

Abstract
The native state of α1-antitrypsin (α1AT), a member of the serine protease inhibitor (serpin) family, is considered a kinetically trapped folding intermediate that converts to a more stable form upon complex formation with a target protease. Although previous structural and mutational studies of α1AT revealed the structural basis of the native strain and the kinetic trap, the mechanism of how the native molecule overcomes the kinetic barrier to reach the final stable conformation during complex formation remains unknown. We hypothesized that during complex formation, a substantial portion of the molecule undergoes unfolding, which we dubbed functional unfolding. Hydrogen-deuterium exchange coupled with ESI-MS was used to analyze this serpin in three forms: native, complexing, and complexed with bovine β-trypsin. Comparing the deuterium content at the corresponding regions of these three samples, we probed the unfolding of α1AT during complex formation. A substantial portion of the α1AT molecule unfolded transiently during complex formation, including not only the regions expected from previous structural studies, such as the reactive site loop, helix F, and the following loop, but also regions not predicted previously, such as helix A, strand 6 of β-sheet B, and the N terminus. Such unfolding of the native interactions may elevate the free energy level of the kinetically trapped native serpin sufficiently to cross the transition state during complex formation. In the current study, we provide evidence that protein unfolding has to accompany functional execution of the protein molecule.
The native strain of serine protease inhibitors (serpins)1 is considered to be crucial to their biological functions, such as plasma protease inhibition (1, 2) and hormone delivery (3). Functional execution of serpins is accompanied by the conversion of the strained native structure into a more stable conformation (4). Because some of the strained native serpin structures are spontaneously converted into a more relaxed stable latent form under physiological conditions (5–7), the native structure is not the thermodynamically most stable conformation but is a kinetically trapped conformation. Upon binding a target protease, the scissile bond of the reactive site loop (RSL) is cleaved while the protease is covalently attached to the N-terminal part of the RSL (8, 9). During the conversion of the strained structure into the stable complex conformation (Fig. 1), RSL is inserted into the central β-sheet (A sheet) between strands 3 and 5 (s3A and s5A) to form strand 4 (s4A), and the covalently attached protease is concomitantly translocated to the opposite pole (10). Serpin inhibition occurs via a suicide substrate mechanism (4, 11, 12) in which serpins, upon binding proteases, partition between cleaved serpins and stable serpin-enzyme complexes.
Fig. 1.

Structures of native α1AT and α1AT-trypsin complex. Left, structure of native α1AT (Protein Data Bank code 1QLP) illustrated with secondary structural elements (1). Right, structure of α1AT-trypsin complex (Protein Data Bank code 1EZX). The nine α-helices are colored dark gray, and the 16 β-strands are colored light gray.
As a member of the serpin family, α1-antitrypsin (α1AT), which serves to modulate the activity of human leukocyte elastase in the lung, has been most extensively studied with regard to both structure and inhibition mechanism. Our previous studies with stabilizing mutations of α1AT showed that the native strain is distributed throughout the molecule and that various unfavorable structural motifs, such as hydrophobic packing, cavity in the core, and surface hydrophobic patch, appear to maintain the native strain (13, 14). Indeed stabilizing mutations localized in the region of RSL insertion during complex formation affected the inhibitory function individually by retarding the loop insertion (15). Mutations in other regions did not affect the inhibitory function individually, but collectively these mutations affected the inhibitory function when the stabilization effect reached a certain threshold (16). Maintaining the kinetic trap appears to require sustaining RSL at the hydrophobic β-barrel composed of sheet B and sheet C (B/C β-barrel) because the conversion into the stable latent conformation occurs by destabilization of the B/C β-barrel (6) as well as by the extension of RSL length (17). Thus, upon binding a target protease, RSL cleavage appears to induce a conformational conversion, and the resultant strain throughout the molecule facilitates the opening of β-sheet A and the insertion of the RSL, which is critical for the inhibitory pathway as opposed to the substrate pathway (10).
Although these structural and mutational studies revealed the structural basis for maintaining the kinetic trap and its relation to the inhibitory mechanism, several questions still remain. For example, what structural changes does the native serpin molecule undergo to overcome the kinetic barrier and reach the final stable conformation during the complex formation? In the present study, to probe the structural process of overcoming the kinetic barrier during complex formation with a target protease, amide hydrogen exchange (hydrogen-deuterium exchange (H/D-EX)) was explored during the conversion of the native α1AT to the stable complex. H/D-EX coupled with ESI-MS is a powerful analytical tool for observing protein dynamics, transient conformational changes, and protein-protein interactions (18–22). These experiments demonstrated that transient structural unfolding occurred in many regions in the α1AT molecule during formation of the complex with β-trypsin, and some of this unfolding was unpredicted from previous structural studies.
EXPERIMENTAL PROCEDURES
Materials—
N-Tosyl-l-phenylalanine chloromethyl ketone-treated bovine trypsin, porcine pepsin, 99.9% deuterium oxide (D2O), and urea-d4 (D2NCOND2) were purchased from Sigma-Aldrich. Immobilized soybean trypsin inhibitor was purchased from Pierce. All other chemicals were reagent grade. The Zorbax C18, 10-port valve, manual sample injector, and HPLC pumps were purchased from Agilent Technologies. Self Pack POROS-20AL support was purchased from Applied Biosystems. The threaded stainless steel column (50 × 2.1 mm) was purchased from Alltech. Magic C18AQ was purchased from Michrome.
