

Abstract
Recent phosphoproteome analyses using mass spectrometry-based technologies have provided new insights into the extensive presence of protein phosphorylation in various species and have raised the interesting question of how this protein modification was gained evolutionarily on such a large scale. We investigated this issue by using human and mouse phosphoproteome data. We initially found that phosphoproteins followed a power-law distribution with regard to their number of phosphosites: most of the proteins included only a few phosphosites, but some included dozens of phosphosites. The power-law distribution, unlike more commonly observed distributions such as normal and log-normal distributions, is considered by the field of complex systems science to be produced by a specific rich-get-richer process called preferential attachment growth. Therefore, we explored the factors that may have promoted the rich-get-richer process during phosphosite evolution. We conducted a bioinformatics analysis to evaluate the relationship of amino acid sequences of phosphoproteins with the positions of phosphosites and found an overconcentration of phosphosites in specific regions of protein surfaces and implications that in many phosphoproteins these clusters of phosphosites are activated simultaneously. Multiple phosphosites concentrated in limited spaces on phosphoprotein surfaces may therefore function biologically as cooperative modules that are resistant to selective pressures during phosphoprotein evolution. We therefore proposed a hypothetical model by which the modularization of multiple phosphosites has been resistant to natural selection and has driven the rich-get-richer process of the evolutionary growth of phosphosite numbers.
Protein phosphorylation is an important and ubiquitous post-translational modification that regulates a variety of biological processes in various organisms (1–4). Reversible phosphorylations of serine, threonine, and tyrosine residues are critical steps in the control of signal transduction pathways (1–4). Recent advances in MS-based technologies and phosphopeptide enrichment methods have allowed high throughput and large scale in vivo phosphosite mapping for a wide variety of organisms such as human (5–8), mouse (9), yeast (10–12), fly (13,14), bacteria (15,16), and plants (17–19). Moreover information on several hundred to several thousand phosphosites from each study has been gathered in public databases such as Phospho.ELM (20), PhosphoSitePlus, PHOSIDA (21), and UniProt (22). However, the total number of phosphosites and most of their biological functions are still unknown. Similarly only about 10% of the estimated 500–600 human kinases target known phosphosite consensus sequences within their substrate proteins (23). Although the tyrosine phosphoproteome in Arabidopsis was recently published (24), the corresponding tyrosine kinases have not been identified because of the lack of known consensus sequences activated by tyrosine kinases.
Computational data-mining approaches have been required to extract information from the large amount of accumulated phosphosite data obtained from experimental approaches. These approaches have also been used to add more meaningful information about each of the phosphosites to understand the proteome-wide protein phosphorylation in various organisms. One of the most useful strategies of computational data mining is to identify phosphorylated sequence motifs by extracting consensus sequences from the sets of amino acid sequences clustered around phosphorylated residues (25). A number of kinases and their corresponding recognition substrate motifs have been successfully identified by the experimental approach of incubating each target kinase with a combinatorial substrate peptide library and ATP, and these data are registered in various databases, including the Human Protein Reference Database (HPRD)1 (26). With this knowledge of documented kinases and their related sequence motifs, we can use computational biology techniques to discover additional phosphorylated motifs in the numerous substrates shown in phosphoproteomics studies to be biologically phosphorylated. This has allowed us to reconstruct the kinome on a large scale (27–29).
A comparative study of phosphoproteome data in multiple species has revealed that a wide range of phosphoproteins are relatively well conserved relative to non-phosphoproteins, and similarly many phosphoserine (Ser(P)), phosphothreonine (Thr(P)), and phosphotyrosine (Tyr(P)) phosphosites are well conserved compared with non-phosphorylated sites (21). Under natural selection, the emergence of phosphoproteins and the gain and loss of phosphosites should have changed the regulation of many intracellular systems, such as kinetic pathways, subcellular protein localization, and protein interactions and stabilization. This triggered our interest in the evolution of phosphoproteins and their phosphosites.
In this study, we combined statistical physics and computational biology to investigate the role of selective pressure in the evolution of phosphoproteins and to create a model of the evolutionary gain of phosphosites. First using the human and mouse phosphoproteome data registered in public databases, we discovered that the number of phosphosites in each phosphoprotein follows a power-law distribution, which has been shown in complex systems science and statistical physics to emerge through a specific rich-get-richer process called preferential attachment growth (30–32). We therefore hypothesized that phosphoproteins may have evolved through a rich-get-richer process, gaining new phosphosites according to a probability density proportional to their current number of phosphosites. Starting from this hypothesis, we then explored how this particular evolutionary pattern may have arisen during natural selection and suggested that sets of phosphosites localized in limited spaces on protein surfaces may function as cooperative modules that are resistant to selective pressures. Therefore, to explain phosphosite evolution, we proposed a model in which the evolutionary gain of phosphosites follows a rich-get-richer process and evolution is promoted by the development of cooperative functional modules on protein surfaces.
EXPERIMENTAL PROCEDURES
Phosphosite Data—
We initially obtained the phosphosite data and phosphoproteome sequences from the UniProt database (22), which incorporates large scale data from many high quality phosphoproteomics studies. The complete list of UniProtKB/Swiss-Prot (Release 12.8) protein entries involving at least one phosphosite identified in high throughput phosphoproteomics studies was downloaded via the Protein Knowledgebase (UniProtKB) (33) in Extensible Markup Language (XML) format by querying the term scope: “PHOSPHORYLATION [LARGE SCALE ANALYSIS] AT.” Within each entry, Swiss-Prot accession IDs, protein name, positions of phosphosites, amino acid sequence of the protein, and functional annotations were included. When multiple Swiss-Prot IDs were included in an entry, throughout this study we used the first one.
The downloaded phosphoproteome data were obtained mainly from human (Homo sapiens), mouse (Mus musculus), and yeast (Saccharomyces cerevisiae). In human, among a total of 10,463 phosphosites in 3,290 phosphoproteins, there were 8,043 Ser(P) sites (in a total of 2,769 proteins), 1,496 Thr(P) sites (in 1,005 proteins), and 892 Tyr(P) sites (in 620 proteins). In mouse, within a total of 4,894 phosphosites (in 2,089 proteins), there were 3,466 Ser(P) sites (in 1,648 proteins), 744 Thr(P) sites (in 573 proteins), and 668 Tyr(P) sites (in 462 proteins). In yeast, within a total of 4,982 phosphosites (in 1,656 proteins), there were 4,069 Ser(P) sites (1,528 proteins), 887 Thr(P) sites (616 proteins), and 25 Tyr(P) sites (25 proteins).
