

Abstract
We describe the Phase II HapMap, which characterizes over 3.1 million human single nucleotide polymorphisms (SNPs) genotyped in 270 individuals from four geographically diverse populations and includes 25–35% of common SNP variation in the populations surveyed. The map is estimated to capture untyped common variation with an average maximum r2 of between 0.9 and 0.96 depending on population. We demonstrate that the current generation of commercial genome-wide genotyping products captures common Phase II SNPs with an average maximum r2 of up to 0.8 in African and up to 0.95 in non-African populations, and that potential gains in power in association studies can be obtained through imputation. These data also reveal novel aspects of the structure of linkage disequilibrium. We show that 10–30% of pairs of individuals within a population share at least one region of extended genetic identity arising from recent ancestry and that up to 1% of all common variants are untaggable, primarily because they lie within recombination hotspots. We show that recombination rates vary systematically around genes and between genes of different function. Finally, we demonstrate increased differentiation at non-synonymous, compared to synonymous, SNPs, resulting from systematic differences in the strength or efficacy of natural selection between populations.
Advances made possible by the Phase I haplotype map
The International HapMap Project was launched in 2002 with the aim of providing a public resource to accelerate medical genetic research. The objective was to genotype at least one common SNP every 5 kilobases (kb) across the euchromatic portion of the genome in 270 individuals from four geographically diverse populations1,2: 30 mother–father–adult child trios from the Yoruba in Ibadan, Nigeria (abbreviated YRI); 30 trios of northern and western European ancestry living in Utah from the Centre d'Etude du Polymorphisme Humain (CEPH) collection (CEU); 45 unrelated Han Chinese individuals in Beijing, China (CHB); and 45 unrelated Japanese individuals in Tokyo, Japan (JPT). The YRI samples and the CEU samples each form an analysis panel; the CHB and JPT samples together form an analysis panel. Approximately 1.3 million SNPs were genotyped in Phase I of the project, and a description of this resource was published in 2005 (ref. 3).
The initial HapMap Project data had a central role in the development of methods for the design and analysis of genome-wide association studies. These advances, alongside the release of commercial platforms for performing economically viable genome-wide genotyping, have led to a new phase in human medical genetics. Already, large-scale studies have identified novel loci involved in multiple complex diseases4,5. In addition, the HapMap data have led to novel insights into the distribution and causes of recombination hotspots3,6, the prevalence of structural variation7,8 and the identity of genes that have experienced recent adaptive evolution3,9. Because the HapMap cell lines are publicly available, many groups have been able to integrate their own experimental data with the genome-wide SNP data to gain new insight into copy-number variation10, the relationship between classical human leukocyte antigen (HLA) types and SNP variation11, and heritable influences on gene expression12-14. The ability to combine genome-wide data on such diverse aspects of genetic variation with molecular phenotypes collected in the same samples provides a powerful framework to study the connection of DNA sequence to function.
In Phase II of the HapMap Project, a further 2.1 million SNPs were successfully genotyped on the same individuals. The resulting HapMap has an SNP density of approximately one per kilobase and is estimated to contain approximately 25–35% of all the 9–10 million common SNPs (minor allele frequency (MAF)≥0.05) in the assembled human genome (that is, excluding gaps in the reference sequence alignment; see Supplementary Text 1), although this number shows extensive local variation. This paper describes the Phase II resource, its implications for genome-wide association studies and additional insights into the fine-scale structure of linkage disequilibrium, recombination and natural selection.
Construction of the Phase II HapMap
Most of the additional genotype data for the Phase II HapMap were obtained using the Perlegen amplicon-based platform15. Briefly, this platform uses custom oligonucleotide arrays to type SNPs in DNA segmentally amplified via long-range polymerase chain reaction (PCR). Genotyping was attempted at 4,373,926 distinct SNPs, which corresponds, with exceptions (see Methods), to nearly all SNPs in dbSNP release 122 for which an assay could be designed. Additional submissions were included from the Affymetrix GeneChip Mapping Array 500K set, the Illumina HumanHap100 and HumanHap300 SNP assays, a set of ~11,000 non-synonymous SNPs genotyped by Affymetrix (ParAllele) and a set of ~4,500 SNPs within the extended major histocompatibility complex (MHC)11. Genotype submissions were subjected to the same quality control (QC) filters as described previously (see Methods) and mapped to NCBI build 35 (University of California at Santa Cruz (UCSC) hg17) of the human genome. The re-mapping of SNPs from Phase I of the project identified 21,177 SNPs that had an ambiguous position or some other feature indicative of low reliability; these are not included in the filtered Phase II data release. All genotype data are available from the HapMap Data Coordination Center (http://www.hapmap.org) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP); analyses described in this paper refer to release 21a. Three data sets are available: ‘redundant unfiltered’ contains all genotype submissions, ‘redundant filtered’ contains all submissions that pass QC, and ‘non-redundant filtered’ contains a single QC+ submission for each SNP in each analysis panel.
The QC filters remove SNPs showing gross errors. However, it is also important to understand the magnitude and structure of more subtle genotyping errors among SNPs that pass QC. We therefore carried out a series of analyses to assess the influence of the long-range PCR amplicon structure on genotyping error, the concordance rates between genotype calls from different genotyping platforms and between those platforms and re-sequencing assays, as well as the rates of false monomorphism and mis-mapping of SNPs (see Supplementary Text 2, Supplementary Figs 1–3 and Supplementary Tables 1–4). We estimate that the average per genotype accuracy is at least 99.5%. However, there are higher rates of missing data and genotype discrepancies at non-reference alleles, with some clustering of errors resulting from the amplicon design and a few incorrectly mapped SNPs.
Table 1 shows the numbers of SNPs attempted and converted to QC+ SNPs in each analysis panel (Supplementary Table 5 shows a breakdown by each major submission). Haplotypes and missing data were estimated for each analysis panel separately using both trio information and statistical methods based on the coalescent model (see Methods). To enable cross-population comparisons, a consensus data set was created consisting of 3,107,620 SNPs that were QC+ in all analysis panels and polymorphic in at least one analysis panel. The equivalent figure from Phase I was 931,340 SNPs. Unless stated otherwise, all analyses have been carried out on the consensus data set. An additional set of haplotypes was created for those SNPs in the consensus where a putative ancestral state could be assigned by comparison of the human alleles to the orthologous position in the chimpanzee and rhesus macaque genomes.
Table 1.
Summary of Phase II HapMap data (release 21)
| Phase | SNP categories | Analysis panel |
||
|---|---|---|---|---|
| YRI | CEU | CHB+JPT | ||
| I | Assays submitted | 1,304,199 | 1,344,616 | 1,306,125 |
| Passed QC | 1,177,312 (90%) | 1,217,902 (91%) | 1,187,800 (91%) | |
| Did not pass QC | 126,887 (10%) | 126,714 (9%) | 118,325 (9%) | |
| >20% missing | 82,463 (65%) | 95,684 (76%) | 78,323 (66%) | |
| >1 duplicate inconsistent | 6,049 (5%) | 5,126 (4%) | 9,242 (8%) | |
| >1 mendelian error | 18,916 (15%) | 11,310 (9%) | N/A | |
| <0.001 Hardy–Weinberg P -value | 10,265 (8%) | 8,922 (7%) | 13,722 (12%) | |
| Other failures | 19,345 (15%) | 13,858 (11%) | 20,674 (17%) | |
|
| ||||
| II | Assays submitted | 5,044,989 | 5,044,996 | 5,043,775 |
| Passed QC | 3,150,433 (62%) | 3,204,709 (64%) | 3,244,897 (64%) | |
| Did not pass QC | 1,894,556 (38%) | 1,840,287 (36%) | 1,798,878 (36%) | |
| >20% missing | 1,419,000 (75%) | 1,398,166 (76%) | 1,403,543 (78%) | |
| >1 duplicate inconsistent | 0 (0%) | 0 (0%) | 6,617 (0%) | |
| >1 mendelian error | 172,339 (9%) | 127,923 (7%) | N/A | |
| <0.001 Hardy–Weinberg P -value | 96,231 (5%) | 82,268 (4%) | 108,880 (6%) | |
| Other failures | 334,511 (18%) | 337,906 (18%) | 340,370 (19%) | |
|
| ||||
| Overall | Assays submitted | 6,349,188 | 6,389,612 | 6,349,900 |
| Passed QC | 4,327,745 (68%) | 4,422,611 (69%) | 4,432,697 (70%) | |
| Did not pass QC | 2,021,443 (32%) | 1,967,001 (31%) | 1,917,203 (30%) | |
| >20% missing | 1,501,463 (74%) | 1,493,850 (76%) | 1,481,866 (77%) | |
| >1 duplicate inconsistent | 6,049 (0%) | 5,126 (0%) | 15,859 (1%) | |
| >1 mendelian error | 191,255 (9%) | 139,233 (7%) | N/A | |
| <0.001 Hardy–Weinberg P -value | 106,496 (5%) | 91,190 (5%) | 122,602 (6%) | |
| Other failures | 353,856 (18%) | 351,764 (18%) | 361,044 (19%) | |
| Non-redundant (unique) SNPs | 3,796,934 | 3,868,157 | 3,890,416 | |
| Monomorphic | 861,299 (23%) | 1,246,183 (32%) | 1,410,152 (36%) | |
| Polymorphic | 2,935,635 (77%) | 2,621,974 (68%) | 2,480,264 (64%) | |
|
| ||||
| SNP categories | All analysis panels | |||
|
| ||||
| Unique QC-passed SNPs | 4,000,107 | |||
| Passed in one analysis panel | 88,140 (2%) | |||
| Passed in two analysis panels | 268,534 (7%) | |||
| Passed in three analysis panels (QC+3) | 3,643,433 (91%) | |||
| QC+3 and monomorphic across three analysis panels |
535,813 | |||
| QC+3 and polymorphic in at least one analysis panel | 3,107,620 | |||
| QC+3 and polymorphic in all three analysis panels | 2,006,352 | |||
|
| ||||
| QC+3 and MAF≥0.05 in at least one of three analysis panels |
2,819,322 | |||
The variation in SNP density within the Phase II HapMap is shown in Fig. 1. On average there are 1.14 genotyped polymorphic SNPs per kilobase (average spacing is 875 base pairs (bp)) and 98.6% of the assembled genome is within 5 kb of the nearest polymorphic SNP. Still, there is heterogeneity in genotyped SNP density at both broad (Fig. 1a) and fine (Fig. 1b) scales. Furthermore, there are systematic changes in genotyped SNP density around genomic features including genes (Fig. 1c).