H/D-EX Sample Preparation—
Plasmids for α1AT expression in Escherichia coli and purification of recombinant α1AT protein were described previously (23). Protein concentration was determined by measuring absorbance at 280 nm in 6 m guanidine hydrochloride and calculated from tyrosine and tryptophan content of the α1AT protein (24). Bovine β-trypsin was purified from a commercial N-tosyl-l-phenylalanine chloromethyl ketone-treated product by chromatography on soybean trypsin inhibitor-immobilized agarose (25, 26). To complete 1:1 complex formation, equimolar active β-trypsin was used with 1 μm α1AT. Complex formation of α1AT with β-trypsin was carried out at 25 °C for 10 min and quenched with 5 mm PMSF, and the final product formation was confirmed by monitoring SDS-resistant α1AT-protease complex (supplemental Fig. 1). The H/D-EX reaction of the native (D1) and complexed α1AT (D3) was initiated by diluting α1AT and α1AT-β-trypsin complex into deuterium buffer (20 mm phosphate buffer, pD 7.0), respectively. Residual activity of β-trypsin in D3 sample was quenched by adding 5 mm PMSF prior to H/D-EX. The samples (91.6% final deuterium content) were incubated at 25 °C for 10 min. For α1AT complexing with β-trypsin (D2), complex formation and H/D-EX were concomitantly performed by adding native α1AT into the deuterium buffer containing equimolar active β-trypsin. The sample (91.6% final deuterium content) was incubated at 25 °C for 10 min, and residual activity of β-trypsin was quenched by adding 5 mm PMSF. The H/D-EX of D1, D2, and D3 samples was quenched by adding chilled quenching buffer (500 mm phosphate buffer, pH 2.4, 50% glycerol), which was followed by rapid freezing in liquid nitrogen. The samples were stored at −80 °C. Hydrated native α1AT protein (H1) and hydrated α1AT-β-trypsin complex (H3) were prepared with H2O buffer instead of deuterium buffer. To determine the tolerable back-exchange rate during digestion and LC/MS analysis, fully deuterated and denatured α1AT (D0) was prepared after 1-h incubation at 37 °C in deuterium buffer containing 7.2 m urea-d4 (5 mm NH4HCO3 buffer, pD 8.0; 89.6% final deuterium content). The H/D-EX of D0 sample was also quenched by adding quenching buffer prior to analysis.
H/D-EX and ESI-MS Spectrometry—
H/D-EX experiment was carried out using an LC/MS system submerged in a 0–4 °C ice bath (supplemental Fig. 2). All aqueous solvents contained 0.72% formic acid (pH 2.4 at 0 °C), and the organic solvent (acetonitrile) contained 0.1% formic acid. The H/D-EX samples were rapidly thawed and digested by an on-line immobilized pepsin column at a flow rate of 150 μl/min. Peptides were retained on the C18 trap column and eluted within 10 min with a fast linear gradient (4 min) of 15–50% acetonitrile from the analytical C18 column (Magic C18AQ, 50 × 0.3 mm) at a flow rate of 20 μl/min. A Thermo Finnigan LTQ linear ion trap mass spectrometer was used to obtain ultrazoom scan spectra (MS) or tandem mass spectra (MS2). For more details, see the supplemental data.
Data Processing of H/D-EX Data—
The dta files for tandem mass spectra (H1 and H3) were generated by Extract-msn program (v.3) of Bioworks software (v3.2) with the following parameters: minimum ion count threshold, 15; minimum intensity, 100. The SEQUEST searching (TurboSequest v.27, revision 12) was performed without enzymatic restriction for a 184-protein sequence database containing human α1AT (Swiss-Prot accession number P01009), bovine trypsin (Swiss-Prot accession number P00760), porcine pepsin A (Swiss-Prot accession number P00791), and 181 contaminant sequences provided by Thermo Finnigan Corp. Mass tolerance for precursor ions with average mass type was set to 3.0 amu, and the mass tolerance for fragment ions with monoisotopic mass type was set to 1 amu. Search criteria included a variable modification of 16 Da for methionine oxidation. The Trans Proteomic Pipeline (v.3.5) software that includes a peptide probability score (p) program, PeptideProphet (27), was used to validate peptide identification. The validation has 88.9% sensitivity and 1.8% error at p ≥ 0.8. Deuterated peptic fragments of the H/D-EX samples (D0–D3) were identified by cross-matching with retention time, charge state, and fragmentation pattern of the previously identified peptides (H1 and H3) through SEQUEST searching of tandem mass spectra and ultrazoom scans. The mass of the hydrated and deuterated peptides was determined as the weighted average mass of isotopic peaks. To adjust for the loss of deuterium content during H/D-EX and ESI-MS analysis, the following calculation was performed. The deuterium content, D, in each fragment was calculated from Equation 1,
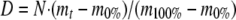 |
(Eq. 1) |
where N is the number of exchangeable amide hydrogens, mt is the experimentally determined mass, m0% is the average mass of the 0% deuterium control, and m100% is the average mass of the 100% deuterium control (28). The back-exchange value was determined according to the method reported previously (29). To analyze the D2-specific changes in the H/D-EX level, the difference was calculated by subtracting the percent exchange of D1 or D3 from that of D2.
RESULTS
H/D-EX and Mass Spectrometry of α1AT—
To measure the structural changes in α1AT during complex formation with β-trypsin, an H/D-EX experiment coupled with mass spectrometry was applied to distinct states of α1AT (Fig. 2). The deuterium incorporation of the native form (indicated as D1) and the α1AT-β-trypsin complex (indicated as D3) was performed as the controls for before and after complex formation, respectively. To detect the H/D-EX caused by transitional structural change during complex formation with β-trypsin (indicated as D2), deuterium incorporation and complex formation of α1AT was performed simultaneously following addition of α1AT to β-trypsin in deuterium buffer. Deuterium incubation for probing transient structural change (D1, D2, and D3) was identically performed for 10 min. The 10 min was chosen for the reaction because shorter time points (2 or 5 min) were relatively not enough in magnitude of change presumably because less of the population participated in complex formation (the rate of loop insertion was estimated to be 1–2 s−1 (30)). The 10 min was long enough to complete 1:1 complex formation for most α1AT molecules with β-trypsin (supplemental Fig. 1). D1, D2, and D3 samples incubated in deuterium buffer (final deuterium content, 91.6%) and two replicates were performed for each sample. LC/MS with tandem mass scan and LC/MS with ultrazoom scan were used for mass analysis of each duplicate of the deuterated peptic fragments. The accuracy of the ultrazoom scan (average of differences between theoretical and experimental m/z values in the identified H1 peptides) was −0.04 ± 0.06. The error for replicates in each sample was ±0.18 m/z (D1), ±0.12 m/z (D2), ±0.15 m/z (D3), and ±0.31 m/z (D0). Tandem mass spectrometry using LC-MS/MS of the native α1AT (indicated as H1) and α1AT complexed with β-trypsin (indicated as H3) in H2O identified 169 peptic fragments of α1AT (98.2% coverage; 387 of 394 residues) (supplemental Fig. 3). The number of peptic fragments detected in all three H/D-EX α1AT samples (native (D1), complexing (D2), and complexed (D3)) was small (39 peptic fragments) presumably due to distinct preference in peptic digestion among the three states (D1 sample with α1AT molecule alone and D2 and D3 in the presence of both α1AT and β-trypsin), but these commonly detected fragments covered 83.5% (314 of 376 exchangeable amides) of the α1AT sequence (Fig. 3, lines under the sequence). Many of these fragments exhibited overlap with other peptic fragments. The H/D-EX data for overlapping peptides from the same secondary structure were very similar (supplemental Table 1), indicating high confidence in the results of the current method. The systematic back-exchange determined with fully deuterated α1AT (indicated as D0) in 7.2 m urea-d4 (D2NCOND2) was ∼28.3%.