Proteome Sequences—
The human proteome sequences were downloaded from the International Protein Index (IPI) of the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI) database (34), which provides minimally redundant but maximally complete sets of proteins for registered species. We obtained a set of 69,731 amino acid sequences.
Phosphoserine-based Motifs—
From the HPRD (26), we obtained a total of 171 Ser(P)-based human sequence motifs. Among these motifs, 158 were kinase and phosphatase motifs, and the others were protein-binding motifs.
Distribution of Phosphosite Numbers among Proteins—
We counted the numbers of phosphosites in each protein by using prepared sets of phosphoproteins that included any type of phosphosite (i.e. all phosphoproteins), Ser(P) sites, Thr(P) sites, or Tyr(P) sites in human, mouse, and yeast. Then the cumulative probability distributions of phosphoproteins in each set were calculated with regard to the numbers of all types of phosphosites, “only Ser(P) sites,” “only Thr(P) sites,” and “only Tyr(P) sites”; in each distribution, the numbers of phosphosites were denoted on the horizontal axis (X), and the proportion having more than X phosphosites of all the phosphoproteins in the set was indicated on the vertical axis (P>(X)). Each distribution was then approximated by a power-law function. When a probability distribution density follows a power law, the probability of variable X (i.e. p(X)) is inversely proportional to the power of the variable X with a power-law exponent −μ, and its cumulative probability distribution density P>(X) also follows a power law with the power-law exponent −μ + 1.
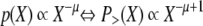 |
(Eq. 1) |
Therefore, to ascertain whether a distribution followed a power law, we analyzed its cumulative probability distribution because the cumulative probability distribution can normalize the effect of noise where the variable X is large with low p(X) arising from a small number of samples; this region is called the long tail region of the power-law distribution.
Characteristics of Phosphoprotein Sequences—
To investigate whether the characteristics of phosphoproteins affect their distribution with regard to the number of phosphosites they contain, we observed the lengths and compositions of the amino acid sequences of phosphoproteins. In both human and mouse, we calculated the Pearson's correlation coefficients between the number of Ser(P) sites and protein lengths and between the number of Ser(P) sites and number of Ser residues.
Probabilistic Logic-based Reannotation of Phosphosite Numbers—
To control for possible redundant annotations in the UniProt data, we newly estimated the number of phosphosites in the proteome sequences. We adopted a probabilistic logic model of the proteome-wide mapping of phosphopeptides and stochastically estimated the number of phosphosites within each protein where at least one of its peptide sources matched multiple other protein sequences. We obtained human phosphopeptide data from the PHOSIDA database (21) and downloaded the human proteome sequences from IPI. Of all phosphosites within the phosphopeptides in the PHOSIDA database, we selected only the Ser(P) sites that scored highly reliable experimental post-translational modification probabilities (p > 0.75) (6).
According to the experimental procedure of PHOSIDA in which trypsin is used to convert the protein mixture into a more analyzable population of peptides (21), we integrated the redundancy of phosphopeptides at the sequence level and matched them to proteome sequences on the basis of the following rules: the exclusive cleavage of a C-terminal Arg or Lys and the possible cleavage of the weakest peptide bond between Asp (on the N-terminal side) and Pro (on the C-terminal side) were induced in the trypsin digestion in the MS-based proteome analysis (35). When a phosphopeptide fragment harboring k Ser(P) sites was redundantly matched to n protein sequences, we counted the expectation value of k divided by n (i.e. k/n) for each of n proteins. Using this procedure, we created a probabilistic logic-based reannotated set of the number of Ser(P) sites in each phosphoprotein. Then we analyzed the cumulative probability density distribution of all phosphoproteins included in the reannotated set according to their estimated number of Ser(P) sites.
Finally by downloading the cross-reference file of the protein IDs of multiple databases from the IPI, we integrated the information on the stochastically estimated number of Ser(P) sites with data from each corresponding phosphoprotein obtained from UniProt. Then using the phosphoproteins present in both the UniProt set and the reannotated set, we demonstrated the respective distributions of phosphoproteins according to the Ser(P) site numbers in the UniProt data and those in the reannotated set. Within the intersection set, the Pearson's correlation coefficient between the number of Ser(P) sites of respective proteins annotated in UniProt and those in the reannotated set was also calculated.
Localization of Phosphosites on Protein Surfaces—
We evaluated the locations of phosphosites on the protein surfaces in the human and mouse phosphoproteome data from UniProt. For each set of all amino acid sequences of phosphoproteins in human and mouse, we used a sliding window of a certain size (see below) with a 1 − amino acid (AA) displacement and counted all the possible distances between two Ser(P) sites within the window when more than two Ser(P) sites were included in the window. Negative controls were generated by a similar procedure; when multiple Ser(P) sites were observed within the sliding window, we moved them to serine residues randomly selected from all the serine residues, including the actual Ser(P) sites, within the window and counted all the possible distances between two Ser(P) sites set randomly in silico.
The SURFACE database of protein surfaces (36) contains the distribution of residue lengths of amino acid segments appearing on protein surfaces. According to the distribution, 85% of the patches exposed on protein surfaces are composed of between 10 AA and 40 AA residues, and the peak of the distribution is around 20 AA. Therefore, to estimate the concentration of multiple phosphosites on the protein surface, we adopted sliding window sizes of 40, 20, and 10 AA using human and mouse phosphoproteomics data, and we compared the probability distributions of all the possible distances between pairs of Ser(P) sites with those of the negative controls. For each of the analyses, the window size and object species were varied, the negative control analyses were repeated 1,000 times, and the average and the 95% confidence intervals of the probability distributions of the negative controls were calculated.
Sequence Consensus around Phosphosites—
We used the data set of Ser(P) sites obtained from UniProt to evaluate the sequence consensus of the amino acids surrounding the phosphosites within each phosphoprotein of human and mouse. For each Ser(P) site of each phosphoprotein, we identified the amino acids between the relative positions of −6 and +6. Then by using each set of 13 AA including the surrounding sequences, the information entropy of every relative position was first calculated on the basis of information theory, such as used in the sequence logo program (37). Let A be a class of 20 amino acids, and a is a given amino acid included within A. The occurrence ratio of a at position i can be represented as pi,a. The information entropy of a relative position i is defined as Hi (bit) and is calculated as follows.