Figure 1. SNP density in the Phase II HapMap.

a, SNP density across the genome. Colours indicate the number of polymorphic SNPs per kb in the consensus data set. Gaps in the assembly are shown as white. b, Example of the fine-scale structure of SNP density for a 100-kb region on chromosome 17 showing Perlegen amplicons (black bars), polymorphic Phase I SNPs in the consensus data set (red triangles) and polymorphic Phase II SNPs in the consensus data set (blue triangles). Note the relatively even spacing of Phase I SNPs. c, The distribution of polymorphic SNPs in the consensus Phase II HapMap data (blue line and left-hand axis) around coding regions. Also shown is the density of SNPs in dbSNP release 125 around genes (red line and right-hand axis). Values were calculated separately 5′ from the coding start site (the left dotted line) and 3′ from the coding end site (right dotted line) and were joined at the median midpoint position of the coding unit (central dotted line).
The Phase II HapMap differs from the Phase I HapMap not only in SNP spacing, but also in minor allele frequency distribution and patterns of linkage disequilibrium (Supplementary Fig. 4). Because the criteria for choosing additional SNPs did not include consideration of SNP spacing or preferential selection for high MAF, the SNPs added in Phase II are, on average, more clustered and have lower MAF than the Phase I SNPs. Because MAF predictably influences the distribution of linkage disequilibrium statistics, the average r2 at a given physical distance is typically lower in Phase II than in Phase I; conversely, the |D′| statistic is typically higher (data not shown). One notable consequence is that the Phase II HapMap includes a better representation of rare variation than the Phase I HapMap.
The increased resolution provided by Phase II of the project is illustrated in Fig. 2. Broadly, an additional SNP added to a region shows one of three patterns. First, it may be very similar in distribution to SNPs present in Phase I. Second, it may provide detailed resolution of haplotype structure (for example, a group of chromosomes with identical local haplotypes in Phase I can be shown in Phase II to carry multiple related haplotypes). Third, the novel SNP (or group of added SNPs) may reveal previously missed recombinant haplotypes. The extent to which each type of event occurs varies among populations and chromosomal regions. The greatest gains in resolution, in terms of identifying new recombinant haplotypes and haplotype groupings, occur in YRI. Consequently, the Phase II HapMap provides increased resolution in the estimated fine-scale genetic map and improved power to detect and localize recombination hotspots (Fig. 2b).
Figure 2. Haplotype structure and recombination rate estimates from the Phase II HapMap.

a, Haplotypes from YRI in a 100 kb region around the β-globin (HBB) gene. SNPs typed in Phase I are shown in dark blue. Additional SNPs in the Phase II HapMap are shown in light blue. Only SNPs for which the derived allele can be unambiguously identified by parsimony (by comparison with an outgroup sequence) are shown (89% of SNPs in the region); the derived allele is shown in colour. b, Recombination rates (lines) and the location of hotspots (horizontal blue bars) estimated for the same region from the Phase I (dark blue) and Phase II HapMap (light blue) data. Also shown are the location of genes within the region (grey bars) and the location of the experimentally verified recombination hotspot57,58 at the 59′ end of the HBB gene (black bar).
The use of the Phase II HapMap in association studies
The increased SNP density of the Phase II HapMap has already been extensively exploited in genome-wide studies of disease association. In this section, we quantify the gain in resolution and outline how the HapMap data can be used to improve the power of association studies.
Improved coverage of common variation
We previously predicted that the vast majority of common SNPs would be correlated to Phase II HapMap SNPs by extrapolation from the ten HapMap ENCODE regions3. Using the actual Phase II marker spacing and frequency distributions (Table 2), we repeated the simulations and estimate that Phase II HapMap marker sets capture the overwhelming majority of all common variants at high r2. For common variants (MAF≥0.05) the mean maximum r2 of any SNP to a typed one is 0.90 in YRI, 0.96 in CEU and 0.95 in CHB+JPT. The impact of the increased density of the Phase II HapMap is most notable in YRI (in the Phase I HapMap the mean maximum r2 was 0.67). Similar results are found if a threshold of r2≥0.8 is used to determine whether an SNP is captured (Table 2). As expected, very common SNPs with MAF>0.25 are captured extremely well (mean maximum r2 of 0.93 in YRI to 0.97 in CEU), whereas rarer SNPs with MAF<0.05 are less well covered (mean maximum r2 of 0.74 in CHB+JPT to 0.76 in YRI). The latter figure is probably an overestimate because it is based on lower frequency SNPs discovered via re-sequencing 48 HapMap individuals, and does not include a much larger number of very rare SNPs. We also assessed the increase in coverage provided by using two-SNP haplotypes as proxies for SNPs that are poorly captured by single SNPs16 (Table 2). These two-SNP haplotypes lead to a modest increase in mean maximum r2 of 0.01 to 0.03 across all allele frequencies. However, in some regions, particularly where marker density is low, gains from multi-marker and imputation approaches in practical situations can be substantial (see below).
Table 2.
Estimated coverage of the Phase II HapMap in the ten HapMap ENCODE regions
| Panel | MAF bin | Phase I HapMap3 | Phase II HapMap |
||||
|---|---|---|---|---|---|---|---|
|
|
Pairwise linkage disequilibrium |
Additional 2-SNP tests |
|||||
| r2≥ 0.8 (%) | Mean maximum r2 | r2≥ 0.8 (%) | Mean maximum r2 | r2≥ 0.8 (%) | Mean maximum r2 | ||
| YRI | ≥0.05 | 45 | 0.67 | 82 | 0.90 | 87 | 0.93 |
| <0.05 | 61 | 0.76 | 62 | 0.78 | |||
| 0.05–0.10 | 81 | 0.89 | 81 | 0.89 | |||
| 0.10–0.25 | 90 | 0.94 | 90 | 0.95 | |||
| 0.25–0.50 | 87 | 0.93 | 92 | 0.96 | |||
|
| |||||||
| CEU | ≥0.05 | 74 | 0.85 | 93 | 0.96 | 95 | 0.97 |
| <0.05 | 70 | 0.79 | 72 | 0.81 | |||
| 0.05–0.10 | 87 | 0.92 | 88 | 0.93 | |||
| 0.10–0.25 | 94 | 0.96 | 95 | 0.97 | |||
| 0.25–0.50 | 95 | 0.97 | 97 | 0.98 | |||
|
| |||||||
| CHB+JPT | ≥0.05 | 72 | 0.83 | 92 | 0.95 | 95 | 0.97 |
| <0.05 | 65 | 0.74 | 65 | 0.74 | |||
| 0.05–0.10 | 81 | 0.89 | 82 | 0.89 | |||
| 0.10–0.25 | 90 | 0.94 | 90 | 0.95 | |||
| 0.25–0.50 | 94 | 0.96 | 97 | 0.98 | |||
2-SNP tests, linkage disequilibrium to haplotypes formed from two nearby SNPs.
Currently, the Phase II HapMap provides the most complete available resource for selecting tag SNPs genome-wide. Using a simple pairwise tagging approach, we find that 1.09 million SNPs are required to capture all common Phase II SNPs with r2≥0.8 in YRI, with slightly more than 500,000 required in CEU and CHB+JPT (Table 3). These numbers are approximately twice those required to capture SNPs in the Phase I HapMap (which has one-third as many SNPs). The number of SNPs required to achieve perfect tagging (r2=1.0) in each analysis panel is almost double that required to achieve the r2≥0.8 threshold. It becomes increasingly expensive to improve the coverage afforded by tags from the Phase I and, now, the Phase II HapMap, because additional tag SNPs are unlikely to capture large groups of additional SNPs.
Table 3.
Number of tag SNPs required to capture common (MAF≥0.05) Phase II SNPs
| Threshold | YRI | CEU | CHB+JPT |
|---|---|---|---|
| r2 ≥ 0.5 | 627,458 | 290,969 | 277,831 |
| r2 ≥ 0.8 | 1,093,422 | 552,853 | 520,111 |
| r2 = 1.0 | 1,616,739 | 1,024,665 | 1,078,959 |
Phase II HapMap and genome-wide association studies
Although the efficient choice of tag SNPs is one use of the Phase II HapMap, for most disease studies the tag SNPs genotyped will be primarily determined by the choice of a commercial platform for the experiment17,18. Using Phase II data, we estimated the coverage of several available products on which genome-wide association studies are already underway (Table 4). Similar to earlier estimates17,18, these products typically perform well in CEU and CHB+JPT, and some also perform well in YRI. For example, arrays of approximately 500,000 SNPs capture 68–88% (depending on selection method) of all HapMap Phase II variation with r2≥0.8 in CEU. SNPs that are not included in the Phase II HapMap will be covered more poorly because most genotyping products were designed using HapMap data.
Table 4.