Fig. 2.

Energy diagram for the conformational change of α1AT during complex formation with a target protease. Functional unfolding during complex formation is illustrated as a process to overcome the highest energy barrier. D0 is a fully deuterated α1AT, D1 is the deuterated native form of α1AT, D2 is α1AT that is deuterated during complex formation with β-trypsin, and D3 is the deuterated α1AT-β-trypsin complex in D2O. H1 and H3 indicate the native α1AT and the α1AT-β-trypsin complex in H2O, respectively.
Fig. 3.

Peptide map of α1AT peptic fragments. The 39 peptic fragments that were commonly detected in all samples are indicated by underlined protein sequence. Lines with arrowheads indicate the selected 23 representative peptic fragments. Elements of secondary structure are shown (cylinders represent α-helices, and boxed arrows represent β-strands). The small vertical arrow indicates the site (C terminus of Met358) that is covalently linked to a target protease by proteolytic cleavage. Colors indicate the four groups in H/D-EX. Red and orange lines indicate group A peptides showing more than a 15 and a 5–15% increase in D2-specific H/D-EX, respectively. Blue lines indicate group B peptides showing less than a 5% change in D2-specific H/D-EX. Black lines indicate the stabilized region in the complex (group C); the green line indicates an open region in the complex (group D).
H/D-EX Pattern of α1AT during Complex Formation with β-Trypsin—
The commonly detected peptic fragments showed a distinct state-dependent exchange pattern. For example, the peptic fragment Phe227–Trp238 (s1B-s2B) showed little change in H/D-EX among the D1, D2, and D3 samples (Fig. 4A). The peptic fragment Thr339–Phe352 (s5A-RSL) and Met1–Leu30 (N-term-hA) showed a D2-specific mass shift (increase) in H/D-EX (Fig. 4, B and C).
Fig. 4.

H/D-EX measured by mass spectrometry. Ultrazoom spectra of the Phe227–Trp238 (s1B-s2B) fragment (A), the Thr339–Phe352 (s5A-RSL) fragment (B), and the Met1–Leu30 (N-term-hA) fragment (C) are shown (see “Results”). Dashed lines indicate manually determined average m/z of peptide ions (see “Experimental Procedures”).
To minimize redundancy and provide a summary of structural change at the level of secondary structure, 23 representative peptic fragments were selected from 39 commonly detected peptides (80.1% coverage; 301 of 376 exchangeable amides). Table I shows the summary of deuterium contents measured for the 23 representative peptides (data for all 39 commonly detected peptides are shown in supplemental Table 1). According to the state-dependent exchange patterns, the 23 representative peptic fragments were classified into four groups. (i) Group A (nine peptides) showed a deuterium content increase in D2 compared with D1 and D3 (Fig. 5A). (ii) Group B (nine peptides) showed similar H/D-EX among all three states (Fig. 5B). (iii) Group C (four peptides) showed a decreasing trend in deuterium contents from D1 to D2 to D3 (Fig. 5C). (iv) Group D (one peptide) showed an increasing pattern in deuterium contents from D1 to D2 to D3 (Fig. 5D). The extent of H/D-EX in each state relative to the theoretical number of exchangeable amide hydrogen is shown in Fig. 5.
Table I.
Properties of H/D-EX for 23 representative peptic fragments of α1AT
Note that the average of percent change error (n = 2) in 23 representative peptic fragments was ±3.81% (D1, ±5.08%; D2, ±2.97%; D3, ±3.42%).