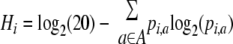 |
(Eq. 2) |
Finally we defined the mean information entropy Hmean (bit) for each phosphoprotein by dividing the total information entropies of all the relative positions between −6 and +6 by 13. Larger values of the Hmean score express a higher consensus of sequences surrounding Ser(P) sites in a protein; the maximum score is ∼4.32, derived from log2(20), and a score of 0 means no consensus.
We also analyzed whether multiple phosphosites in a single protein matched the same previously documented phosphosite-based motif. By using the human Ser(P)-based motifs obtained from the HPRD, we searched for the motifs that most commonly matched the Ser(P) sites within the phosphoproteins. For each human phosphoprotein having more than 10 Ser(P) sites, we calculated the percentage of Ser(P) sites that matched the most common motif in the protein. The negative control for each phosphoprotein was prepared by randomly selecting the same number of phosphosites from the total pool of human Ser(P) sites in UniProt. Then the occurrence percentage of the most common motif among the randomly selected set was calculated. This random procedure was repeated 1,000 times, and its mean and S.D. were computed.
RESULTS
Power-law Rule in Phosphoproteins—
We analyzed the distribution of phosphoproteins in human and mouse with regard to their number of Ser(P) sites using phosphoprotein data obtained from the UniProt database. The cumulative probability density of each human phosphoprotein was approximated by a power-law exponent of −1.92, and the r2 value of Pearson's correlation coefficient was 0.98 (Fig. 1A). The cumulative probability density of mouse proteins was approximated with a power-law exponent of −2.00, and the r2 value was 0.98 (Fig. 1B). Human and mouse phosphoproteins also followed power-law distributions with high correlations with regard to their number of Ser(P)/Thr(P)/Tyr(P) phosphosites, only Thr(P) sites, and only Tyr(P) sites (see supplemental Figs. S1–S3) as did yeast phosphoproteins for all types of phosphosites (data not shown).
Fig. 1.

Cumulative probability distribution of number of Ser(P) sites within phosphoproteins obtained from UniProt. Distributions of cumulative probabilities of human (A) and mouse (B) phosphoproteins obtained from the UniProt database are shown according to their number of Ser(P) sites (“# of pS”); each plot shows the proportion of phosphoproteins having more Ser(P) sites than the corresponding variable indicated on the horizontal axis (see “Experimental Procedures”). In each figure, both axes are on log scales, and the optimal approximate line of the power-law distribution is shown as a gray dotted line with the r2 value of the Pearson's correlation coefficient. The approximate formula of each distribution is shown in the dotted box; “C” means constant.
Next to investigate whether the power-law distributions were potentially affected by the characteristics of the phosphoprotein sequences, we examined the correlations between the numbers of Ser(P) sites and the lengths of the phosphoproteins in human and mouse. The r2 values calculated from the linear approximation were 0.02 in human and 0.05 in mouse (supplemental Fig. S4, A and B), indicating that there was no correlation between the Ser(P) site numbers and the lengths of the phosphoproteins. Similarly the number of Ser(P) sites and the number of Ser residues in the human and mouse phosphoproteins had r2 values of 0.05 and 0.10, respectively (supplemental Fig. S4, C and D). In fact, the probability distributions of human and mouse phosphoproteins with regard to the number of Ser residues were log-normal distributions (Fig. 2). Therefore, the power-law rule linking the phosphosite numbers to protein probabilities was not caused by the characteristics of the primary sequences of amino acids within the phosphoproteins.
Fig. 2.

Probability distribution of number of Ser(P) sites and Ser residues within phosphoproteins obtained from UniProt. Probability distributions of the number of Ser(P) sites (“# of pS”) within the human (A) and mouse (B) phosphoproteins obtained from the UniProt database are shown; each point shows the proportion of phosphoproteins with the number of Ser(P) sites indicated on the horizontal axis. Using the same phosphoprotein set, the probability distributions of the number of Ser residues (“# of S”) within each human (C) and mouse (D) phosphoprotein were calculated. In each figure, both axes are log scale.
More than one protein may have identical stretches of amino acids, an issue that is not addressed in the UniProt database, and we were concerned about the possible effect of redundant or duplicative annotation on the distribution pattern of phosphosite numbers of individual proteins. Therefore, we used PHOSIDA phosphopeptide data obtained with an MS-based analysis to conduct a probabilistic logic-based estimation of the numbers of phosphosites for each protein within the IPI database, which consists of minimally redundant but maximally complete sets of documented proteins. From this, we generated a stochastically reannotated set of phosphoproteins along with their estimated number of Ser(P) sites (see “Experimental Procedures”). The distribution of the reannotated phosphoproteins with regard to their number of Ser(P) sites was also proportional to a power law with an exponent of −1.73 and an r2 value of 0.99 (Fig. 3A). When we focused only on phosphoproteins included in both UniProt and the reannotated data, a power-law distribution of phosphoproteins with regard to the number of Ser(P) sites annotated in UniProt was observed with an exponent of −2.00 and an r2 value of 0.98 (Fig. 3B). In this population, a power-low distribution was also observed according to the number of Ser(P) sites estimated in the reannotated set with a power-law exponent of −1.76 and an r2 value of 0.97 (Fig. 3C). Moreover the number of Ser(P) sites estimated in the reannotated set was correlated with the number in the UniProt database with an r2 value of 0.51 (Fig. 3D). Thus, the power-law rule in the number of phosphosites was not likely to arise from redundant proteome-wide mapping of phosphopeptides.
Fig. 3.

Cumulative probability distribution of number of Ser(P) sites within phosphoproteins of the reannotated set. The distribution of cumulative probabilities of phosphoproteins according to their estimated number of Ser(P) sites (“# of pS”) in the human phosphoproteins reannotated by using PHOSIDA and IPI data (A) is shown. The distribution of phosphoproteins in the intersection between UniProt and the reannotated set according to the number of Ser(P) sites annotated in the UniProt data (B) and in the reannotated set (C) is shown. For a detailed explanation of the cumulative probability distribution, see the legend to Fig. 1. Within the intersection set, the correlation between the number of Ser(P) sites annotated in UniProt and those estimated in the reannotated set was calculated with the r2 value of the Pearson's correlation coefficient (D). The dotted gray line indicates its straight line approximation.