Estimated coverage of commercially available fixed marker arrays
| Platform* | YRI |
CEU |
CHB+JPT |
|||
|---|---|---|---|---|---|---|
| r2≥0.8 (%) | Mean maximum r2 | r2≥0.8 (%) | Mean maximum r2 | r2≥0.8 (%) | Mean maximum r2 | |
| Affymetrix GeneChip 500K | 46 | 0.66 | 68 | 0.81 | 67 | 0.80 |
| Affymetrix SNP Array 6.0 | 66 | 0.80 | 82 | 0.90 | 81 | 0.89 |
| Illumina HumanHap300 | 33 | 0.56 | 77 | 0.86 | 63 | 0.78 |
| Illumina HumanHap550 | 55 | 0.73 | 88 | 0.92 | 83 | 0.89 |
| Illumina HumanHap650Y | 66 | 0.80 | 89 | 0.93 | 84 | 0.90 |
| Perlegen 600K | 47 | 0.68 | 92 | 0.94 | 84 | 0.90 |
Assuming all SNPs on the product are informative and pass QC; in practice these numbers are overestimates.
HapMap data have several additional roles in the analysis of disease-association studies using fixed marker sets. For example, the high-quality haplotype information within the Phase II HapMap can be used to aid the phasing of genotype data from new samples because additional haplotypes are likely to be locally very similar to at least one haplotype in the Phase II data. By a similar argument, missing genotypes can potentially be inferred through comparison to the Phase II haplotypes. Genotypes may be missing either because of genotyping failure or because the SNP was not assayed within the experiment. Therefore, the HapMap haplotypes provide a way of in silico genotyping Phase II SNPs that were not included in the experiment.
Although there is no clear consensus yet about the role of SNP imputation in the analysis of genome-wide association studies, high imputation accuracy can be achieved using model-based methods19-23 and can lead to an increase in power23,24. To illustrate the possibilities, in the 500-kb HapMap ENCODE region on 8q24.11 (Supplementary Fig. 5) we evaluated imputation of Phase II SNPs from the Affymetrix GeneChip 500K array. To do this, we used a leave-one-out procedure to assess the accuracy of genotype prediction in the YRI. For SNPs with MAF≥0.2, the average maximum r2 to a typed SNP in the region is 0.59 compared to an average genotype prediction r2 of 0.86. Furthermore, whereas 44% of such SNPs in the region have no single-marker proxy with r2≥0.5, fewer than 6% of the SNPs have a genotype imputation accuracy of r2<0.5, establishing that accurate imputation can be achieved even in the population where linkage disequilibrium is the weakest.
New insights into linkage disequilibrium structure
The paradigm underlying association studies is that linkage disequilibrium can be used to capture associations between markers and nearby untyped SNPs. However, the Phase II HapMap has revealed several properties of linkage disequilibrium that illustrate the full complexity of empirical patterns of genetic variation. Two striking features are the long-range similarity among haplotypes, and SNPs that show almost no linkage disequilibrium with any other SNP.
The extent of recent common ancestry and segmental sharing
A simplified view of linkage disequilibrium is that genetic variation is organized in relatively short stretches of strong linkage disequilibrium (haplotype blocks), each containing only a few common haplotypes and separated by recombination hotspots across which little association remains25. Although this view has heuristic value, if chromosomes share a recent common ancestor then similarity between chromosomes can extend over considerable genetic distance and span multiple recombination hotspots26. The extent of such recent ancestry in the four populations surveyed here has not been characterized previously. Therefore we identified stretches of identity between pairs of chromosomes, both within and across individuals, reflecting autozygosity and identity-by-descent (IBD) (Fig. 3a). After first checking for stratification within each analysis panel (see Supplementary Text 3; none was found for YRI, CEU and JPT, and only small stratification was found for CHB), we calculated genome-wide probabilities of sharing 0, 1 or 2 chromosomes identical by descent for each pair of individuals (see Supplementary Text 4). In addition to identifying a few close relationships (as reported in HapMap Phase I3), we estimate that, on average, any two individuals from the same population share approximately 0.5% of their genome through recent IBD (Table 5). Using a hidden Markov model approach27 (see Supplementary Text 5), we searched for such shared segments over 1-megabase (Mb) long and containing at least 50 SNPs, after first pruning the list of SNPs to remove local linkage disequilibrium. We find that 10–30% of pairs in each analysis panel share regions of extended identity resulting from sharing a common ancestor within 10–100 generations. These regions typically span hundreds of SNPs and can extend over tens of megabases (Table 5).
Figure 3. The extent of recent co-ancestry among HapMap individuals.
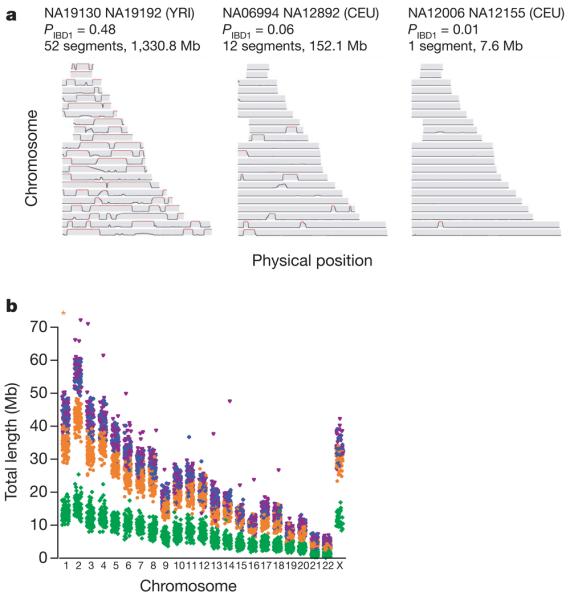
a, Three pairs of individuals with varying levels of identity-by-descent (IBD) sharing illustrate the continuum between very close and very distant relatedness and its relation to segmental sharing. The three pairs are: high sharing (NA19130 and NA19192 from YRI; previously identified as second-degree relatives3), moderate sharing (NA06994 and NA12892 from CEU) and low sharing (NA12006 and NA12155 from CEU). Along each chromosome, the probability of sharing at least one chromosome IBD is plotted, based on the HMM method described in Supplementary Text 5. Red sections indicate regions called as segments: in general, the proportion of the genome in segments is similar to each pair's estimated global relatedness. b, The extent of homozygosity on each chromosome for each individual in each analysis panel. Excludes segments <106 kb and chromosome X in males. Asterisk, NA12874, length=107 Mb. YRI, green; CEU, orange; CHB, blue; JPT, magenta.
Table 5.
Relatedness, extended segmental sharing and homozygosity
| Property | YRI | CEU | CHB | JPT |
|---|---|---|---|---|
| Number of pairs included | 1,767 | 1,708 | 990 | 861 |
| Mean identity by state (IBS) (%) | 81.9 | 83.7 | 85.0 | 85.1 |
| Mean identity by descent (IBD) (%) | 0.04 | 0.34 | 0.36 | 0.42 |
| Number of pairs with >1% IBD (%) | 8.8 | 20.4 | 21.1 | 29.7 |
| Number of pairs with one or more segment (%) |
195 (11.0) |
350 (20.5) |
135 (13.6) |
216 (25.1) |
| Total number of segments | 250 | 427 | 146 | 273 |
| Total distance spanned (Mb) | 1,416 | 2,336 | 704 | 1,301 |
| Mean segment length (Mb) | 5.7 | 5.5 | 4.8 | 4.8 |
| Maximum segment length (Mb) | 51.7 | 56.2 | 15.0 | 25.3 |
| Maximum segment length (Mb) (including close relatives) |
141.4 | 128.5 | N/A | N/A |
| Total number of 2-SNPs | 6,219 | 9,220 | 8,174 | 8,750 |
| Number of 2-SNPs in segments | 109 | 162 | 116 | 132 |
| 2-SNP fold increase | 6.7 | 7.3 | 7.6 | 7.0 |
| Number of homozygous segments (×103)* |
0.9 | 2.2 | 2.6 | 2.6 |
| SNPs in homozygous segments (×105) | 1.6 | 4.2 | 5.3 | 5.4 |
| Total length of homozygous segments (Mb) |
160 | 410 | 510 | 520 |
2-SNP, SNPs where only two copies of the minor allele are present.
Homozygous segments >106 kb.
Similarly, extended stretches of homozygosity are indicative of recent inbreeding within populations28,29. Although short runs of homozygosity are commonplace, covering up to one-third of the genome and showing population differences reflective of ancient linkage disequilibrium patterns (Table 5 and Fig. 3b), very long homozygous runs exist that are clearly distinct from this process. Including two JPT individuals who have unusually high levels of homozygosity (NA18987 and NA18992) and one CEU individual (NA12874), we identified 79 homozygous regions over 3 Mb in 51 individuals, with many segments extending over 10 Mb (Supplementary Tables 7 and 8). Segments intersecting with suspected deletions were first removed from the analysis (Supplementary Text 6).
In studies of rare mendelian diseases, the extended haplotype sharing surrounding recent mutations, usually with a frequency of much less than 1%, has been exploited to great advantage through homo-zygosity mapping30,31 and haplotype sharing32 methods. In studies of common disease, extended haplotype sharing among patients potentially offers a route for identifying rare variants (MAF in the range of 1–5%) of high penetrance33,34, which tend to be poorly captured through single-marker association with genome-wide arrays. To illustrate the idea, we identified SNPs where only two copies of the minor allele are present (referred to as ‘2-SNPs’), which have minor allele frequencies of 1–2%. We find that these are enriched approximately sevenfold (Table 5) among regions of IBD identified by the hidden Markov model approach. Notably, identification of IBD regions can be performed with the same genome-wide SNP data being collected in large-scale association studies, making haplotype-sharing approaches an attractive and complementary analysis to standard SNP association tests, with the potential to identify rare variants associated with complex disease.
The distribution and causes of untaggable SNPs
Despite the SNP density of the Phase II HapMap, there are high-frequency SNPs for which no tag can be identified. Among high-frequency SNPs (MAF≥0.2), we marked as untaggable SNPs to which no other SNP within 100 kb has an r2 value of at least 0.2. In Phase II, approximately 0.5–1.0% of all high-frequency SNPs are untaggable and the proportion in YRI is approximately twice as high as in the other panels. Similar proportions are observed across the ten HapMap ENCODE regions.