| Regiona | Position | Sequence | H/D-EX
|
D2-specific H/D-EX
|
Groupf (colors in Figs. 3 and 6) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nb | D1c | D2c | D3c | D2(%) − D1 (%)d | D2(%) − D3 (%)d | Averagee | ||||
| % | ||||||||||
| N-term-hA | 1–32 | MDPQGDAAQKTDTSHHDQDHPTFNKITPNLAE | 28 | 20.6 | 24.8 | 19.5 | 15.0 | 18.8 | 16.9 | A (red) |
| hA-s6B | 38–51 | YRQLAHQSNSTNIF | 13 | 6.2 | 9.3 | 6.8 | 24.4 | 19.6 | 22.0 | A (red) |
| hB | 52–60 | FSPVSIATA | 7 | 0.5 | 0.2 | 0.1 | −5.2 | 0.4 | B | |
| hB-hC | 65–77 | LSLGTKADTHDEIL | 13 | 4.9 | 5.7 | 5.4 | 6.7 | 2.1 | 4.4 | B |
| hC1-hD | 83–99 | NLTEIPEAQIHEGFQEL | 15 | 8.5 | 8.2 | 8.1 | −2.1 | 0.7 | B | |
| s2A-hE-s1A | 120–142 | LSEGLKLVDKFLEDVKKLYHSEA | 22 | 4.5 | 12.2 | 12.1 | 34.9 | 0.4 | D | |
| s1A-hF | 143–159 | FTVNFGDTEEAKKQIND | 16 | 9.2 | 9.5 | 8.2 | 2.1 | 8.1 | 4.9 | B |
| hF-thFs3A | 160–172 | YVEKGTQGKIVDL | 12 | 9.0 | 10.2 | 9.5 | 9.9 | 6.0 | 7.9 | A (orange) |
| thFs3A | 172–182 | LVKELDRDTVF | 10 | 8.3 | 9.0 | 7.8 | 6.7 | 11.9 | 9.3 | A (orange) |
| s3A | 183–189 | ALVNYIF | 6 | 0.4 | 0.0 | 0.0 | 7.3 | 0.0 | B | |
| s3A-s4C | 190–205 | FKGKWERPFEVKDTEE | 14 | 11.1 | 13.5 | 10.1 | 17.1 | 24.2 | 20.7 | A (red) |
| s4C-s3C | 208–227 | FHVDQVTTVKVPMMKRLGMF | 18 | 8.1 | 8.2 | 8.0 | 0.7 | 1.3 | 1.0 | B |
| s1B-s2B | 227–238 | FNIQHCKKLSSW | 11 | 3.5 | 3.4 | 2.6 | −0.9 | 6.9 | 3.0 | B |
| s2B-s3B | 242–251 | MKYLGNATAI | 9 | 3.9 | 5.0 | 3.8 | 11.8 | 12.9 | 12.3 | A (orange) |
| s3B-hG | 253–266 | FLPDEGKLQHLENE | 12 | 6.7 | 8.1 | 7.9 | 11.2 | 1.4 | 6.3 | A (orange) |
| hH | 267–275 | LTHDIITKF | 8 | 4.4 | 3.8 | 3.0 | −7.4 | 9.7 | C | |
| hH-s2C | 276–291 | LENEDRRSASLHLPKL | 14 | 5.4 | 4.7 | 3.5 | −5.1 | 8.7 | C | |
| s6A | 291–300 | LSITGTYDLK | 9 | 5.9 | 7.2 | 7.0 | 14.1 | 1.9 | 8.0 | A (orange) |
| hI-hI1 | 304–317 | GQLGITKVFSNGAD | 13 | 7.0 | 7.4 | 6.6 | 2.9 | 5.7 | 4.3 | B |
| thI1s5A-s5A | 318–338 | LSGVTEEAPLKLSKAVHKAVL | 19 | 9.9 | 10.1 | 8.8 | 0.9 | 7.1 | 4.0 | B |
| s5A | 325–338 | APLKLSKAVHKAVL | 12 | 5.4 | 4.2 | 3.1 | −10.3 | 8.9 | C | |
| s5A-RSL | 339–352 | TIDEKGTEAAGAMF | 13 | 9.0 | 11.0 | 10.1 | 15.4 | 6.3 | 10.8 | A (orange) |
| s4B-C-term | 373–394 | LMIDQNTKSPLFMGKVVNPTQK | 19 | 7.0 | 5.2 | 4.4 | −9.6 | 4.1 | C | |
The nomenclature of Huber and Carrell (1) was followed.
The number of theoretically exchangeable amide hydrogens. Proline residues have no exchangeable amide hydrogens on the peptide bond.
D1 shows exchanged deuterium content in native α1AT, whereas D2 and D3 show exchanged deuterium content during complex formation and after complex formation, respectively.
D1(%), D2(%), and D3(%) indicate the portion of H/D-EX in relation to the number of theoretically exchangeable amide hydrogens in each peptic fragment. For instance, D2(%) = D2/N × 100. D2(%) − D1(%) or D2(%) − D3(%) indicates the difference in percent exchange for D2 in relation to D1 or D3.
Average of percent differences for D2-specific H/D-EX (average of D2(%) − D1(%) and D2(%) − D3(%)).
Each group (A–D) was categorized according to the pattern of H/D-EX for D1 to D2 to D3. The groups are the same as in Fig. 5 (see “Results” for explanation).
Fig. 5.

H/D-EX pattern of 23 representative peptic fragments. A, group A showed a D2-specific exchange pattern. Solid lines indicate peptides with a greater than 15% increase in D2-specific H/D-EX, and dotted lines indicate peptides with a 5–15% increase in D2-specific H/D-EX. B, group B showed little change in H/D-EX among the three states. C, group C showed a decreasing pattern in H/D-EX from D1 to D2 to D3. D, group D showed an increasing pattern in H/D-EX from D1 to D2 to D3. Note that the average of percent change error (n = 2) in 23 representative peptic fragments was ±3.81% (D1, ±5.08%; D2, ±2.97%; D3, ±3.42%).
Among the peptides in group A, the peptides with more than 15% D2-specific H/D-EX (D2 relative to D1 or D3) were found mainly in three regions (Fig. 5A, solid lines): N-term-hA, hA-s6B, and s3A-s4C (Fig. 6, colored red except for the N terminus (residues 1–22), which is not shown because of a lack of three-dimensional structural information). Six peptides that showed 5–15% more D2-specific H/D-EX were found at the hF and flanking sequence region, s2B-s3B, s3B-hG, s6A, and s5A-RSL (Fig. 5A, dotted line; Fig. 6, colored orange). Peptic fragments of group B showed less than 5% D2-specific H/D-EX and were found at hB, hB-hC, hC1-hD, s1A-hF, s3A, s4C-s3C, s1B-s2B, hI-hI1, and thI1s5A-s5A (Fig. 5B and Fig. 6, colored blue). Peptides in group C showed decreased H/D-EX in D3 (>10% lower exchange in D3 than D1) and were mainly found at the hydrophobic core region of the α1AT molecule: hH, hH-s2C, s5A, and s4B-C-term (Fig. 5C and Fig. 6, colored black). The peptides in group D showed increased H/D-EX in D3 (higher exchange in the complex) and were only found at s2A-hE-s1A (Fig. 5D and Fig. 6, colored green).
Fig. 6.