Furthermore we demonstrated that the observed power-law distribution was independent of protein abundance. We were concerned that the phosphosites of abundant proteins might be more likely to be identified by MS, thereby resulting in the power-law distribution. Therefore, we estimated the absolute abundances of proteins in a lysate of HeLa cells at the proteome level by calculating the exponentially modified protein abundance index (emPAI (38)) in an LC-MS/MS experiment. We also used phosphoproteome data we obtained from the same cell lysate in our previous studies (5, 39,40) to count the numbers of phosphosites of individual proteins. We found no correlation between the number of phosphosites within respective proteins and their copy number (for details, see supplemental Fig. S5). In fact, in this analysis, proteins containing many phosphosites seemed to be less abundant than other proteins (supplemental Fig. S5).
Localization of Phosphosites on Protein Surfaces—
We hypothesized that the power-law distribution of phosphoproteins emerged during evolution according to the preferential attachment growth principle whereby a phosphoprotein gains new phosphosites according to a probability that is proportional to its current phosphosite number. To investigate what might have promoted the rich-get-richer growth of phosphosites during evolution, we conducted a bioinformatics analysis using human and mouse data from UniProt to estimate phosphosite localization on the protein surface. By sliding a window of a given size along the phosphoprotein sequences, we were able to measure the distances between all possible pairs of Ser(P) sites within each sliding window. According to the SURFACE database, most stretches of amino acids appearing on protein surfaces range from 10 to 40 AA with a peak at 20 AA. Therefore, we used these three values as the window sizes to predict the localization of phosphosites on protein surfaces. Negative controls were generated by randomly placing Ser(P) sites within the sliding windows onto any serine residues within the respective windows (see “Experimental Procedures”).
In all the results obtained by displacing sliding windows of 40, 20, and 10 AA by 1 AA on the sequences of human and mouse phosphoproteins, the distances between pairs of Ser(P) sites within the windows were significantly shorter than those generated by negative control trials (Fig. 4). For each window size in each species, we compared the probability distribution of distances between two actual Ser(P) sites with the higher confidence bound of the probability distribution estimated by 1,000 repetitions of the negative controls. When the sliding window size of 40 AA was used on the human data, the probabilities of distances of two Ser(P) sites under 5 were significantly higher than those estimated for the negative controls in human (p < 0.05) (Fig. 4A), and the probabilities of distances under 4 were significantly higher than those of the negative controls in mouse (p < 0.05) (Fig. 4D). Similarly the probabilities of distances under 3 were significantly higher than those of the negative controls when the sliding window of 20 AA was used in both human and mouse (p < 0.05) (Fig. 4, B and E), and the probabilities of distances under 2 were significantly higher than those of the negative controls when the sliding window of 10 AA was used in both species (p < 0.05) (Fig. 4, C and F). Although some probability differences between the positive and negative controls are hard to observe in the figures when a 10-AA sliding window was used in either species, especially at the distance of 1 AA in mouse, all of the probabilities for the positive controls were significantly higher than those for the negative controls (detailed data not shown). These results suggest that multiple phosphosites within a phosphoprotein tend to localize within the sequences located on the protein surfaces.
Fig. 4.

Distribution of distances between two Ser(P) sites within limited spaces of phosphoprotein sequences. Using sliding windows of 40, 20, and 10 AA on the phosphoproteome sequences with a 1-AA displacement, the probability density distributions of distances between all possible pairs of Ser(P) (pS) sites included within the sliding windows are shown as the black-filled graphs in A–C for human and D–F for mouse. Sequence distance (d) between two Ser(P) sites is shown by the number of amino acid residues between the two sites; thus, d = 0 means the two sites are next to each other. In each figure, the gray solid line indicates the average of distributions calculated from 1,000 random trials as negative controls, and both sides of the 95% confidence intervals estimated by the negative trials are indicated by gray dotted lines (see “Experimental Procedures”).
Sequence Consensus around Phosphosites with Each Protein—
Next we analyzed the sequence consensus around phosphosites in human proteins. Using information theory, we calculated the mean information entropy of Hmean for the amino acids between the positions of −6 and +6 relative to each Ser(P) within the same phosphoprotein. Within the 20 phosphoproteins with the largest number of Ser(P) sites, most of the Hmean scores were above 1, indicating that sequences surrounding Ser(P) sites in the respective phosphoproteins are strongly biased toward specific patterns (Table I). Moreover in sequence logo representations of the sequences surrounding each Ser(P) site within some example phosphoproteins, we also observed remarkable sequence biases around the Ser(P) sites (Fig. 5). However, we could not rule out the possibility that this result was caused by bias in the occurrence of amino acid residues within each protein rather than by the relationship of these residues to phosphosites.
Table I.
The 20 human phosphoserine proteins containing the highest numbers of Ser(P) sites
Proteins are sorted by number of Ser(P) sites annotated in the UniProt database; the top 20 are listed (for the full list, see the supplemental Table S1). The column “Name” indicates the protein names obtained from the UniProt database. Numbers of Ser(P) sites in each protein are shown under “No. of Ser(P)”; the column “UniProt” indicates those annotated in UniProt, and “Reannotated” indicates those estimated using the PHOSIDA and IPI databases. In the column “Length,” the number of amino acid residues in each protein is shown, and the number of Ser residues is in parentheses. Each value in the column “Hmean” is the mean information entropy calculated by using all the sequences surrounding Ser(P) sites within a given protein. The occurrence percentage of the most common motif of each protein is represented in subcolumn “P” of the column “Percentage of the most common motif.” The subcolumn “N” denotes the mean and S.D. calculated from 1,000 trials of the negative control. Selected annotations from UniProt are listed in the column “Comments.”