To identify factors influencing the location of untaggable SNPs we considered their distribution relative to segmental duplications, repeat sequence, CpG dinucleotide density, regions of low SNP density, unusual allele frequency distribution, linkage disequilibrium patterns and recombination hotspots. We find no evidence for an enrichment of untaggable SNPs in segmental duplications or repeat sequence, as would be expected from mis-mapping of SNPs (2% and 35% of common SNPs lie in segmental duplications and repeat sequence, respectively, compared to 1.8% and 29%, respectively, of untaggable SNPs). Untaggable SNPs are slightly enriched in CpG islands (0.37% of common SNPs are in CpG islands compared to 1.4% of untaggable SNPs) and have slightly reduced MAF (Fig. 4). Most notably, untaggable SNPs are strongly enriched in regions of low linkage disequilibrium, particularly in recombination hotspots. To test whether these untaggable SNPs are themselves responsible for the identification of recombination hotspots, we eliminated them from 100 randomly chosen recombination hotspots and reassessed the evidence for a local peak in recombination. In all cases we still find evidence for a considerable increase in local recombination rate.
Figure 4. Properties of untaggable SNPs.

a–e, Properties of the genomic regions surrounding untaggable SNPs in terms of: a, the density of polymorphic SNPs within the consensus data set; b, mean minor allele frequency of polymorphic SNPs; c, maximum r2 of SNPs to any others in the Phase II data; d, the density of estimated recombination hotspots (defined from hotspot centres); and e, the estimated mean recombination rate. YRI, green; CEU, orange; CHB+JPT, purple.
Over 50% of all untaggable SNPs lie within 1 kb of the centre of a detected recombination hotspot and over 90% are within 5 kb. Because only 3–4% of all SNPs lie within 1 kb from the centre of a detected recombination hotspot (16% are within 5 kb), this constitutes a marked enrichment and implies that at least 10% of all SNPs within 1 kb of hotspots are untaggable. The implication for association mapping is that when a region of interest contains a known hotspot it may be prudent to perform additional sequencing within the hotspot. Many of the variants identified in this manner will be untaggable SNPs that should be genotyped directly in association studies. From a biological perspective, the proximity of untaggable SNPs to the centre of hotspots suggests that they may lie within gene conversion tracts associated with the repair of double-strand breaks. Double-strand breaks are thought to resolve as crossover events only 5–25% of the time35. Consequently, SNPs lying near the centre of a hotspot are liable to be included within gene conversion tracts and will experience much higher effective recombination rates than predicted from crossover rates alone.
The distribution of recombination
In the Phase II HapMap we identified 32,996 recombination hotspots3,6,36 (an increase of over 50% from Phase I) of which 68% localized to a region of≤5 kb. The median map distance induced by a hotspot is 0.043 cM (or one crossover per 2,300 meioses) and the hottest identified, on chromosome 20, is 1.2 cM (one crossover per 80 meioses). Hotspots account for approximately 60% of recombination in the human genome and about 6% of sequence (Supplementary Fig. 6). We do not find marked differences among chromosomes in the concentration of recombination in hotspots, which implies that obligate differences in recombination among chromosomes of different size result from differences in hotspot density and intensity6.
The increased number of well-defined hotspots allows us to understand better the influence of genomic features on the distribution of recombination. Previous work identified specific DNA motifs that influence hotspot location6,37 as well as additional influences of local sequence context including the location of genes6 and base composition38. The Phase II HapMap provides the resolution to separate these influences. Figure 5a shows the distribution of recombination, hotspot motifs and base composition around genes. Within the transcribed region of genes there is a marked decrease in the estimated recombination rate. However, 5′ of the transcription start site is a peak in recombination rate with a corresponding local increase in the density of hotspot motifs. This region also shows a marked increase in G+C content, reflecting the presence of CpG islands in promoter regions. There is also an asymmetry in recombination rate across genes, with recombination rates 3′ of transcribed regions being elevated (as are motif density and G+C content) compared to regions 5′ of genes. Studies in yeast have previously suggested an association between promoter regions and recombination hotspots39. Our results suggest a significant, although weak, relationship between promoters and recombination in humans. Nevertheless, the vast majority of hotspots in the human genome are not in gene promoters. The association may reflect a general association between regions of accessible chromatin and crossover activity.
Figure 5. Recombination rates around genes.

a, The recombination rate, density of recombination-hotspot-associated motifs (all motifs with up to 1 bp different from the consensus CCTCCCTNNCCAC) and G+C content around genes. The blue line indicates the mean. For the recombination rate, grey lines indicate the quartiles of the distribution. Values were calculated separately 5′ from the transcription start site (the first dotted line) and 3′ from the transcription end site (third dotted line) and were joined at the median midpoint position of the transcription unit (central dotted line). Note the sharp drop in recombination rate within the transcription unit, the local increase around the transcription start site and the broad decrease away from the 3′ end of genes. These patterns only partly reflect the distribution of G+C content and the hotspot-associated motif, suggesting that additional factors influence recombination rates around genes. b, Recombination rates within genes of different molecular function41. The chart shows the increase or decrease for each category compared to the genome average. P values were estimated by permutation of category; numbers of genes are shown in parentheses.
Systematic differences in recombination rate by gene class
Previous work has demonstrated differences in the magnitude of linkage disequilibrium, as measured at a megabase scale, among genes associated with different functions3,40. Using the fine-scale genetic map estimated from the Phase II HapMap data we can quantify local increases in recombination rate associated with genes of different function using the Panther gene ontology annotation41. Average recombination rates vary more than sixfold among such gene classes (Fig. 5b), with defence and immunity genes showing the highest rates (1.9 cM Mb−1) and chaperones showing the lowest rates (0.3 cM Mb−1). Gene functions associated with cell surfaces and external functions tend to show higher recombination rates (immunity, cell adhesion, extracellular matrix, ion channels, signalling) whereas those with lower recombination rates are typically internal to cells (chaperones, ligase, isomerase, synthase). Controlling for systematic differences between gene classes in base composition and gene clustering, the differences between groups remain significant. We also find that the density of hotspot-associated DNA motifs varies systematically among gene classes and that variation in motif density explains over 50% of the variance in recombination rate among gene functions (Supplementary Fig. 7).
These results pose interesting evolutionary questions. Because recombination involves DNA damage through double-strand breaks, hotspots may be selected against in some highly conserved parts of the genome. In regions exposed to recurrent selection (for example, from changes in environment or pathogen pressure) it is plausible that recombination may be selected for. However, because the fine-scale structure of recombination seems to evolve rapidly42,43 it will be important to learn whether patterns of recombination rate heterogeneity among molecular functions are conserved between species.
Natural selection
The Phase I HapMap data have been used to identify genomic regions that show evidence for the influence of adaptive evolution3,9, primarily through extended haplotype structure indicative of recent positive selection. Using two established approaches9,44, we identified approximately 200 regions with evidence of recent positive selection from the Phase II HapMap (Supplementary Table 9). These regions include many established cases of selection, such as the genes HBB and LCT, the HLA region, and an inversion on chromosome 17. Many other regions have been previously identified in HapMap Phase I including LARGE, SYT1 and SULT1C2 (previously called SULT1C1). A detailed description of the findings from the Phase II HapMap is published elsewhere45.
The Phase II HapMap also provides new insights into the forces acting on SNPs in coding regions. Effort was made to genotype as many known or putative non-synonymous SNPs as possible. Of the 56,789 non-synonymous SNPs identified in dbSNP release 125, attempts were made to genotype 36,777, which resulted in 17,427 that are QC+ in all three analysis panels and polymorphic. We selected only those SNPs for which ancestral allele information was available (approximately 90%). For comparison, we used patterns of variation at synonymous SNPs. As previously reported46,47, non-synonymous SNPs show an increase in frequency of rare variants and a slight decrease of common variants compared to synonymous SNPs, compatible with widespread purifying selection against non-synonymous mutations (Fig. 6a). In contrast, we find no excess of high-frequency derived non-synonymous mutations, as might be expected if positive selection were widespread.
Figure 6. Properties of non-synonymous and synonymous SNPs.

a, The derived allele frequency (DAF) spectrum in each analysis panel for all SNPs (black), synonymous SNPs (green) and non-synonymous SNPs (red). Note the excess of rare variants for coding sequence SNPs but no excess of high-frequency derived variants. b, Enrichment of non-synonymous SNPs among genic SNPs showing high differentiation. For each of ten classes of derived allele frequency (averaged across analysis panels) the fraction of non-synonymous (red) and synonymous (green) variants in that class that show FST > 0.5 is shown. Note the strong enrichment of non-synonymous SNPs among SNPs of moderate to high derived-allele frequency (asterisk, P < 0.05; double asterisk, P < 0.01). c, Lack of enrichment of non-synonymous SNPs among those showing long-range haplotype structure. The integrated extended haplotype homozygosity (iEHH) statistic9 was calculated for non-synonymous and synonymous SNPs in each analysis panel (YRI, green; CEU, orange; CHB+JPT, purple). For each of ten derived allele frequency classes, the proportion of non-synonymous SNPs among those showing the 5% most extreme statistics (within the allele frequency class) is shown (points). Also shown is the proportion of non-synonymous SNPs among SNPs in the coding sequence for each frequency class (dotted lines). Differences between synonymous and non-synonymous SNPs are tested for using a contingency table test.