Conformational changes in α1AT during complex formation with β-trypsin. The front view (left) and side view (right) of the native α1AT structure (Protein Data Bank code 1QLP) are shown. Colors indicate H/D-EX patterns of 23 representative peptic fragments. For the group A peptides, regions showing more than a 15% increase in D2-specific H/D-EX are shown in red, and regions showing a 5–15% increase in D2-specific H/D-EX are shown in orange. For the group B peptides, regions showing less than a 5% change in D2-specific H/D-EX are shown in blue. The stabilized region in the complex is colored black (group C); an open region of the complex is colored green (group D). Gray regions represent those not detected in the current study or not considered in the analysis with the subset of 23 representative peptic fragments.
DISCUSSION
Many previous structural and biochemical studies revealed that serpins are designed to have the proper fold to perform biological functions (13, 14, 31, 32). The native form, which is a kinetically trapped fold, converts to a more stable conformation through a drastic structural change (33, 34); however, the details of how the trapped fold overcomes the kinetic barrier to convert into a very stable serpin-protease complex has not been elucidated. Because the native serpin structure is considered a trapped folding intermediate, an analogy between overcoming the kinetic barrier of the serpin native structure and passing the transition state during protein folding may exist. The transition state of protein folding is considered to involve a substantial unfolding of initial intermediates formed by hydrophobic coalescence (35–37). Here we accumulated evidence for unfolding in a substantial portion of the serpin molecule during complex formation. H/D-EX coupled with ESI-MS illuminated the structural changes of the protein during the functional execution of α1AT.
D2-specific H/D-EX: Regions Unfolded during the Complex Formation—
To focus on the transient structural changes of α1AT during complex formation with β-trypsin, three samples (D1, before complex formation; D2, during complex formation; and D3, after complex formation) were compared for H/D-EX. The overall H/D-EX level of α1AT was slightly higher than that of a previous report (38, 39) presumably because of the non-salt condition of the current H/D-EX study. It was reported that the presence of salts stabilized protein conformation and suppressed H/D-EX (40). Nonetheless exchangeable hydrogens in D2 sample showed globally ∼22.7 greater deuterium exchange than in D1 or D3 sample (data not shown). D2-specific H/D-EX, corresponding to higher exchange at D2 relative to that at D1 or D3 (Fig. 5, group A), reflects structural changes of α1AT during the complex formation. Remarkably D2-specific H/D-EX occurred for a substantial portion of the peptic fragments (Fig. 5A, nine of 23 representative peptic fragments, covering 133 of 301 residues). The current results revealed a D2-specific increase in H/D-EX at some expected regions, such as RSL, hF, thFs3A, and s3A-s4C (Fig. 5A and Table I). Crystal structures of the native serpin and serpin-protease complex (Fig. 1) predict the regions of α1AT that should be mobilized during the complex formation, such as RSL, s3A, s5A, hF, and thFs3A. Also mobilization of cleaved RSL should allow solvent exposure of s3A-s4C. Thus all the expected regions besides s3A and s5A showed D2-specific H/D-EX. For s3A and s5A strands, additional D2-specific H/D-EX was not observed during complex formation (Fig. 5B and Table I), and these results will be discussed in the next section.
Interestingly a D2-specific increase in H/D-EX was also observed at other regions that were not expected from crystallography, such as the N terminus, hA-s6B, s2B-s3B, and hG. These regions surround the hydrophobic core composed of s4B-s5B (Fig. 6, inset). The C-terminal peptide (s4B-s5B-C-term) surrounded by these peptides showed decreased H/D-EX in the complex as shown in Fig. 5C. Note that this C-terminal peptide is not released out from the complex molecule (Fig. 1B). It may be that regions surrounding the hydrophobic core of β-sheet B need to be unfolded during complex formation. Structural investigation of the region surrounding hA indicates a crucial role of hA in the opening of the major β-sheet, β-sheet A. The location of hA is located at the center that divides the molecule into β-sheet A-containing parts and β-sheet B-containing parts (Fig. 6B). The hA connects the hydrophobic core composed of s4B, s5B, hG, and hH and the so-called “shutter domain” composed of hydrophobic residues Phe52–Leu55 of hB. The “shutter domain region” is located just beneath the major β-sheet, β-sheet A, and was suggested to play a critical role in the structural transition from the native state to a more stable conformation (2). The unfolding of hA thus may perturb a significant portion of the surrounding interactions including those between the hydrophobic core of β-sheet B and β-sheet A. This event may then cause the opening of the gap between s5A and s3A of β-sheet A for RSL insertion. A D2-specific increase in H/D-EX was also observed at s6A (Figs. 5A and 6), but we do not have a good explanation of why this solvent-exposed strand showed D2-specific H/D-EX. The region may just be exposed more during the complex formation. Various levels of D2-specific H/D-EX observed in the current results suggest that transient structural unfolding occurred differentially in many parts of the molecule during complex formation.
Amide Hydrogen Exchange Is Not Allowed in s3A and s5A during Complex Formation—
Most of the rest of the peptic fragments did not show any increase in H/D-EX in D2 state (Fig. 5), and many of them showed no state-specific H/D-EX (Fig. 5, group B). Fig. 6 shows that these are located on β-sheet A (s3A and s5A) and surrounding helices (hB, hC, hD, and hI-hI1) and B/C β-barrel (s1B, s2B, s3C, and s4C). These regions may not participate in transient unfolding during complex formation. Alternatively these regions may undergo a conformational change but without any concomitant solvent exposure. Particularly s3A (Ala183–Phe189) and s5A (Ala325–Leu338) of β-sheet A did not show any significant increase in H/D-EX during complex formation, and the exchange of s5A was even further decreased in the complex state (Fig. 5C). Because complete opening of the two strands is required for the full insertion of RSL and complex formation, the strand opening of s3A and s5A seemingly does not involve solvent exposure. In a recent study of serpin polymerization using H/D-EX, the increase in deuterium exchange was not observed at these sites during polymerization (39). The serpin polymerization also accompanies the opening event of the β-strands (s3A and s5A) with RSL insertion of one molecule into another.