| Name | No. of Ser(P)
|
Length | Hmean | Percentage of the most common motif
|
Comments | ||
|---|---|---|---|---|---|---|---|
| UniProt | Reannotated | P | N | ||||
| SRRM2 | 142 | 45.78 | 2752 (642) | 1.27 | 36.62 | 21.52 ± 3.15 | Serine/arginine repetitive matrix protein 2 |
| SRRM1 | 60 | 31.96 | 904 (151) | 1.72 | 48.33 | 22.31 ± 4.24 | Serine/arginine repetitive matrix protein 1 |
| TCOF1 | 59 | 8.94 | 1488 (203) | 1.44 | 27.12 | 22.47 ± 4.61 | Treacle protein; Treacher Collins syndrome protein |
| TP53BP1 | 49 | 11.45 | 1972 (246) | 0.99 | 26.53 | 22.94 ± 4.87 | Tumor suppressor p53-binding protein 1 |
| IWS1 | 44 | 14.00 | 819 (112) | 1.81 | 38.64 | 22.72 ± 5.01 | IWS1 homolog |
| BCLAF1 | 39 | 5.62 | 920 (151) | 1.14 | 30.77 | 23.15 ± 5.27 | Bcl-2-associated transcription factor 1 |
| MAP1B | 33 | 50.00 | 2468 (295) | 1.26 | 24.24 | 23.22 ± 5.39 | Microtubule-associated protein 1B |
| ACIN1 | 33 | 8.17 | 1341 (152) | 1.33 | 54.55 | 23.22 ± 5.39 | Apoptotic chromatin condensation inducer in the nucleus |
| MKI67 | 31 | 4.00 | 3256 (280) | 1.47 | 41.94 | 23.40 ± 5.57 | Antigen KI-67 |
| HIRIP3 | 28 | 11.00 | 556 (70) | 1.67 | 25.00 | 23.70 ± 6.00 | HIRA-interacting protein 3 |
| KIAA1802 | 27 | 21.00 | 812 (113) | 1.62 | 14.81 | 24.17 ± 6.08 | Zinc finger protein KIAA1802 |
| PRPF4B | 26 | 27.00 | 1007 (117) | 1.49 | 50.00 | 24.20 ± 6.34 | Serine/threonine-protein kinase PRP4 homolog; PRP4 kinase |
| NUMA1 | 26 | 1.75 | 2115 (162) | 1.32 | 23.08 | 24.20 ± 6.34 | Nuclear mitotic apparatus protein 1 |
| THRAP3 | 25 | 28.67 | 955 (156) | 1.33 | 40.00 | 24.22 ± 6.20 | Thyroid hormone receptor-associated protein 3 |
| MDC1 | 25 | 2.50 | 2089 (201) | 1.30 | 12.00 | 24.22 ± 6.20 | Mediator of DNA damage checkpoint protein 1; nuclear factor with BRCT domains 1 |
| TNKS1BP1 | 23 | 3.50 | 1729 (217) | 1.37 | 26.09 | 24.81 ± 6.73 | 182-kDa tankyrase 1-binding protein |
| MAP4 | 23 | 1.58 | 1152 (114) | 1.30 | 34.78 | 24.81 ± 6.73 | Microtubule-associated protein 4; MAP 4 |
| ZC3H13 | 22 | 10.67 | 1668 (195) | 1.44 | 31.82 | 25.15 ± 6.67 | Zinc finger CCCH domain-containing protein 13 |
| INCENP | 22 | 2.67 | 923 (74) | 1.43 | 13.64 | 25.15 ± 6.67 | Inner centromere protein |
| AHNAK | 21 | 13.50 | 5890 (323) | 1.55 | 14.29 | 24.67 ± 6.70 | Neuroblast differentiation-associated protein; AHNAK |
| LEO1 | 21 | 10.50 | 666 (78) | 1.90 | 23.81 | 24.67 ± 6.70 | RNA polymerase-associated protein LEO1 |
| SFRS2IP | 21 | 6.33 | 1148 (164) | 1.42 | 23.81 | 24.67 ± 6.70 | SFRS2-interacting protein; splicing factor, arginine/serine-rich 2-interacting protein; SC35-interacting protein 1 |
| NUCKS1 | 21 | 4.80 | 243 (32) | 1.58 | 38.10 | 24.67 ± 6.70 | Nuclear ubiquitous casein and cyclin-dependent kinases substrate |
Fig. 5.

Sequence logo representation of sequences surrounding the Ser(P) sites within example phosphoproteins. Sequence logo representations of all the sequences surrounding the Ser(P) sites of the SRRM1, IWS1, MKI67, HIRIP3, KIAA1802, and LEO1 phosphoproteins are shown in A–F, respectively. The surrounding sequences include the relative positions of −6 to +6 from the Ser(P) (pS) site. The height of each symbol represents the occurrence percentage of the corresponding amino acid in the relative position indicated on the horizontal axis. The name of each protein is denoted in the left side of the black box on the upper side of each figure, and the mean information entropy (Hmean) calculated by using the sequences surrounding all of the Ser(P) sites in each protein is indicated on the right side with the number of surrounding sequences (equal to the number of Ser(P) sites in the protein) in parentheses.
On the other hand, many phosphorylation sequence motifs have been documented in previous studies, and substrate proteins including phosphorylation motifs are thought to be the targets of specific kinases (26). We therefore next examined whether multiple phosphosites in a single protein tended to be targeted by some of the common kinases. We obtained the documented Ser(P)-based motifs in human proteins from the HPRD, and for each phosphoprotein that included more than 10 Ser(P) sites we extracted the motif that most commonly matched the Ser(P) sites and calculated the percentage of Ser(P) sites represented by the most common motif. For each phosphoprotein, we calculated the occurrence percentage of the most common motif and the mean and S.D. calculated from 1,000 repetitions of the negative control trials (Table I and Fig. 6). The occurrence percentages of the most common motifs were much higher than those of the negative controls, especially in proteins with larger numbers of Ser(P) sites (Fig. 6). Although it is possible that the specific sequence pattern biases around Ser(P) sites within the respective phosphoproteins are caused by pattern biases that exist naturally in the whole proteins, we suggest that specific observed biases in the sequences surrounding sets of Ser(P) sites might promote the targeting of phosphosites within a protein by common kinase proteins.
Fig. 6.

Occurrence percentages of the most common motifs in phosphoproteins having many Ser(P) sites. The occurrence percentages of the most common motifs in each phosphoprotein are plotted as black-filled circles corresponding to the number of Ser(P) (pS) sites within the protein. Phosphoproteins with ≥10 Ser(P) sites were used in this analysis. Negative controls are represented as gray crosses; for each plot, we used the mean of the occurrence percentages of the most common motifs calculated by 1,000 negative trials (see “Experimental Procedures”).
Model of Phosphoprotein Evolution—
We further developed a computational program simulating the evolution of the phosphoprotein population according to the preferential attachment growth rule (supplemental Fig. S6). The model generated power-law distributions of phosphoproteins when several parameters were adopted (supplemental Fig. S7). These findings from our model might explain why phosphoproteins containing many phosphosites are likely to be conserved among multiple species (supplemental Fig. S8).