Natural selection also influences the extent to which allele frequencies differ between populations, not only through local selective pressures that drive alleles to different frequencies48,49, but also through local variation in the strength of purifying selection. We compared the distribution of population differentiation (as measured by FST, the proportion of total variation in allele frequency that is due to differences between populations) at non-synonymous SNPs and synonymous SNPs matched for allele frequency (Fig. 6b). We find a systematic bias for non-synonymous SNPs to show stronger differentiation than synonymous SNPs. Among SNPs showing high levels of differentiation there is a strong tendency for the derived allele to be at higher frequency in non-YRI populations. Among SNPs with FST>0.5 between CEU and YRI, in 79% and 75% of non-synonymous and synonymous variants, respectively, the derived allele is more common in CEU. Although this difference between non-synonymous and synonymous SNPs is not significant, among the eight exonic SNPs with FST>0.95, all are non-synonymous. We see no such bias towards increased MAF in CEU at high-differentiation SNPs, indicating that SNP ascertainment is unlikely to explain the difference. Rather, this effect can largely be explained by more genetic drift in the non-African populations, as confirmed by simulations (data not shown). In addition, reduced selection against deleterious mutations and local adaptation within non-African populations will both act to increase the frequency of derived variants in non-African populations.
To assess the evidence for widespread local adaptation influencing non-synonymous mutations we considered the distribution of integrated extended haplotype homozygosity (iEHH) statistics9,44 (Fig. 6c). We find no evidence for systematic differences between non-synonymous and synonymous SNPs, suggesting that local adaptation does not explain their higher differentiation. Although hitch-hiking effects will tend to obscure differences between selected and neutral SNPs, these results are consistent with a scenario in which the higher differentiation of non-synonymous SNPs is primarily driven by a reduction in the strength or efficacy of purifying selection in non-African populations.
Discussion and prospects
The International HapMap Project has been instrumental in making well-powered, large-scale, genome-wide association studies a reality. It is now clear that the HapMap can be a useful resource for the design and analysis of disease association studies in populations across the world50-53. Furthermore, the decreasing costs and increasing SNP density of standard genotyping panels mean that the focus of attention in disease association studies is shifting from candidate gene approaches towards genome-wide analyses. Alongside developments in technology, new statistical methodologies aimed at improving aspects of analysis, such as genotype calling21,54, the identification of and correction for population stratification and relatedness55,56, and imputation of untyped variants21-23, are increasing the accuracy and reliability of genome-wide association studies.
Within this context, it is important to consider the future of the HapMap Project. Currently, additional samples from the populations used to develop the initial HapMap, as well as samples from seven additional populations (Luhya in Webuye, Kenya; Maasai in Kinyawa, Kenya; Tuscans in Italy; Gujarati Indian in Houston, Texas, USA; Denver (Colorado) metropolitan Chinese community; people of Mexican origin in Los Angeles, California, USA; and people with African ancestry in the southwestern United States; http://ccr.coriell.org/Sections/Collections/NHGRI/?SsId=11) will be sequenced and genotyped extensively to extend the HapMap, providing information on rarer variants and helping to enable genome-wide association studies in additional populations. There are also ongoing efforts by many groups to characterize additional forms of genetic variation, such as structural variation, and molecular phenotypes in the HapMap samples. Finally, in the future, whole-genome sequencing will provide a natural convergence of technologies to type both SNP and structural variation. Nevertheless, until that point, and even after, the HapMap Project data will provide an invaluable resource for understanding the structure of human genetic variation and its link to phenotype.
METHODS SUMMARY
Of approximately 6.9 million SNPs in dbSNP release 122 approximately 4.7 million were selected for genotyping by Perlegen. 2.5 million SNPs were excluded because no assay could be designed and a further 350,000 were excluded for other reasons (see Methods). Perlegen performed genotyping using custom high-density oligonucleotide arrays as previously described15. Additional genotype submissions are described in the text. QC filters were applied as previously described3. Where multiple submissions met the QC criteria the submission with the lowest missing data rate was chosen for inclusion in the non-redundant filtered data set. Haplotypes were estimated from genotype data as described previously3. Ancestral states at SNPs were inferred by parsimony by comparison to orthologous bases in the chimpanzee (panTro2) and rhesus macaque (rheMac2) assemblies. Recombination rates and the location of recombination hotspots were estimated as described previously3. Additional details can be found in the Methods section and the Supplementary Information. The data described in this paper are in release 21 of the International HapMap Project.
METHODS
SNP selection and genotyping
All SNPs in dbSNP release 122 were considered for genotyping by Perlegen. Among these the following were excluded: SNPs for which no assay could be designed (primarily through location in repeat-rich regions; approximately 2.5 million); SNPs shown previously in samples from related populations15 to be most probably in perfect association (r2 = 1) with a Phase I SNP (approximately 122,000); all but one of SNPs shown previously15 to be most probably in perfect association (r2 = 1) with each other but not with a Phase I SNP (approximately 62,000); and SNPs shown previously15 to have MAF < 0.05 (approximately 119,000). In addition, a few SNPs were excluded for efficiency (for example, if an amplicon contained a single SNP). Approximately 30,000 SNPs that had been typed in Phase I were deliberately retyped in Phase II to allow detailed comparisons of data quality, and an additional 15,000 SNPs that showed discrepancies between multiple genotyping attempts in Phase I were re-typed in Phase II. A further 2,000 SNPs identified by the Mammalian Gene Collection were also typed.
Perlegen performed genotyping using custom high-density oligonucleotide arrays as previously described15. Initially, a pilot phase was carried out on chromosome 2p to optimize experimental workflow and data handling. Details of amplicons used in the experiment and PCR primers can be found at http://genome.perlegen.com/pcr/ and also on the HapMap website. The arrays were tiled with sets of 25-bp probes for each SNP, with either 40 or 24 probes per SNP. These consisted of four sets of features, corresponding to forward and reverse strand tilings of sequences complementary to each of the two SNP alleles. Within a feature set, the position of the SNP within the oligonucleotide varied from position 11 to position 15. Mismatch probes were used to measure background, and by comparison with the perfect match probes, to detect the presence or absence of a specific PCR product. The 40-feature and 24-feature tilings both provided 10 perfect-match features for each SNP allele and differed only in the number of mismatch probes.
Genotypes were scored by clustering intensity measurements as previously described15. In addition, quality scores similar to Phred scores were computed for each genotype call, based on a combination of experimental metrics correlated to data quality. Assays with overall call rates less than 80% or with poor average quality scores were flagged as failed. About 38% of the tiled assays failed these basic criteria, and the remainder were processed using the more rigorous HapMap Project data quality control filters. For analysis of the whole genome, probes for 4,373,926 distinct SNPs were tiled onto 32 chip designs, with 32 SNPs tiled in replicate onto each chip design for quality control (QC). Perlegen did not type the samples by plates as had been done for the Phase I genotyping, instead typing large numbers of SNPs one sample at a time. Consequently, blank wells on each plate were not included as a component of QC for this genotyping. In the Phase I HapMap a single JPT sample had been excluded because of technical problems. Perlegen typed a replacement sample (from the original JPT collection) for all new SNPs. This sample was not specifically genotyped on the Phase I SNPs, although a substantial fraction of these was typed in Phase II.
Additional genotype submissions came from the Affymetrix GeneChip Human Mapping 500K array called with the BRLMM algorithm. In release 21a additional genotype submissions were incorporated from the MHC haplotype consortium11, the Illumina HumanHap300 BeadChip, the Illumina Human-1 Genotyping BeadChip and the 10K non-synonymous SNP set from Affymetrix (ParAllele).
Details of primer design, DNA amplification, DNA labelling and hybridization and signal detection for the Perlegen platform can be found in Supplementary Text 7.
QC analyses
Genotype submissions were assessed for mendelian errors (where possible), missing data rates and Hardy–Weinberg proportions. QC filters were applied as previously described3; to achieve QC1 status a SNP had to have fewer than two mendelian errors, less than 20% missing data and P > 0.001 for Hardy–Weinberg analysis. The consensus data set consists only of SNPs for which QC+ submissions were available from all analysis panels. Where multiple submissions met the QC criteria the submission with the lowest missing data rate was chosen for inclusion in the non-redundant filtered data set. Comparison of the Phase II HapMap with the Affymetrix 500K genotypes has shown approximately 20 SNPs where the reported minor allele is discrepant (referred to as ‘allele-flipping’). Over the entire data set, we expect that 500–2,000 SNPs have this problem and the vast majority will occur in SNPs from Phase I of the project. The Data Coordination Center (DCC) is working to resolve as many of these as possible.
Analyses of data quality
See Supplementary Text 2.
Analyses of population stratification, relatedness and homozygosity
Analysis of recombination rate and gene ontology
We used the Panther Database41 to obtain details of the gene molecular function and biological process. Genes are grouped into 28 top-level molecular function groups and 30 top-level biological process groups, with each gene allowed to exist in more than one group. We identified 14,979 non-overlapping autosomal genes from the Panther RefSeq Annotation for which we could obtain recombination rates. Of these, 9,735 had at least one assigned molecular function and 9,432 had at least one assigned biological process. Genes without a molecular function or biological process were removed from the corresponding analysis. To control for gene size, we estimated the mean recombination rate over a 20-kb region centred on the mid-point of each gene transcription region.
Genes were grouped based on molecular function and biological process. A mean recombination rate was calculated for each group. The significance of the result from each group was calculated via a permutation test involving 105 random groupings of genes. No correction was made for multiple testing. To account for the effect of G+C content on recombination, we performed a linear regression between the G+C content and recombination rate of all genes in each sample. Using the estimated regression parameters, the proportion of recombination explained by G+C content was subtracted from each gene.
Identification of non-synonymous SNPs and tests for natural selection
Using annotations from dbSNP release 125 we identified 17,427 polymorphic non-synonymous SNPs in release 21 and 15,976 polymorphic synonymous SNPs. Of these, 15,583 non-synonymous and 14,324 synonymous SNPs were autosomal and could have ancestral allele status unambiguously assigned by parsimony through comparison to the chimpanzee and macaque genomes. We used the phased haplotypes for analysis in which missing data had been imputed. FST was calculated using the method of Weir and Cockerham59.