Our results also showed that the C-terminal portion (upper part from the view of Fig. 6) of the same strands was significantly more accessible for H/D-EX during complex formation as observed with the peptides Phe190–Glu205 of s3A and Thr339–Phe352 of s5A. Because these peptic fragments happened to include the following loop regions, the observed D2-specific H/D-EX of the C-terminal portion of s3A and s5A may have been due to a higher D2-specific exchange of the following loop regions. However, it is tempting to suggest that the upper portion of s3A and s5A indeed undergoes D2-specific unfolding during complex formation because several previous studies including crystallography suggested that RSL insertion occurs via a two-step process: the partial insertion of RSL into the upper part of β-sheet A was demonstrated with crystal structures of an α1-antichymotrypsin variant and latent antithrombin (41–44). In any case, our results suggest two distinct (and possibly consecutive) steps during complex formation: one that consists of unfolding to allow amide hydrogen exchange at various regions that may or may not accompany opening of the C-terminal portion of s3A and s5A and a second that consists of fast displacement of strand interaction without accompanying solvent exposure (like “zipping”). These two steps may correspond to ascending and descending the hill of the transition state (Fig. 2).
Regions Stabilized or Exposed in the Complex—
Four peptic fragments showed a decreasing pattern of H/D-EX from D1 to D2 to D3 (Fig. 5C and Fig. 6, colored black), and they correspond to hH, s2C, s5A, and s4B-C-term. The s4B-C-term peptide belongs to the hydrophobic core of the α1AT molecule (1). The decreased H/D-EX in the complex for the region is likely to reflect increased conformational stability of α1AT in the final complex conformation because the unfolding transition midpoints of native α1AT were significantly shifted from 0.8 to 6.7 m guanidine HCl after RSL insertion (45). Likewise the decreased H/D-EX of hH, located just underneath β-sheet B, may reflect the increased stability of the complex. The strand s5A showed decreased exchange in the complex (Fig. 5C and Table I), and s3A also showed decreased exchange, although the data were within the error range (Fig. 5B and Table I). It is likely that the amide proton exchange is indeed decreased for these strands because they form an antiparallel β-sheet in the complex with the newly inserted RSL (note that they are parallel in the native serpin). It is well known that antiparallel β-sheet is more stable than parallel β-sheet because of better alignment of backbone hydrogen bonds.
The current results also suggested that the s2C strand is stabilized in the complex (Fig. 5C and Fig. 6, colored black). This may be due to better strand pairing of s1C to s2C without constraint from the preceding RSL in the complex (Fig. 1B). Our results also implicate a distinct role of β-sheet C for various conformational changes of the serpin molecule. The B/C β-barrel composed of s1B, s2B, s3C, and s4C did not show any significant increase in H/D-EX during complex formation (Fig. 6, colored blue). Although we could not analyze the peptides spanning s1C due to the absence of a common peptide between D1, D2, and D3, the current results suggest that β-sheet C maintains its structure during complex formation. This is quite a contrast to the recent report of Wintrode and co-workers (39) in which β-sheet C appears to be destabilized first during polymerization. Our previous mutational studies on α1AT (6) showed that destabilization of β-sheet C caused the molecule to convert into the latent form (RSL-inserted monomer) and suggested that the integrity of β-sheet C is crucial for maintaining the kinetic trap of the native form. These different results may appear contradictory, but the difference between the current case and the others was that s1C (immediately C-terminal of the scissile Met-Ser bond) stays with the B/C β-barrel in the complex whereas it leaves the original position in the other cases (inserted into β-sheet A of another molecule in polymers and moves out as a loop in the latent form).
One peptic fragment, Leu120–Ala142, spanning s2A, hE, and s1A showed a drastic increase (38% more) in H/D-EX during complex formation, and the increased state was maintained in the complex (Fig. 5D and Fig. 6, colored green). This peptide includes Leu131 and Val134 of hE and Ala142 of s1A, which is equivalent to the Leu159, Ile162, and Ile170 residues of tengpin, a thermophilic bacterial serpin that contains an additional 56-residue N-terminal extension. These residues compose a hydrophobic patch (46). The region in tengpin is covered by Met42 of the N-terminal region (46). Mutational and biochemical studies on tengpin indicated that these interactions are important for maintaining the kinetic trap of the native tengpin structure (46). Thus, this region of α1AT is likely covered by the N-terminal region in the native structure, and our current results (Fig. 5D) suggest that the region is also exposed in α1AT during complex formation.
Mechanism of Functional Unfolding of α1AT—
The amide proton exchange pattern observed in the current study suggests a picture of conformational changes in serpin during complex formation: β-sheet A including surrounding helices and β-sheet C do not appear to undergo significant structural changes, but surrounding regions of β-sheet B including hA appear to unfold substantially. This unfolding propagates into the shutter domain and various loop regions including the N terminus (whose decapping from hE and s1A possibly occurs). This event may then cause the opening of the gap between s5A and s3A of β-sheet A for the RSL insertion, which occurs in a manner of fast displacement of strand interaction without accompanying solvent exposure. The unfolding of these native interactions may mainly elevate the free energy level of the kinetically trapped native serpin sufficiently to cross the transition state. Remarkably during this process, the hydrophobic core of the molecule (strand 4 and 5 B, hH, and the shutter domain of hB) does not undergo any structural change (Fig. 6, black and blue), and only the surrounding regions unfold. In particular, unfolding of hA appears to play a pivotal role in overcoming the kinetic barrier. In the active native state of the serpin molecule, much of the energy is loaded as strain, and this kinetic barrier is necessary to execute inhibitory function. This barrier, however, must be overcome easily when inhibitory function is required. Our previous studies showed that initial formation of the B/C β-barrel hydrophobic core and stability of β-sheet C are critical for maintaining the kinetic trap of the native structure (6). Taken together, our current study suggests that the role of the B/C β-barrel can be further dissected into maintaining the kinetic trap through stabilizing interactions in β-sheet C and overcoming the kinetic trap through unfolding in the surrounding regions of β-sheet B.
In summary, we provided evidence for protein unfolding that is necessary for functional execution of the protein molecule, thus dubbed “functional unfolding.” The results from the H/D-EX experiments indicate a dynamic change of the molecule in solution, and these data are particularly powerful because some of the regions could not be predicted from x-ray crystal structures. This method has some limitations because time-resolved conformational changes cannot be probed because of the reaction time required to complete complex formation for most α1AT molecules, but the method still provides powerful evidence for dynamics of regions undergoing conformational change.