DISCUSSION
When the probability of measuring a particular value of some quantity varies inversely as a power of that value, the quantity is said to follow a power law, also known as Zipf's law or the Pareto distribution (30). Notably in the field of statistical physics, it has been shown that this distribution emerges by a rich-get-richer process (31,32) known as preferential attachment growth. A simple example of this process is the growing network of Web page links, which follow a power-law distribution (32). In this rich-get-richer generative model, each new Web page creates links to existing Web pages with a probability distribution that is not uniform or normal but is proportional to the current in degrees of Web pages (32). Power-law distributions have also been observed in some biological phenomena, such as the distribution of the node degrees in a protein-protein interaction network (41,42) and the distribution of the gene expression levels of various species (43). These findings suggest that genome evolution might be promoted by some extremely general and simple mechanisms based on the preferential attachment growth principle (41, 44–46). Here we hypothesized that the distribution of phosphosites and phosphoproteins has evolved through the rich-get-richer process of preferential attachment growth whereby the phosphoprotein population regularly grows and each phosphoprotein gains new phosphosites according to a probability that is proportional to its current phosphosite number.
We initially found that human and mouse phosphoproteins identified in the UniProt database followed a power-law distribution with regard to their numbers of phosphosites: whereas most of the phosphoproteins included only a few phosphosites, there were some that contained many tens of phosphosites. Because every category of phosphoprotein data (all types of phosphosites, only Ser(P) sites, only Thr(P) sites, and only Tyr(P) sites) exhibited a power-law distribution and the data were most abundant for Ser(P) sites and Ser(P) proteins in both the human and mouse (giving the most support for further statistical analyses), we focused on Ser(P) sites throughout this study to further investigate the power-law rule in phosphoproteins and their biological and evolutionary significance.
In the human phosphoprotein data in UniProt, we observed phosphoproteins with up to 142 Ser(P) sites (Table I). Although the overall content of Ser residues within a few of these proteins, such as the SRRM2 protein, was also high (Table I), we demonstrated that the power-law rule did not arise from the amino acid contents or any sequence characteristics of the phosphoproteins. We also eliminated the possibility that the power-law distribution of phosphoproteins was affected by redundant annotation in the UniProt database. In MS-based phosphoproteome analyses, proteins are digested to peptides, and then proteome-wide mapping of identified phosphopeptides is conducted to search for their source phosphoproteins and to determine the positions of phosphosites on the protein sequences. However, a fragment ion spectrum of a unique phosphopeptide can sometimes be matched to, and potentially attributed to, multiple proteins. Even a single gene may produce multiple proteins that contain similar protein fragments through alternative splicing (47). This problem has not been addressed in the UniProt data. To rule out the effects of redundant annotations of phosphosites possibly included in the UniProt data, we reannotated the phosphosites by using data from PHOSIDA and IPI and probabilistic logic, and we determined that the experimental and annotation procedures used for the phosphoprotein data did not affect the bias of phosphosite number or the power-law distribution of the phosphoproteins. However, the possibility remained that the phosphosite numbers of individual proteins were correlated with the abundance of the protein. We further demonstrated that phosphosite numbers were not related to protein abundance and that the power-law rule was not affected by the abundance of each protein (supplemental Fig. S5). The evolution of the phosphoprotein population according to the preferential attachment growth rule was further supported by our computational model (see supplemental Figs. S6 and S7).
Protein phosphorylation is an important post-translational modification that regulates a variety of biological processes, such as cellular signaling pathways, subcellular protein localizations, and protein interactions and stabilization (1–4). Among the list in Table I of human phosphoproteins that include many phosphosites, there are several that have notable functions. For example, TP53BP1 is a DNA damage checkpoint protein that binds to the DNA-binding domain of p53 and enhances p53-mediated transcriptional activation (48). TP53BP1 is known to be hyperphosphorylated in response to DNA damage (48). MDC1 is another DNA damage checkpoint protein that contributes to the early cellular responses to DNA damage by inhibiting phosphorylation of p53 to protect cells from apoptotic cell death (49). Additionally the phosphorylation of INCENP, an inner centromere protein, by Aurora B kinase regulates mitosis (50). Because the functional behaviors of these proteins might be highly related to their phosphorylation status, improving our understanding of the evolution of phosphosites will help us to understand the important functions of these and other phosphoproteins.
We then explored what factors can lead to the preferential attachment of new phosphosites within the growing phosphoprotein population. In our two bioinformatics sequence analyses (of phosphosite localization and the consensus sequence around phosphosites), phosphosites within a phosphoprotein tended to be concentrated in limited regions of the protein surface and to be targeted together by several common kinases. We therefore suggest that groups of phosphosites concentrated in specific regions of protein surfaces tend to be activated simultaneously by their respective kinases and act biologically as functional modules. Recently simultaneous phosphorylation of closely concentrated Ser(P) sites was identified in the amino acid sequence of the Amphiphysin I protein from rat brain nerve terminals (51). The simultaneous activation of localized phosphosites and their cooperativity may provide some benefits for the cellular system. In some cases, phosphorylation of multiple amino acid residues enables protein interaction or stabilization that is not achieved by single phosphorylation. For example, multiple phosphorylations in some components of the postsynaptic density, a protein complex lining the postsynaptic membranes of neurons, regulate synaptic structure and function (52). Furthermore when multiple phosphosites that are closely concentrated on a protein surface have similar effects on the cellular system, each might be able to amplify the effect of the phosphorylation or act as a backup for the others. For example, in cell cycle regulation of mitogen-activated protein kinase signaling in yeast, phosphorylation of multiple phosphosites on Ste5, a mitogen-activated protein kinase (MAPK) cascade scaffold protein, results in substantial accumulation of negative charges and is required for the effective inhibition of mitogen-activated protein kinase signaling (53).
In the evolution of a living system, biological merit is directly linked to the robustness of resistance to genetic changes. Phosphosites are significantly more conserved than non-phosphorylated sites (21). Here we have suggested that multiple phosphosites concentrated on a protein surface form functional modules and act cooperatively, and we consequently propose that they may be evolutionarily robust and resistant to natural selection pressure. Moreover if a phosphoprotein contains a larger number of phosphosites, the protein is more likely to form phosphosite modules, and the phosphosites are more likely to be robust; this increases the chance that each member of a larger cluster of phosphosites in a protein will survive over evolution. This logic is consistent with the rich-get-richer generative model of phosphosite evolution (see supplemental Figs. S6 and S7) and the result that phosphoproteins containing many phosphosites are likely to be conserved among multiple species (see supplemental Fig. S8). Thus, the functional and cooperative organization of multiple phosphosites that are concentrated in a limited space on the protein surface is a possible factor that has promoted the preferential attachment growth of phosphoproteins. Although the increasing amount of phosphoproteomics data is remarkable because of breakthroughs in many experimental techniques, our knowledge of the functions and evolution of phosphoproteins has not kept pace. We believe that our proposed hypothesis and findings will provide useful tools for uncovering the role of phosphorylation in living cellular systems.
