

Abstract
During the past decade, we have witnessed an explosive increase in generation of large proteomics data sets, not least in cancer research. There is a growing need to extract and correctly interpret information from such data sets to generate biologically relevant hypotheses. A pathway search engine (PSE) has recently been developed as a novel tool intended to meet these requirements. Ionizing radiation (IR) is an anticancer treatment modality that triggers multiple signal transduction networks. In this work, we show that high linear energy transfer (LET) IR induces apoptosis in a non-small cell lung cancer cell line, U-1810, whereas low LET IR does not. PSE was applied to study changes in pathway status between high and low LET IR to find pathway candidates of importance for high LET-induced apoptosis. Such pathways are potential clinical targets, and they were further validated in vitro. We used an unsupervised shotgun proteomics approach where high resolution mass spectrometry coupled to nanoflow liquid chromatography determined the identity and relative abundance of expressed proteins. Based on the proteomics data, PSE suggested the JNK pathway (p = 6·10−6) as a key event in response to high LET IR. In addition, the Fas pathway was found to be activated (p = 3·10−5) and the p38 pathway was found to be deactivated (p = 0.001) compared with untreated cells. Antibody-based analyses confirmed that high LET IR caused an increase in phosphorylation of JNK. Moreover pharmacological inhibition of JNK blocked high LET-induced apoptotic signaling. In contrast, neither an activation of p38 nor a role for p38 in high LET IR-induced apoptotic signaling was found. We conclude that, in contrast to conventional low LET IR, high LET IR can trigger activation of the JNK pathway, which in turn is critical for induction of apoptosis in these cells. Thus PSE predictions were largely confirmed, and PSE was proven to be a useful hypothesis-generating tool.
Radiotherapy is important in curative treatment of primary inoperable tumors and for palliation of metastatic disease. The two major classes of ionizing radiation (IR)1 quality are low and high linear energy transfer (LET) IR. Currently low LET γ-IR (photons) is the standard quality used for radiotherapy. However, high LET IR (accelerated ions) has recently been suggested to be therapeutically superior because of its physical as well as biological properties (1, 2). A large fraction of tumors show resistance to low LET IR by mechanisms that are only partly elucidated (3–7). Non-small cell lung carcinoma (NSCLC) is one example of such a resistance-prone tumor type (4, 5). Here high LET IR offers a possible alternative and has been used with promising results (8–10). To date, however, the signaling transduction events that are critical for the improved efficacy of high LET IR remain elusive.
In the present study we used methods for global analysis of cellular pathways to obtain more insight into signaling events triggered in NSCLC after high LET IR. Results from such analyses may provide biomarkers of high LET IR efficacy and possibly even markers for selection of patients that would benefit from treatment with such IR quality.
Conventional low LET IR is known to cause relatively well separated ionizations, resulting in non-clustered DNA double strand breaks (11). In contrast, high LET ions release energy densely along their track through the cell nucleus, creating several double strand breaks in a narrow region, resulting in more complex and less repairable DNA damages (11). The signal transduction events triggered by these clustered lesions are anticipated to be critical for the increased biological effectiveness observed with high LET (12, 13).
It has been suggested that non-repairable DNA lesions and/or lesions with ongoing repair most likely will induce different types of cell death including apoptosis. We have previously shown that insufficient activation of mitochondria may play a role in the impaired apoptotic signaling observed in response to low LET photon IR in NSCLC cell lines. This lack of apoptotic signaling may contribute to the emergence of an intrinsic radioresistant phenotype of NSCLC (4, 6).
One important group of modulators of apoptotic signaling is the stress-activated protein kinases (SAPKs) (14–17). Activation of the SAPK JNK in response to DNA damage is mainly associated with increased cell death, i.e. increased induction of apoptosis (6, 14, 16, 18, 19). Moreover the DNA damage-induced apoptotic response has been found to be reduced in JNK-deficient cells, in cells expressing a kinase-dead JNK mutant, and in cells treated with SP600125, a pharmacological inhibitor of JNK (6, 17, 19). In agreement with this, we have shown that impaired activation of JNK is involved in resistance to low LET IR in NSCLC cells (6). A role for p38 in DNA damage-induced apoptotic signaling is less conclusive as p38 activation has been shown both to promote and prevent apoptotic signaling (6, 20).