Supplementary Material
Footnotes
Published, MCP Papers in Press, January 11, 2009, DOI 10.1074/mcp.M800365-MCP200
The abbreviations used are: serpin, serine protease inhibitor; RSL, reactive site loop; α1AT, α1-antitrypsin; B/C β-barrel, β-barrel composed of sheet B and sheet C; H/D-EX, hydrogen-deuterium exchange; D2O, deuterium oxide.
This work was supported by Korean Ministry of Education, Science and Technology Grant FPR08A1-030 of the 21C Frontier Functional Proteomics Program.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Huber, R., and Carrell, R. W. ( 1989) Implications of the three-dimensional structure of α1AT for structure and function of serpins. Biochemistry 28, 8951–8966 [DOI] [PubMed] [Google Scholar]
- 2.Stein, P. E., and Carrell, R. W. ( 1995) What do dysfunctional serpins tell us about molecular mobility and disease? Nat. Struct. Biol. 2, 96–113 [DOI] [PubMed] [Google Scholar]
- 3.Pemberton, P. A., Stein, P. E., Pepys, M. B., Potter, J. M., and Carrell, R. W. ( 1988) Hormone binding globulins undergo serpin conformational change in inflammation. Nature 336, 257–258 [DOI] [PubMed] [Google Scholar]
- 4.Engh, R. A., Huber, R., Bode, W., and Schulze, A. J. ( 1995) Divining the serpin inhibition mechanism: a suicide substrate ‘springe’? Trends Biotechnol. 13, 503–510 [DOI] [PubMed] [Google Scholar]
- 5.Berkenpas, M. B., Lawrence, D. A., and Ginsburg, D. ( 1995) Molecular evolution of plasminogen activator inhibitor-1 functional stability. EMBO J. 14, 2969–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Im, H., Woo, M. S., Hwang, K. Y., and Yu, M. H. ( 2002) Interactions causing the kinetic trap in serpin protein folding. J. Biol. Chem. 277, 46347–46354 [DOI] [PubMed] [Google Scholar]
- 7.Mottonen, J., Strand, A., Symersky, J., Sweet, R. M., Danley, D. E., Geoghegan, K. F., Gerard, R. D., and Goldsmith, E. J. ( 1992) Structural basis of latency in plasminogen activator inhibitor-1. Nature 355, 270–273 [DOI] [PubMed] [Google Scholar]
- 8.Wei, A., Rubin, H., Cooperman, B. S., and Christianson, D. W. ( 1994) Crystal structure of an uncleaved serpin reveals the conformation of an inhibitory reactive loop. Nat. Struct. Biol. 1, 251–258 [DOI] [PubMed] [Google Scholar]
- 9.Wilczynska, M., Fa, M., Karolin, J., Ohlsson, P. I., Johansson, L. B., and Ny, T. ( 1997) Structural insights into serpin-protease complexes reveal the inhibitory mechanism of serpins. Nat. Struct. Biol. 4, 354–357 [DOI] [PubMed] [Google Scholar]
- 10.Huntington, J. A., Read, R. J., and Carrell, R. W. ( 2000) Structure of a serpin-protease complex shows inhibition by deformation. Nature 407, 923–926 [DOI] [PubMed] [Google Scholar]
- 11.Patston, P. A., Gettins, P., Beechem, J., and Schapira, M. ( 1991) Mechanism of serpin action: evidence that C1 inhibitor functions as a suicide substrate. Biochemistry 30, 8876–8882 [DOI] [PubMed] [Google Scholar]
- 12.Wright, H. T., and Scarsdale, J. N. ( 1995) Structural basis for serpin inhibitor activity. Proteins 22, 210–225 [DOI] [PubMed] [Google Scholar]
- 13.Lee, K. N., Park, S. D., and Yu, M. H. ( 1996) Probing the native strain in α1AT. Nat. Struct. Biol. 3, 497–500 [DOI] [PubMed] [Google Scholar]
- 14.Seo, E. J., Im, H., Maeng, J. S., Kim, K. E., and Yu, M. H. ( 2000) Distribution of the native strain in human α1AT and its association with protease inhibitor function. J. Biol. Chem. 275, 16904–16909 [DOI] [PubMed] [Google Scholar]
- 15.Lee, C., Park, S. H., Lee, M. Y., and Yu, M. H. ( 2000) Regulation of protein function by native metastability. Proc. Natl. Acad. Sci. U. S. A. 97, 7727–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo, E. J., Lee, C., and Yu, M. H. ( 2002) Concerted regulation of inhibitory activity of α1AT by the native strain distributed throughout the molecule. J. Biol. Chem. 277, 14216–14220 [DOI] [PubMed] [Google Scholar]
- 17.Im, H., Ahn, H. Y., and Yu, M. H. ( 2000) Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci. 9, 1497–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Englander, S. W., Sosnick, T. R., Englander, J. J., and Mayne, L. ( 1996) Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 6, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoofnagle, A. N., Resing, K. A., Goldsmith, E. J., and Ahn, N. G. ( 2001) Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. U. S. A. 98, 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan, X., Watson, J., Ho, P. S., and Deinzer, M. L. ( 2004) Mass spectrometric approaches using electrospray ionization charge states and hydrogen-deuterium exchange for determining protein structures and their conformational changes. Mol. Cell. Proteomics 3, 10–23 [DOI] [PubMed] [Google Scholar]
- 21.Kang, S., and Prevelige, P. E., Jr. ( 2005) Domain study of bacteriophage p22 coat protein and characterization of the capsid lattice transformation by hydrogen/deuterium exchange. J. Mol. Biol. 347, 935–948 [DOI] [PubMed] [Google Scholar]
- 22.Mandell, J. G., Baerga-Ortiz, A., Croy, C. H., Falick, A. M., and Komives, E. A. ( 2005) Curr. Protoc. Protein Sci. Unit20.9, pp. 1–20, John Wiley & Sons, Inc. NJ [DOI] [PubMed]