To detect recent partial selective sweeps we used the long-range haplotype (LRH) test44,49 and the integrated haplotype score (iHS) test9. On simulated data45, we found that the tests have similar power to detect recent selection but the iHS test has slightly lower power at low haplotype frequency and the LRH test has slightly lower power at high frequency. This can be seen in applications to HapMap Phase I data3,9, where the iHS test misses the well-known cases of HBB and CD36 and the LRH test misses the SULT1C2 region. Although both tests are based on the concept of EHH44, we observed that the false positives produced by the two tests tend not to overlap and thus that signals detected by both tests have a very low false-positive rate.
Supplementary Material
Acknowledgements
We thank many people who contributed to this project: all members of the genotyping laboratory and the sample, primer, bioinformatics, data quality and IT groups at Perlegen Sciences for technical and infrastructural support; J. Beck, C. Beiswanger, D. Coppock, A. Leach, J. Mintzer and L. Toji for transforming the Yoruba, Japanese and Han Chinese samples, distributing the DNA and cell lines, storing the samples for use in future research, and producing the community newsletters and reports; J. Greenberg and R. Anderson for providing funding and support for cell line transformation and storage in the NIGMS Human Genetic Cell Repository at the Coriell Institute; T. Dibling, T. Ishikura, S. Kanazawa, S. Mizusawa and S. Saito for help with genotyping; C. Hind and A. Moghadam for technical support in genotyping and all members of the subcloning and sequencing teams at the Wellcome Trust Sanger Institute; X. Ke for help with data analysis; Oxford E-Science Centre for provision of high-performance computing resources; H. Chen, W. Chen, L. Deng, Y. Dong, C. Fu, L. Gao, H. Geng, J. Geng, M. He, H. Li, H. Li, S. Li, X. Li, B. Liu, Z. Liu, F. Lu, F. Lu, G. Lu, C. Luo, X. Wang, Z. Wang, C. Ye and X. Yu for help with genotyping and sample collection; X. Feng, Y. Li, J. Ren and X. Zhou for help with sample collection; J. Fan, W. Gu, W. Guan, S. Hu, H. Jiang, R. Lei, Y. Lin, Z. Niu, B. Wang, L. Yang, W. Yang, Y. Wang, Z. Wang, S. Xu, W. Yan, H. Yang, W. Yuan, C. Zhang, J. Zhang, K. Zhang and G. Zhao for help with genotyping; P. Fong, C. Lai, C. Lau, T. Leung, L. Luk and W. Tong for help with genotyping; C. Pang for help with genotyping; K. Ding, B. Qiang, J. Zhang, X. Zhang and K. Zhou for help with genotyping; Q. Fu, S. Ghose, X. Lu, D. Nelson, A. Perez, S. Poole, R. Vega and H. Yonath for help with genotyping; C. Bruckner, T. Brundage, S. Chow, O. Iartchouk, M. Jain, M. Moorhead and K. Tran for help with genotyping; N. Addleman, J. Atilano, T. Chan, C. Chu, C. Ha, T. Nguyen, M. Minton and A. Phong for help with genotyping, and D. Lind for help with quality control and experimental design; R. Donaldson and S. Duan for help with genotyping, and J. Rice and N. Saccone for help with experimental design; J. Wigginton for help with implementing and testing QA/QC software; A. Clark, B. Keats, R. Myers, D. Nickerson and A. Williamson for providing advice to NIH; C. Juenger, C. Bennet, C. Bird, J. Melone, P. Nailer, M. Weiss, J. Witonsky and E. DeHaut-Combs for help with project management; M. Gray for organizing phone calls and meetings; D. Leja for help with figures; the Yoruba people of Ibadan, Nigeria, the people of Tokyo, Japan, and the community at Beijing Normal University, who participated in public consultations and community engagements; the people in these communities who donated their blood samples; and the people in the Utah CEPH community who allowed the samples they donated earlier to be used for the Project. This work was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology, the Wellcome Trust, Nuffield Trust, Wolfson Foundation, UK EPSRC, Genome Canada, Génome Québec, the Chinese Academy of Sciences, the Ministry of Science and Technology of the People's Republic of China, the National Natural Science Foundation of China, the Hong Kong Innovation and Technology Commission, the University Grants Committee of Hong Kong, the SNP Consortium, the US National Institutes of Health (FIC, NCI, NCRR, NEI, NHGRI, NIA, NIAAA, NIAID, NIAMS, NIBIB, NIDA, NIDCD, NIDCR, NIDDK, NIEHS, NIGMS, NIMH, NINDS, NLM, OD), the W.M. Keck Foundation, and the Delores Dore Eccles Foundation. All SNPs genotyped within the HapMap Project are available from dbSNP (http://www.ncbi.nlm.nih.gov/SNP); all genotype information is available from dbSNP and the HapMap website (http://www.hapmap.org).
The International HapMap Consortium
The International HapMap Consortium (Participants are arranged by institution and then alphabetically within institutions except for Principal Investigators and Project Leaders, as indicated.)
Genotyping centres: Perlegen Sciences Kelly A. Frazer (Principal Investigator)1, Dennis G. Ballinger2, David R. Cox2, David A. Hinds2, Laura L. Stuve2; Baylor College of Medicine and ParAllele BioScience Richard A. Gibbs (Principal Investigator)3, John W. Belmont3, Andrew Boudreau4, Paul Hardenbol5, Suzanne M. Leal3, Shiran Pasternak6, David A. Wheeler3, Thomas D. Willis4, Fuli Yu7; Beijing Genomics Institute Huanming Yang (Principal Investigator)8, Changqing Zeng (Principal Investigator)8, Yang Gao8, Haoran Hu8, Weitao Hu8, Chaohua Li8, Wei Lin8, Siqi Liu8, Hao Pan8, Xiaoli Tang8, Jian Wang8, Wei Wang8, Jun Yu8, Bo Zhang8, Qingrun Zhang8, Hongbin Zhao8, Hui Zhao8, Jun Zhou8; Broad Institute of Harvard and Massachusetts Institute of Technology Stacey B. Gabriel (Project Leader)7, Rachel Barry7, Brendan Blumenstiel7, Amy Camargo7, Matthew Defelice7, Maura Faggart7, Mary Goyette7, Supriya Gupta7, Jamie Moore7, Huy Nguyen7, Robert C. Onofrio7, Melissa Parkin7, Jessica Roy7, Erich Stahl7, Ellen Winchester7, Liuda Ziaugra7, David Altshuler (Principal Investigator)7,9; Chinese National Human Genome Center at Beijing Yan Shen (Principal Investigator)10, Zhijian Yao10; Chinese National Human Genome Center at Shanghai Wei Huang (Principal Investigator)11, Xun Chu11, Yungang He11, Li Jin12, Yangfan Liu11, Yayun Shen11, Weiwei Sun11, Haifeng Wang11, Yi Wang11, Ying Wang11, Xiaoyan Xiong11, Liang Xu11; Chinese University of Hong Kong Mary M. Y. Waye (Principal Investigator)13, Stephen K. W. Tsui13; Hong Kong University of Science and Technology Hong Xue (Principal Investigator)14, J. Tze-Fei Wong14; Illumina Luana M. Galver (Project Leader)15, Jian-Bing Fan15, Kevin Gunderson15, Sarah S. Murray1, Arnold R. Oliphant16, Mark S. Chee (Principal Investigator)17; McGill University and Génome Québec Innovation Centre Alexandre Montpetit (Project Leader)18, Fanny Chagnon18, Vincent Ferretti18, Martin Leboeuf18, Jean-François Olivier4, Michael S. Phillips18, Stéphanie Roumy15, Clémentine Sallée19, Andrei Verner18, Thomas J. Hudson (Principal Investigator)20; University of California at San Francisco and Washington University Pui-Yan Kwok (Principal Investigator)21, Dongmei Cai21, Daniel C. Koboldt22, Raymond D. Miller22, Ludmila Pawlikowska21, Patricia Taillon-Miller22, Ming Xiao21; University of Hong Kong Lap-Chee Tsui (Principal Investigator)23, William Mak23, You Qiang Song23, Paul K. H. Tam23; University of Tokyo and RIKEN Yusuke Nakamura (Principal Investigator)24,25, Takahisa Kawaguchi25, Takuya Kitamoto25, Takashi Morizono25, Atsushi Nagashima25, Yozo Ohnishi25, Akihiro Sekine25, Toshihiro Tanaka25, Tatsuhiko Tsunoda25; Wellcome Trust Sanger Institute Panos Deloukas (Project Leader)26, Christine P. Bird26, Marcos Delgado26, Emmanouil T. Dermitzakis26, Rhian Gwilliam26, Sarah Hunt26, Jonathan Morrison27, Don Powell26, Barbara E. Stranger26, Pamela Whittaker26, David R. Bentley (Principal Investigator)28
Analysis groups: Broad Institute Mark J. Daly (Project Leader)7,9, Paul I. W. de Bakker7,9, Jeff Barrett7,9, Yves R. Chretien7, Julian Maller7,9, Steve McCarroll7,9, Nick Patterson7, Itsik Pe'er29, Alkes Price7, Shaun Purcell9, Daniel J. Richter7, Pardis Sabeti7, Richa Saxena7,9, Stephen F. Schaffner7, Pak C. Sham23, Patrick Varilly7, David Altshuler (Principal Investigator)7,9; Cold Spring Harbor Laboratory Lincoln D. Stein (Principal Investigator)6, Lalitha Krishnan6, Albert Vernon Smith6, Marcela K. Tello-Ruiz6, Gudmundur A. Thorisson30; Johns Hopkins University School of Medicine Aravinda Chakravarti (Principal Investigator)31, Peter E. Chen31, David J. Cutler31, Carl S. Kashuk31, Shin Lin31; University of Michigan Gonçalo R. Abecasis (Principal Investigator)32, Weihua Guan32, Yun Li32, Heather M. Munro33, Zhaohui Steve Qin32, Daryl J. Thomas34; University of Oxford Gilean McVean (Project Leader)35, Adam Auton35, Leonardo Bottolo35, Niall Cardin35, Susana Eyheramendy35, Colin Freeman35, Jonathan Marchini35, Simon Myers35, Chris Spencer7, Matthew Stephens36, Peter Donnelly (Principal Investigator)35; University of Oxford, Wellcome Trust Centre for Human Genetics Lon R. Cardon (Principal Investigator)37, Geraldine Clarke38, David M. Evans38, Andrew P. Morris38, Bruce S. Weir39; RIKEN Tatsuhiko Tsunoda (Principal Investigator)25, Todd A. Johnson25; US National Institutes of Health James C. Mullikin40; US National Institutes of Health National Center for Biotechnology Information Stephen T. Sherry41, Michael Feolo41, Andrew Skol42