Supplementary Material
Acknowledgments
We are grateful to Dr. Naoyuki Sugiyama for helpful discussions and Hiroyuki Nakamura for creating the graphical sequence logo representations.
Footnotes
Published, MCP Papers in Press, January 9, 2009, DOI 10.1074/mcp.M800466-MCP200
The abbreviations used are: HPRD, Human Protein Reference Database; IPI, International Protein Index; AA, amino acid(s).
This work was supported by research funds from Yamagata Prefecture and Tsuruoka City to Keio University and a grant from the Japan Society for the Promotion of Science (to N. Y.).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Hunter, T. ( 2000) Signaling—2000 and beyond. Cell 100, 113–127 [DOI] [PubMed] [Google Scholar]
- 2.Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. ( 2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 3.Cohen, P. ( 2000) The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 4.Pawson, T., and Nash, P. ( 2000) Protein-protein interactions define specificity in signal transduction. Genes Dev. 14, 1027–1047 [PubMed] [Google Scholar]
- 5.Sugiyama, N., Masuda, T., Shinoda, K., Nakamura, A., Tomita, M., and Ishihama, Y. ( 2007) Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol. Cell. Proteomics 6, 1103–1109 [DOI] [PubMed] [Google Scholar]
- 6.Olsen, J. V., Blagoev, B., Gnad, F., Macek, B., Kumar, C., Mortensen, P., and Mann, M. ( 2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 7.Beausoleil, S. A., Jedrychowski, M., Schwartz, D., Elias, J. E., Villen, J., Li, J., Cohn, M. A., Cantley, L. C., and Gygi, S. P. ( 2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U. S. A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molina, H., Horn, D. M., Tang, N., Mathivanan, S., and Pandey, A. ( 2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villen, J., Beausoleil, S. A., Gerber, S. A., and Gygi, S. P. ( 2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U. S. A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ficarro, S. B., McCleland, M. L., Stukenberg, P. T., Burke, D. J., Ross, M. M., Shabanowitz, J., Hunt, D. F., and White, F. M. ( 2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 11.Chi, A., Huttenhower, C., Geer, L. Y., Coon, J. J., Syka, J. E., Bai, D. L., Shabanowitz, J., Burke, D. J., Troyanskaya, O. G., and Hunt, D. F. ( 2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 104, 2193–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson-Grady, J. T., Villen, J., and Gygi, S. P. ( 2008) Phosphoproteome analysis of fission yeast. J. Proteome Res. 7, 1088–1097 [DOI] [PubMed] [Google Scholar]
- 13.Bodenmiller, B., Malmstrom, J., Gerrits, B., Campbell, D., Lam, H., Schmidt, A., Rinner, O., Mueller, L. N., Shannon, P. T., Pedrioli, P. G., Panse, C., Lee, H. K., Schlapbach, R., and Aebersold, R. ( 2007) PhosphoPep—a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol. Syst. Biol. 3, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhai, B., Villen, J., Beausoleil, S. A., Mintseris, J., and Gygi, S. P. ( 2008) Phosphoproteome analysis of Drosophila melanogaster embryos. J. Proteome Res. 7, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macek, B., Gnad, F., Soufi, B., Kumar, C., Olsen, J. V., Mijakovic, I., and Mann, M. ( 2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 16.Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., and Mann, M. ( 2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 17.Nuhse, T. S., Stensballe, A., Jensen, O. N., and Peck, S. C. ( 2003) Large-scale analysis of in vivo phosphorylated membrane proteins by immobilized metal ion affinity chromatography and mass spectrometry. Mol. Cell. Proteomics 2, 1234–1243 [DOI] [PubMed] [Google Scholar]
- 18.Gruhler, A., Schulze, W. X., Matthiesen, R., Mann, M., and Jensen, O. N. ( 2005) Stable isotope labeling of Arabidopsis thaliana cells and quantitative proteomics by mass spectrometry. Mol. Cell. Proteomics 4, 1697–1709 [DOI] [PubMed] [Google Scholar]
- 19.Benschop, J. J., Mohammed, S., O'Flaherty, M., Heck, A. J., Slijper, M., and Menke, F. L. ( 2007) Quantitative phosphoproteomics of early elicitor signaling in Arabidopsis. Mol. Cell. Proteomics 6, 1198–1214 [DOI] [PubMed] [Google Scholar]
- 20.Diella, F., Gould, C. M., Chica, C., Via, A., and Gibson, T. J. ( 2008) Phospho.ELM: a database of phosphorylation sites—update 2008. Nucleic Acids Res. 36, D240–D244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gnad, F., Ren, S., Cox, J., Olsen, J. V., Macek, B., Oroshi, M., and Mann, M. ( 2007) PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8, R250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UniProt Consortium ( 2008) The universal protein resource (UniProt). Nucleic Acids Res. 36, D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Obenauer, J. C., Cantley, L. C., and Yaffe, M. B. ( 2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiyama, N., Nakagami, H., Mochida, K., Daudi, A., Tomita, M., Shirasu, K., and Ishihama, Y. ( 2008) Large-scale phosphorylation mapping reveals the extent of tyrosine phosphorylation in Arabidopsis. Mol. Syst. Biol. 4, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwartz, D., and Gygi, S. P. ( 2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 26.Mishra, G. R., Suresh, M., Kumaran, K., Kannabiran, N., Suresh, S., Bala, P., Shivakumar, K., Anuradha, N., Reddy, R., Raghavan, T. M., Menon, S., Hanumanthu, G., Gupta, M., Upendran, S., Gupta, S., Mahesh, M., Jacob, B., Mathew, P., Chatterjee, P., Arun, K. S., Sharma, S., Chandrika, K. N., Deshpande, N., Palvankar, K., Raghavnath, R., Krishnakanth, R., Karathia, H., Rekha, B., Nayak, R., Vishnupriya, G., Kumar, H. G., Nagini, M., Kumar, G. S., Jose, R., Deepthi, P., Mohan, S. S., Gandhi, T. K., Harsha, H. C., Deshpande, K. S., Sarker, M., Prasad, T. S., and Pandey, A. ( 2006) Human protein reference database—2006 update. Nucleic Acids Res. 34, D411–D414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amanchy, R., Periaswamy, B., Mathivanan, S., Reddy, R., Tattikota, S. G., and Pandey, A. ( 2007) A curated compendium of phosphorylation motifs. Nat. Biotechnol. 25, 285–286 [DOI] [PubMed] [Google Scholar]
