulies S, Brinkmann R, Tas P, Halbrugge M, Walter U, Holmes HC, Ter Meulen V. HIV-1 gp120 receptor on CD4-negative brain cells activates a tyrosine kinase. Virology. 1992;191:765–772. doi: 10.1016/0042-6822(92)90252-k. [DOI] [PubMed] [Google Scholar]
- 42.Scorziello A, Meucci O, Florio T, Fattore M, Forloni G, Salmona M, Schettini G. β amyloid 25-35 alters calcium homeostasis and induces neurotoxicity in cerebellar granule cells. J Neurochem. 1996;66:1995–2003. doi: 10.1046/j.1471-4159.1996.66051995.x. [DOI] [PubMed] [Google Scholar]
- 43.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 44.Ushijima H, Nishio O, Klocking R, Perovic S, Muller WEG. Exposure to gp120 of HIV-1 induces an increased release of arachidonic acid in rat primary neuronal cell culture followed by NMDA receptor-mediated neurotoxicity. Eur J Neurosci. 1995;7:1353–1359. doi: 10.1111/j.1460-9568.1995.tb01126.x. [DOI] [PubMed] [Google Scholar]
- 45.van Lookeren Campagne M, Lucassen PJ, Vermeulen JP, Balaz R. NMDA and kainate induce internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the normal rat brain. Eur J Neurosci. 1995;7:1627–1640. doi: 10.1111/j.1460-9568.1995.tb01158.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang L-C, Baird DH, Hatten ME, Mason CA. Astroglia differentiation is required for support of neurite outgrowth. J Neurosci. 1994;14:3195–3207. doi: 10.1523/JNEUROSCI.14-05-03195.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiley CA, Masliah E, Morey M, Lemere C, DeTeresa R, Grafe M, Hansen L, Terry R. Neocortical damage during HIV infection. Ann Neurol. 1991;29:651–657. doi: 10.1002/ana.410290613. [DOI] [PubMed] [Google Scholar]