Community engagement/public consultation and sample collection groups: Beijing Normal University and Beijing Genomics Institute Houcan Zhang43, Changqing Zeng8, Hui Zhao8; Health Sciences University of Hokkaido, Eubios Ethics Institute, and Shinshu University Ichiro Matsuda (Principal Investigator)44, Yoshimitsu Fukushima45, Darryl R. Macer46, Eiko Suda47; Howard University and University of Ibadan Charles N. Rotimi (Principal Investigator)48, Clement A. Adebamowo49, Ike Ajayi49, Toyin Aniagwu49, Patricia A. Marshall50, Chibuzor Nkwodimmah49, Charmaine D. M. Royal48; University of Utah Mark F. Leppert (Principal Investigator)51, Missy Dixon51, Andy Peiffer51
Ethical, legal and social issues: Chinese Academy of Social Sciences Renzong Qiu52; Genetic Interest Group Alastair Kent53; Kyoto University Kazuto Kato54; Nagasaki University Norio Niikawa55; University of Ibadan School of Medicine Isaac F. Adewole49; University of Montréal Bartha M. Knoppers19; University of Oklahoma Morris W. Foster56; Vanderbilt University Ellen Wright Clayton57; Wellcome Trust Jessica Watkin58
SNP discovery: Baylor College of Medicine Richard A. Gibbs (Principal Investigator)3, John W. Belmont3, Donna Muzny3, Lynne Nazareth3, Erica Sodergren3, George M. Weinstock3, David A. Wheeler3, Imtaz Yakub3; Broad Institute of Harvard and Massachusetts Institute of Technology Stacey B. Gabriel (Project Leader)7, Robert C. Onofrio7, Daniel J. Richter7, Liuda Ziaugra7, Bruce W. Birren7, Mark J. Daly7,9, David Altshuler (Principal Investigator)7,9; Washington University Richard K. Wilson (Principal Investigator)59, Lucinda L. Fulton59; Wellcome Trust Sanger Institute Jane Rogers (Principal Investigator)26, John Burton26, Nigel P. Carter26, Christopher M. Clee26, Mark Griffiths26, Matthew C. Jones26, Kirsten McLay26, Robert W. Plumb26, Mark T. Ross26, Sarah K. Sims26, David L. Willey26
Scientific management: Chinese Academy of Sciences Zhu Chen60, Hua Han60, Le Kang60; Genome Canada Martin Godbout61, John C. Wallenburg62; Génome Québec Paul L'Archevêque63, Guy Bellemare63; Japanese Ministry of Education, Culture, Sports, Science and Technology Koji Saeki64; Ministry of Science and Technology of the People's Republic of China Hongguang Wang65, Daochang An65, Hongbo Fu65, Qing Li65, Zhen Wang65; The Human Genetic Resource Administration of China Renwu Wang66; The SNP Consortium Arthur L. Holden15; US National Institutes of Health Lisa D. Brooks67, Jean E. McEwen67, Mark S. Guyer67, Vivian Ota Wang67,68, Jane L. Peterson67, Michael Shi69, Jack Spiegel70, Lawrence M. Sung71, Lynn F. Zacharia67, Francis S. Collins72; Wellcome Trust Karen Kennedy61, Ruth Jamieson58, John Stewart58
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
The Scripps Research Institute, 10550 North Torrey Pines Road MEM275, La Jolla, California 92037, USA.
Perlegen Sciences, Inc., 2021 Stierlin Court, Mountain View, California 94043, USA.
Baylor College of Medicine, Human Genome Sequencing Center, Department of Molecular and Human Genetics, 1 Baylor Plaza, Houston, Texas 77030, USA.
Affymetrix, Inc., 3420 Central Expressway, Santa Clara, California 95051, USA.
Pacific Biosciences, 1505 Adams Drive, Menlo Park, California 94025, USA.
Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, New York 11724, USA.
The Broad Institute of Harvard and Massachusetts Institute of Technology, 1 Kendall Square, Cambridge, Massachusetts 02139, USA.
Beijing Genomics Institute, Chinese Academy of Sciences, Beijing 100300, China.
Massachusetts General Hospital and Harvard Medical School, Simches Research Center, 185 Cambridge Street, Boston, Massachusetts 02114, USA.
Chinese National Human Genome Center at Beijing, 3-707 N. Yongchang Road, Beijing Economic-Technological Development Area, Beijing 100176, China.
Chinese National Human Genome Center at Shanghai, 250 Bi Bo Road, Shanghai 201203, China.
Fudan University and CAS-MPG Partner Institute for Computational Biology, School of Life Sciences, SIBS, CAS, Shanghai 201203, China.
The Chinese University of Hong Kong, Department of Biochemistry, The Croucher Laboratory for Human Genetics, 6/F Mong Man Wai Building, Shatin, Hong Kong.
Hong Kong University of Science and Technology, Department of Biochemistry and Applied Genomics Center, Clear Water Bay, Knowloon, Hong Kong.
Illumina, 9885 Towne Centre Drive, San Diego, California 92121, USA.
Complete Genomics, Inc., 658 North Pastoria Avenue, Sunnyvale, California 94085, USA.
Prognosys Biosciences, Inc., 4215 Sorrento Valley Boulevard, Suite 105, San Diego, California 92121, USA.
McGill University and Génome Québec Innovation Centre, 740 Dr. Penfield Avenue, Montréal, Québec H3A 1A4, Canada.
University of Montréal, The Public Law Research Centre (CRDP), PO Box 6128, Downtown Station, Montréal, Québec H3C 3J7, Canada.
Ontario Institute for Cancer Research, MaRS Centre, South Tower, 101 College Street, Suite 500, Toronto, Ontario M5G 1L7, Canada.
University of California, San Francisco, Cardiovascular Research Institute, 513 Parnassus Avenue, Box 0793, San Francisco, California 94143, USA.
Washington University School of Medicine, Department of Genetics, 660 South Euclid Avenue, Box 8232, St Louis, Missouri 63110, USA.
University of Hong Kong, Genome Research Centre, 6/F, Laboratory Block, 21 Sassoon Road, Pokfulam, Hong Kong.
University of Tokyo, Institute of Medical Science, 4-6-1 Sirokanedai, Minato-ku, Tokyo 108-8639, Japan.
RIKEN SNP Research Center, 1-7-22 Suehiro-cho, Tsurumi-ku Yokohama, Kanagawa 230-0045, Japan.
Wellcome Trust Sanger Institute, The Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA, UK.
University of Cambridge, Department of Oncology, Cambridge CB1 8RN, UK.
Solexa Ltd, Chesterford Research Park, Little Chesterford, Nr Saffron Walden, Essex CB10 1XL, UK.
Columbia University, 500 West 120th Street, New York, New York 10027, USA.
University of Leicester, Department of Genetics, Leicester LE1 7RH, UK.
Johns Hopkins University School of Medicine, McKusick-Nathans Institute of Genetic Medicine, Broadway Research Building, Suite 579, 733 North Broadway, Baltimore, Maryland 21205, USA.
University of Michigan, Center for Statistical Genetics, Department of Biostatistics, 1420 Washington Heights, Ann Arbor, Michigan 48109, USA.
International Epidemiology Institute, 1455 Research Boulevard, Suite 550, Rockville, Maryland 20850, USA.
Center for Biomolecular Science and Engineering, Engineering 2, Suite 501, Mail Stop CBSE/ITI, UC Santa Cruz, Santa Cruz, California 95064, USA.
University of Oxford, Department of Statistics, 1 South Parks Road, Oxford OX1 3TG, UK.
University of Chicago, Department of Statistics, 5734 South University Avenue, Eckhart Hall, Room 126, Chicago, Illinois 60637, USA.
Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, Seattle, Washington 98109, USA.
University of Oxford/Wellcome Trust Centre for Human Genetics, Roosevelt Drive, Oxford OX3 7BN, UK.
University of Washington Department of Biostatistics, Box 357232, Seattle, Washington 98195, USA.
US National Institutes of Health, National Human Genome Research Institute, 50 South Drive, Bethesda, Maryland 20892, USA.
US National Institutes of Health, National Library of Medicine, National Center for Biotechnology Information, 8600 Rockville Pike, Bethesda, Maryland 20894, USA.
University of Chicago, Department of Medicine, Section of Genetic Medicine, 5801 South Ellis, Chicago, Illinois 60637, USA.
Beijing Normal University, 19 Xinjiekouwai Street, Beijing 100875, China.
Health Sciences University of Hokkaido, Ishikari Tobetsu Machi 1757, Hokkaido 061-0293, Japan.
Shinshu University School of Medicine, Department of Medical Genetics, Matsumoto 390-8621, Japan.
United Nations Educational, Scientific and Cultural Organization (UNESCO Bangkok), 920 Sukhumwit Road, Prakanong, Bangkok 10110, Thailand.
University of Tsukuba, Eubios Ethics Institute, PO Box 125, Tsukuba Science City 305-8691, Japan.
Howard University, National Human Genome Center, 2216 6th Street, NW, Washington DC 20059, USA.
University of Ibadan College of Medicine, Ibadan, Oyo State, Nigeria.
Case Western Reserve University School of Medicine,Department of Bioethics, 10900 Euclid Avenue, Cleveland, Ohio 44106, USA.