It is most likely that the observed IR effects in human tumors are the result of multiple signal transduction networks working in concert. To study such signal transduction networks, methods allowing adequate simultaneous analysis of multiple proteins are required. Recent technical developments in the proteomics field allow high throughput identification and characterization of large numbers of proteins from almost any biological material. This has been shown to be greatly useful when studying signal transduction (21, 22). However, although being highly informative, these proteomics strategies often generate large amounts of data that are difficult to interpret in terms of their biological relevance. To deal with these problems, we have recently developed a prototype of a pathway search engine (PSE) that is capable of determining the cellular signaling response from the multitude of changes in global protein expression (23). The PSE performs non-linear analysis of proteomics data using TRANSPATH®, a high quality database focused on signal transduction (24, 25), and TRANSFAC, a collection of experimental data on transcription factors (26).
We consider PSE as a hypothesis-generating discovery tool. The input data for generating hypotheses using PSE is relative protein abundance generated from shotgun high resolution mass spectrometry analysis. One of the basic principles of analytical sciences is to validate a discovery with a complementary technique that independently measures a different parameter compared with the technique used for the discovery. In the case of PSE-based analysis, such a parameter is the phosphorylation levels or expression turnover of proteins potentially involved in the predicted pathway. Furthermore it is fortunate if the validation is performed by a technique complementary to mass spectrometry. In the present study we applied this novel approach to study how high LET IR can trigger proapoptotic signaling where low LET IR cannot.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments—
In this study, a previously characterized NSCLC cell line, U-1810, was used (5). The cells were maintained in a humidified atmosphere of 5% CO2 and cultured in glutamine-containing RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum and antibiotics (100 units/ml penicillin and 100 units/ml streptomycin). For experiments with inhibitors, the cells were preincubated for 1 h prior to IR with either JNK inhibitor SP600125 (10 μm) or the p38 inhibitor SB203850 (10 μm). After IR, fresh medium containing the inhibitor was added, and the cells were kept in the inhibitor-containing medium until harvest. All chemicals were purchased from Sigma-Aldrich if not otherwise stated.
Irradiation Procedure—
Cells were irradiated with either low LET γ-photons (LET = 3 keV/μm; absorbed dose, 8 Gy) or high LET accelerated nitrogen ions (LET = 125 keV/μm; absorbed dose, 4 Gy) at room temperature. Low LET IR was carried out with adherent cells using a 60Co source (dose rate was recalculated every month according to the known decay rate). For high LET IR cells were irradiated in suspension as was demanded by the IR source setup. Before IR, cells were detached and concentrated in 300 μl of culture medium and were thereafter placed in in-house constructed plastic chambers (22 mm in diameter and 1 mm in depth) shielded with a coverslip (Thermanox, Nunc, Neperville, IL). High LET IR was delivered in the form of accelerated 14N7+ ions with a mean energy of 20.9 MeV/nucleon from the Gustaf Werner synchrocyclotron at The Svedberg Laboratory, Uppsala University, Sweden.
Preparation of Protein Extracts—
Low or high LET-irradiated cells were harvested 4 h post-treatment using non-enzymatic cell dissociation solution. At the same time point, untreated cells were harvested and used as a control. Cell pellets were washed in a buffer containing 100 mm Tris-HCl at pH 7.5 and 250 mm sucrose, centrifuged at 1,500 × g for 10 min, snap frozen, and stored at −80 °C until analysis. After thawing, cell pellets were lysed in 200 mm ammonium bicarbonate and 2 m urea, and lysates were completely solubilized by sonication. The protein extracts were separated from insoluble cell debris by centrifugation at 10,000 × g for 10 min, and the protein concentrations of the obtained protein supernatants were determined using the Bradford DC assay (Bio-Rad).