- 23.Kwon, K. S., Kim, J., Shin, H. S., and Yu, M. H. ( 1994) Single amino acid substitutions of α1AT that confer enhancement in thermal stability. J. Biol. Chem. 269, 9627–9631 [PubMed] [Google Scholar]
- 24.Edelhoch, H. ( 1967) Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 25.Robinson, N. C., Tye, R. W., Neurath, H., and Walsh, K. A. ( 1971) Isolation of trypsins by affinity chromatography. Biochemistry 10, 2743–2747 [DOI] [PubMed] [Google Scholar]
- 26.Yung, B. Y., and Trowbridge, C. G. ( 1980) A calorimetric comparison of trypsin and its anhydro modification in complex formation with Kunitz soybean inhibitor. J. Biol. Chem. 255, 9724–9730 [PubMed] [Google Scholar]
- 27.Keller, A., Nesvizhskii, A. I., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 28.Smith, D. L., Deng, Y., and Zhang, Z. ( 1997) Probing the non-covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J. Mass Spectrom. 32, 135–146 [DOI] [PubMed] [Google Scholar]
- 29.Resing, K. A., Hoofnagle, A. N., and Ahn, N. G. ( 1999) Modeling deuterium exchange behavior of ERK2 using pepsin mapping to probe secondary structure. J. Am. Soc. Mass Spectrom. 10, 685–702 [DOI] [PubMed] [Google Scholar]
- 30.Shin, J. S., and Yu, M. H. ( 2002) Kinetic dissection of α1AT inhibition mechanism. J. Biol. Chem. 277, 11629–11635 [DOI] [PubMed] [Google Scholar]
- 31.Im, H., Seo, E. J., and Yu, M. H. ( 1999) Metastability in the inhibitory mechanism of human α1AT. J. Biol. Chem. 274, 11072–11077 [DOI] [PubMed] [Google Scholar]
- 32.Lee, C., Maeng, J. S., Kocher, J. P., Lee, B., and Yu, M. H. ( 2001) Cavities of α1AT that play structural and functional roles. Protein Sci. 10, 1446–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence, D. A. ( 1997) The serpin-proteinase complex revealed. Nat. Struct. Biol. 4, 339–341 [DOI] [PubMed] [Google Scholar]
- 34.Stratikos, E., and Gettins, P. G. ( 1999) Formation of the covalent serpin-proteinase complex involves translocation of the proteinase by more than 70 Å and full insertion of the RSL into β-sheet A. Proc. Natl. Acad. Sci. U. S. A. 96, 4808–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayor, U., Guydosh, N. R., Johnson, C. M., Grossmann, J. G., Sato, S., Jas, G. S., Freund, S. M., Alonso, D. O., Daggett, V., and Fersht, A. R. ( 2003) The complete folding pathway of a protein from nanoseconds to microseconds. Nature 421, 863–867 [DOI] [PubMed] [Google Scholar]
- 36.Religa, T. L., Markson, J. S., Mayor, U., Freund, S. M., and Fersht, A. R. ( 2005) Solution structure of a protein denatured state and folding intermediate. Nature 437, 1053–1056 [DOI] [PubMed] [Google Scholar]
- 37.Schanda, P., Forge, V., and Brutscher, B. ( 2007) Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 104, 11257–11262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsutsui, Y., Liu, L., Gershenson, A., and Wintrode, P. L. ( 2006) The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry 45, 6561–6569 [DOI] [PubMed] [Google Scholar]
- 39.Tsutsui, Y., Kuri, B., Sengupta, T., and Wintrode, P. L. ( 2008) The structural basis of serpin polymerization studied by hydrogen/deuterium exchange and mass spectrometry. J. Biol. Chem. 283, 30804–30811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laurents, D. V., Scholtz, J. M., Rico, M., Pace, C. N., and Bruix, M. ( 2005) Ribonuclease Sa conformational stability studied by NMR-monitored hydrogen exchange. Biochemistry 44, 7644–7655 [DOI] [PubMed] [Google Scholar]
- 41.Wardell, M. R., Chang, W. S., Bruce, D., Skinner, R., Lesk, A. M., and Carrell, R. W. ( 1997) Preparative induction and characterization of l-antithrombin: a structural homologue of latent plasminogen activator inhibitor-1. Biochemistry 36, 13133–13142 [DOI] [PubMed] [Google Scholar]
- 42.Mellet, P., Boudier, C., Mely, Y., and Bieth, J. G. ( 1998) Stopped flow fluorescence energy transfer measurement of the rate constants describing the reversible formation and the irreversible rearrangement of the elastase-α1-proteinase inhibitor complex. J. Biol. Chem. 273, 9119–9123 [DOI] [PubMed] [Google Scholar]
- 43.Gooptu, B., Hazes, B., Chang, W. S., Dafforn, T. R., Carrell, R. W., Read, R. J., and Lomas, D. A. ( 2000) Inactive conformation of the serpin α1-antichymotrypsin indicates two-stage insertion of the reactive loop. Proc. Natl. Acad. Sci. U. S. A. 97, 67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malley, K. M., and Cooperman, B. S. ( 2001) Formation of the covalent chymotrypsin-antichymotrypsin complex involves no large-scale movement of the enzyme. J. Biol. Chem. 276, 6631–6639 [DOI] [PubMed] [Google Scholar]
- 45.Bruch, M., Weiss, V., and Engel, J. ( 1988) Plasma serine proteinase inhibitors exhibit major conformational changes and a large increase in conformational stability upon cleavage at their reactive sites. J. Biol. Chem. 263, 16626–16630 [PubMed] [Google Scholar]
- 46.Zhang, Q., Buckle, A. M., Law, R. H., Pearce, M. C., Cabrita, L. D., Lloyd, G. J., Irving, J. A., Smith, A. I., Ruzyla, K., Rossjohn, J., Bottomley, S. P., and Whisstock, J. C. ( 2007) The N terminus of the serpin, tengpin, functions to trap the metastable native state. EMBO Rep. 8, 658–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.