University of Utah, Eccles Institute of Human Genetics, Department of Human Genetics, 15 North 2030 East, Salt Lake City, Utah 84112, USA.
Chinese Academy of Social Sciences, Institute of Philosophy/Center for Applied Ethics, 2121, Building 9, Caoqiao Xinyuan 3 Qu, Beijing 100067, China.
Genetic Interest Group, 4D Leroy House, 436 Essex Road, London N130P, UK.
Kyoto University, Institute for Research in Humanities and Graduate School of Biostudies, Ushinomiya-cho, Sakyo-ku, Kyoto 606-8501, Japan.
Nagasaki University Graduate School of Biomedical Sciences, Department of Human Genetics, Sakamoto 1-12-4, Nagasaki 852-8523, Japan.
University of Oklahoma, Department of Anthropology, 455 West Lindsey Street, Norman, Oklahoma 73019, USA.
Vanderbilt University, Center for Genetics and Health Policy, 507 Light Hall, Nashville, Tennessee 37232, USA.
Wellcome Trust, 215 Euston Road, London NW1 2BE, UK.
Washington University School of Medicine, Genome Sequencing Center, Box 8501, 4444 Forest Park Avenue, St Louis, Missouri 63108, USA.
Chinese Academy of Sciences, 52 Sanlihe Road, Beijing 100864, China.
Genome Canada, 150 Metcalfe Street, Suite 2100, Ottawa, Ontario K2P 1P1, Canada.
McGill University, Office of Technology Transfer, 3550 University Street, Montréal, Québec H3A 2A7, Canada.
Génome Québec, 630, boulevard René-Lévesque Ouest, Montréal, Québec H3B 1S6, Canada.
Ministry of Education, Culture, Sports, Science, and Technology, 3-2-2 Kasumigaseki, Chiyodaku, Tokyo 100-8959, Japan.
Ministry of Science and Technology of the People's Republic of China, 15 B. Fuxing Road, Beijing 100862, China.
The Human Genetic Resource Administration of China, b7, Zaojunmiao, Haidian District, Beijing 100081, China.
US National Institutes of Health, National Human Genome Research Institute, 5635 Fishers Lane, Bethesda, Maryland 20892, USA.
US National Institutes of Health, Office of Behavioral and Social Science Research, 31 Center Drive, Bethesda, Maryland 20892, USA.
Novartis Pharmaceuticals Corporation, Biomarker Development, One Health Plaza, East Hanover, New Jersey 07936, USA.
US National Institutes of Health, Office of Technology Transfer, 6011 Executive Boulevard, Rockville, Maryland 20852, USA.
University of Maryland School of Law, 500 West Baltimore Street, Baltimore,Maryland 21201, USA.
US National Institutes of Health, National Human Genome Research Institute, 31 Center Drive, Bethesda, Maryland 20892, USA.
References
- 1.The International HapMap Consortium Integrating ethics and science in the International HapMap Project. Nature Rev. Genet. 2004;5:467–475. doi: 10.1038/nrg1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 3.The International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bowcock AM. Genomics: guilt by association. Nature. 2007;447:645–646. doi: 10.1038/447645a. [DOI] [PubMed] [Google Scholar]
- 5.Altshuler D, Daly M. Guilt beyond a reasonable doubt. Nature Genet. 2007;39:813–815. doi: 10.1038/ng0707-813. [DOI] [PubMed] [Google Scholar]
- 6.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 7.McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nature Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 8.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nature Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 9.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Bakker PI, et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nature Genet. 2006;38:1166–1172. doi: 10.1038/ng1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastinen T, et al. Mapping common regulatory variants to human haplotypes. Hum. Mol. Genet. 2005;14:3963–3971. doi: 10.1093/hmg/ddi420. [DOI] [PubMed] [Google Scholar]
- 13.Stranger BE, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung VG, et al. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–1369. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinds DA, et al. Whole-genome patterns of common DNA variation in three human populations. Science. 2005;307:1072–1079. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 16.de Bakker PI, et al. Efficiency and power in genetic association studies. Nature Genet. 2005;37:1217–1223. doi: 10.1038/ng1669. [DOI] [PubMed] [Google Scholar]
- 17.Pe'er I, et al. Evaluating and improving power in whole-genome association studies using fixed marker sets. Nature Genet. 2006;38:663–667. doi: 10.1038/ng1816. [DOI] [PubMed] [Google Scholar]
- 18.Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nature Genet. 2006;38:659–662. doi: 10.1038/ng1801. [DOI] [PubMed] [Google Scholar]
- 19.Burdick JT, Chen WM, Abecasis GR, Cheung VG. In silico method for inferring genotypes in pedigrees. Nature Genet. 2006;38:1002–1004. doi: 10.1038/ng1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Servin BR, Stephens M. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–668. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott LJ, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies via imputation of genotypes. Nature Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 24.Chapman JM, Cooper JD, Todd JA, Clayton DG. Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum. Hered. 2003;56:18–31. doi: 10.1159/000073729. [DOI] [PubMed] [Google Scholar]
- 25.Paabo S. The mosaic that is our genome. Nature. 2003;421:409–412. doi: 10.1038/nature01400. [DOI] [PubMed] [Google Scholar]
- 26.McVean G, Spencer CC, Chaix R. Perspectives on human genetic variation from the HapMap Project. PLoS Genet. 2005;1:e54. doi: 10.1371/journal.pgen.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Purcell S, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broman KW, Weber JL. Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain. Am. J. Hum. Genet. 1999;65:1493–1500. doi: 10.1086/302661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006;15:789–795. doi: 10.1093/hmg/ddi493. [DOI] [PubMed] [Google Scholar]
- 30.Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 31.Leutenegger AL, et al. Using genomic inbreeding coefficient estimates for homozygosity mapping of rare recessive traits: application to Taybi-Linder syndrome. Am. J. Hum. Genet. 2006;79:62–66. doi: 10.1086/504640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Te Meerman GJ, Van der Meulen MA, Sandkuijl LA. Perspectives of identity by descent (IBD) mapping in founder populations. Clin. Exp. Allergy. 1995;25(Suppl 2):97–102. doi: 10.1111/j.1365-2222.1995.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 33.Houwen RH, et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nature Genet. 1994;8:380–386. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 34.Durham LK, Feingold E. Genome scanning for segments shared identical by descent among distant relatives in isolated populations. Am. J. Hum. Genet. 1997;61:830–842. doi: 10.1086/514891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeffreys AJ, May CA. Intense and highly localized gene conversion activity in human meiotic crossover hot spots. Nature Genet. 2004;36:151–156. doi: 10.1038/ng1287. [DOI] [PubMed] [Google Scholar]
- 36.McVean GA, et al. The fine-scale structure of recombination rate variation in the human genome. Science. 2004;304:581–584. doi: 10.1126/science.1092500. [DOI] [PubMed] [Google Scholar]
- 37.Myers S, et al. The distribution and causes of meiotic recombination in the human genome. Biochem. Soc. Trans. 2006;34:526–530. doi: 10.1042/BST0340526. [DOI] [PubMed] [Google Scholar]
- 38.Spencer CC, et al. The influence of recombination on human genetic diversity. PLoS Genet. 2006;2:e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petes TD. Meiotic recombination hot spots and cold spots. Nature Rev. Genet. 2001;2:360–369. doi: 10.1038/35072078. [DOI] [PubMed] [Google Scholar]
- 40.Smith AV, Thomas DJ, Munro HM, Abecasis GR. Sequence features in regions of weak and strong linkage disequilibrium. Genome Res. 2005;15:1519–1534. doi: 10.1101/gr.4421405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas PD, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winckler W, et al. Comparison of fine-scale recombination rates in humans and chimpanzees. Science. 2005;308:107–111. doi: 10.1126/science.1105322. [DOI] [PubMed] [Google Scholar]
- 43.Ptak SE, et al. Fine-scale recombination patterns differ between chimpanzees and humans. Nature Genet. 2005;37:429–434. doi: 10.1038/ng1529. [DOI] [PubMed] [Google Scholar]
- 44.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 45.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. doi: 10.1038/nature06250. doi:10.1038/nature06250 (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bustamante CD, et al. Natural selection on protein-coding genes in the human genome. Nature. 2005;437:1153–1157. doi: 10.1038/nature04240. [DOI] [PubMed] [Google Scholar]
- 47.Cargill M, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nature Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 48.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabeti PC, et al. Positive natural selection in the human lineage. Science. 2006;312:1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 50.de Bakker PI, et al. Transferability of tag SNPs in genetic association studies in multiple populations. Nature Genet. 2006;38:1298–1303. doi: 10.1038/ng1899. [DOI] [PubMed] [Google Scholar]
- 51.Conrad DF, et al. A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nature Genet. 2006;38:1251–1260. doi: 10.1038/ng1911. [DOI] [PubMed] [Google Scholar]
- 52.Service S, Sabatti C, Freimer N. Tag SNPs chosen from HapMap perform well in several population isolates. Genet. Epidemiol. 2007;31:189–194. doi: 10.1002/gepi.20201. [DOI] [PubMed] [Google Scholar]
- 53.Lim J, et al. Comparative study of the linkage disequilibrium of an ENCODE region, chromosome 7p15, in Korean, Japanese, and Han Chinese samples. Genomics. 2006;87:392–398. doi: 10.1016/j.ygeno.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 54.Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. [DOI] [PubMed] [Google Scholar]
- 55.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 57.Smith RA, Ho PJ, Clegg JB, Kidd JR, Thein SL. Recombination breakpoints in the human β-globin gene cluster. Blood. 1998;92:4415–4421. [PubMed] [Google Scholar]
- 58.Holloway K, Lawson VE, Jeffreys AJ. Allelic recombination and de novo deletions in sperm in the human β-globin gene region. Hum. Mol. Genet. 2006;15:1099–1111. doi: 10.1093/hmg/ddl025. [DOI] [PubMed] [Google Scholar]
- 59.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.