Tryptic Digestion—
Additional urea was added to a final concentration of 8 m to an aliquot containing 50 μg of protein from each sample. Thereafter the samples were reduced for 1 h at 37 °C using 10 mm DTT and subsequently alkylated for 30 min in darkness using 20 mm iodoacetamide. Before digestion, the urea concentration was lowered to 2 m by adding 100 mm ammonium bicarbonate and concentrated to a 50-μl volume using a 5-kDa-cutoff filter (Agilent Technologies). Finally 2% (w/w) sequence grade modified trypsin (Promega Corp.) was added, and proteins were allowed to digest for 12 h at 37 °C. The digested samples were purified using Strata X C18 columns (Phenomenex Torrance, CA) according to the manufacturer's instructions. Briefly the columns were equilibrated with 1 ml of methanol and 1 ml of deionized water. The samples were acidified to pH 2 by formic acid and were thereafter loaded onto the Strata X C18 columns. After washing with 10% ACN and 2% formic acid in deionized H2O, the peptides were eluted in three steps by first adding 1 ml of 80% ACN, 2% formic acid, and 18% deionized H2O followed by 2 × 1 ml of 98% ACN and 2% formic acid in deionized H2O. The collected fractions from each sample were pooled and then divided into two aliquots (experiments 1 and 2) that subsequently were lyophilized to dryness. Prior to LC-MS/MS analysis the peptides were redissolved in 12 μl of 0.1% trifluoroacetic acid.
Nanoflow Liquid Chromatography, Mass Spectrometry, and Protein Identification—
For the MS analysis, a 7-tesla hybrid linear ion trap Fourier transform mass spectrometer (LTQ FT, Thermo Fisher Scientific, Bremen, Germany) equipped with a nanoelectrospray ion source (Proxeon Biosystems, Odense, Denmark) was used. For separation of the peptides, a high performance liquid chromatography setup, Agilent 1100 nanoflow system, was applied. As analytical column, a 15-cm-long fused silica emitter (75-μm inner diameter, 375-μm outer diameter; Proxeon Biosystems) was used. The column was packed in house from a slurry of reverse phase material (fully end-capped Reprosil-Pur C18-AQ 3-μm resin from Dr. Maisch GmbH, Ammerbuch-Entrigen, Germany) in methanol using a “packing bomb” (Proxeon Biosystems) at a helium pressure of 50–60 bars. The mobile phases consisted of 0.5% acetic acid and 99.5% HPLC grade H2O (v/v) (A) and 0.5% acetic acid, 90% ACN, and 9.5% HPLC grade H2O (v/v) (B). For the analysis, 8 μl of the peptide solution was loaded onto the analytical column that subsequently was rinsed in 2% B for 30 min at a flow rate of 500 nl/min, and thereafter a gradient from 2 to 60% B at a constant flow rate of 200 nl/min was applied for either 360 or 720 min. For data acquisition, unattended mode was used in which the mass spectrometer automatically alternated between a high resolution (R = 100,000) overview MS scan and a lower resolution (R = 20,000) fragmentation scan (electron capture dissociation followed by collision-associated dissociation) of the two most abundant peptides at any given moment. Peak lists were generated using Xcalibur™ 2.0 SR2 (Thermo Fisher Scientific). For protein identification, the obtained MS/MS peak lists were searched against the International Protein Index (January 19, 2007) human database containing 67,250 sequences using the Mascot search engine (v2.1, Matrix Science, London, UK) in which trypsin specificity was used with up to two missed cleavages allowed. For the searches, methionine oxidation was used as a flexible modification, and cysteine alkylation was used as a fixed modification. The mass tolerance for MS was 10 ppm and for MS/MS was 0.02 Da. A group of peptides were considered as a positive match to a database protein if the confidence interval for the match, as determined by Mascot, was at least 95%. The cutoff scores as determined by Mascot were specific for each run and varied between 25 and 30. The false discovery rate was estimated to be 1.5% by a search in a concatenated reversed database.