- 28.Linding, R., Jensen, L. J., Ostheimer, G. J., van Vugt, M. A., Jørgensen, C., Miron, I. M., Diella, F., Colwill, K., Taylor, L., Elder, K., Metalnikov, P., Nguyen, V., Pasculescu, A., Jin, J., Park, J. G., Samson, L. D., Woodgett, J. R., Russell, R. B., Bork, P., Yaffe, M. B., and Pawson, T. ( 2007) Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linding, R., Jensen, L. J., Pasculescu, A., Olhovsky, M., Colwill, K., Bork, P., Yaffe, M. B., and Pawson, T. ( 2008) NetworKIN: a resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 36, D695–D699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newman, M. E. J. ( 2005) Power laws, Pareto distributions and Zipf's law. Contemp. Phys. 46, 323–351 [Google Scholar]
- 31.Keller, E. F. ( 2005) Revisiting “scale-free” networks. BioEssays 27, 1060–1068 [DOI] [PubMed] [Google Scholar]
- 32.Barabasi, A. L., and Albert, R. ( 1999) Emergence of scaling in random networks. Science 286, 509–512 [DOI] [PubMed] [Google Scholar]
- 33.Quintaje, S. B., and Orchard, S. ( 2008) The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: one small step in manual annotation, one giant leap for full comprehension of genomes. Mol. Cell. Proteomics 7, 1409–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kersey, P. J., Duarte, J., Williams, A., Karavidopoulou, Y., Birney, E., and Apweiler, R. ( 2004) The International Protein Index: an integrated database for proteomics experiments. Proteomics 4, 1985–1988 [DOI] [PubMed] [Google Scholar]
- 35.Olsen, J. V., Ong, S. E., and Mann, M. ( 2004) Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614 [DOI] [PubMed] [Google Scholar]
- 36.Ferrè, F., Ausiello, G., Zanzoni, A., and Helmer-Citterich, M. ( 2004) SURFACE: a database of protein surface regions for functional annotation. Nucleic Acids Res. 32, D240–D244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider, T. D., and Stephens, R. M. ( 1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18, 6097–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishihama, Y., Oda, Y., Tabata, T., Sato, T., Nagasu, T., Rappsilber, J., and Mann, M. ( 2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 39.Kyono, Y., Sugiyama, N., Imami, K., Tomita, M., and Ishihama, Y. ( 2008) Successive and selective release of phosphorylated peptides captured by hydroxy acid-modified metal oxide chromatography. J Proteome Res. 7, 4585–4593 [DOI] [PubMed] [Google Scholar]
- 40.Imami, K., Sugiyama, N., Kyono, Y., Tomita, M., and Ishihama, Y. ( 2008) Automated phosphoproteome analysis for cultured cancer cells by two-dimensional nanoLC-MS using a calcined titania/C18 biphasic column. Anal. Sci. 24, 161–166 [DOI] [PubMed] [Google Scholar]
- 41.Koonin, E. V., Wolf, Y. I., and Karev, G. P. ( 2002) The structure of the protein universe and genome evolution. Nature 420, 218–223 [DOI] [PubMed] [Google Scholar]
- 42.Deeds, E. J., Ashenberg, O., and Shakhnovich, E. I. ( 2006) A simple physical model for scaling in protein-protein interaction networks. Proc. Natl. Acad. Sci. U. S. A. 103, 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueda, H. R., Hayashi, S., Matsuyama, S., Yomo, T., Hashimoto, S., Kay, S. A., Hogenesch, J. B., and Iino, M. ( 2004) Universality and flexibility in gene expression from bacteria to human. Proc. Natl. Acad. Sci. U. S. A. 101, 3765–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisenberg, E., and Levanon, E. Y. ( 2003)Preferential attachment in the protein network evolution. Phys. Rev. Lett. 91, 138701. [DOI] [PubMed] [Google Scholar]
- 45.Light, S., Kraulis, P., and Elofsson, A. ( 2005) Preferential attachment in the evolution of metabolic networks. BMC Genomics 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davids, W., and Zhang, Z. ( 2008) The impact of horizontal gene transfer in shaping operons and protein interaction networks—direct evidence of preferential attachment. BMC Evol. Biol. 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Itoh, H., Washio, T., and Tomita, M. ( 2004) Computational comparative analyses of alternative splicing regulation using full-length cDNA of various eukaryotes. RNA 10, 1005–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rappold, I., Iwabuchi, K., Date, T., and Chen, J. ( 2001) Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell Biol. 30, 613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakanishi, M., Ozaki, T., Yamamoto, H., Hanamoto, T., Kikuchi, H., Furuya, K., Asaka, M., Delia, D., and Nakagawara, A. ( 2007) NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. J. Biol. Chem. 282, 22993–23004 [DOI] [PubMed] [Google Scholar]
- 50.Bishop, J. D., and Schumacher, J. M. ( 2002) Phosphorylation of the carboxyl terminus of inner centromere protein (INCENP) by the Aurora B kinase stimulates Aurora B kinase activity. J. Biol. Chem. 277, 27577–27580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Craft, G. E., Graham, M. E., Bache, N., Larsen, M. R., and Robinson, P. J. ( 2008) The in vivo phosphorylation sites in multiple isoforms of amphiphysin I from rat brain nerve terminals. Mol. Cell. Proteomics 7, 1146–1161 [DOI] [PubMed] [Google Scholar]
- 52.Jaffe, H., Vinade, L., and Dosemeci, A. ( 2004) Identification of novel phosphorylation sites on postsynaptic density proteins. Biochem. Biophys. Res. Commun. 321, 210–218 [DOI] [PubMed] [Google Scholar]
- 53.Strickfaden, S. C., Winters, M. J., Ben-Ari, G., Lamson, R. E., Tyers, M., and Pryciak, P. M. ( 2007) A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell 128, 519–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.