Determination of Pathway Status—
Quantification of the expression level of identified peptides was based on peak intensity and was carried out using the Xtract routine program in the Xcalibur software (Thermo Fisher Scientific). The threshold for a peak to be used for quantification was 10,000 counts. Charge and isotope deconvolution was performed in the high resolution MS scans, and then the shape of the chromatographic peak was extracted using the accurate neutral mass. The summed intensities of the MS peaks originating from the same peptide were taken as the peptide abundance. The sum of the abundances of at least two peptides was attributed to the protein abundance. Peptide expression data were added to the peptide sequences determined by Mascot using in-house programmed scripts. Thereafter lists containing International Protein Index entries and protein abundances were loaded into the ExPlain™ 1.3 tool (BioBase GmbH). The previously described (23) PSE was used to determine the signaling status of the U-1810 cells after treatment with low and high LET IR, respectively.
Briefly when the identified proteins and their corresponding relative abundance were loaded into the ExPlain tool (BioBase), the protein identities were converted to their corresponding gene identities, creating a gene list, GL1. Next the proteins/genes from GL1 were mapped onto a pathway database TRANSPATH. TRANSPATH is one of the largest available databases of signal transduction components, containing proteins involved in cellular signaling such as ligands, receptors, adaptors, transcription factors; how they form complexes; their PTMs; and how they react to and interact with each other. Currently TRANSPATH contains information on about 150,000 signal transduction components of mammalian cells.
When a protein in the input list matched a gene in a network, an upstream analysis was initiated leading from the gene to one or several “key nodes” using the algorithm of searching for shortest path in a directed graph with the purpose to find an upstream node in the graph that was maximally connected to the nodes in the target list (24). Consequently not just one but several key nodes were found. Key nodes are molecules typically found in an intersection of networks and are known from literature to be critical elements in signal regulation. During analysis they were sorted in the output according to the “key node specificity score” K, which was calculated by the following formula.
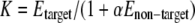 |
(Eq. 1) |
Here Etarget is the sum of expression values of all proteins from the target list that are reachable from the key node in a defined number of consecutive signal transduction reactions (default is set to four reactions); Enon-target is the sum of all other molecules in the whole network that also can be reached from the key node and that are considered as “non-target nodes.” The system assigns each non-target node with the lowest possible expression value, 1, because these nodes are not found as expressed in the target list. For a definition of and a thorough explanation of the penalty levels α, which describe how the user can adjust the balance between true and false positives, see Ref. 24.
Computation of key nodes allows us to extend the initial list of target proteins by considering other proteins that are revealed based on the database knowledge on the protein signaling reactions and that may not be detected in the proteomics experiments but whose activation status can influence a maximal number of the target proteins. The result of the first step of the algorithm is the list of key nodes sorted by the key node specificity score. Therefore at this step we enrich the initial list of proteins by more proteins that are known to be the most frequently connected in the signal transduction network generated from the proteins of the initial list.
The key nodes build networks with the downstream proteins; one example of a network that mapped to the JNK pathway is shown in supplemental Fig. S1. Red nodes (which all have numbers reflecting their relative abundance) are proteins identified from our proteomics experiments, the brown nodes are undetected molecules found in the TRANSPATH database, and finally purple molecules represent the key nodes. The key nodes are sometimes found among the identified proteins from the proteomics experiment (in that case, the relative abundance is written on the node), but more often they are found in the pathway database. At the next step, only 50% of the highest scoring key nodes are retained, and these key nodes together with their downstream proteins (both experimentally detected proteins and molecules detected from the database) are extracted into a new gene list, GL2. This new list is mapped onto all pathways in the TRANSPATH database, which contains 120 major pathways and several hundred subpathways. This time, if an input protein matches a gene in the pathway, it is considered a “hit.” Each pathway is therefore characterized by the number of hits N and the total number of key nodes L (N ≤ L). Additionally ExPlain calculates a p value of the pathway taking into account N, L, the size of the database, and the size of the input protein list GL2. The score S of the obtained pathway candidates was calculated by dividing the number of hits N for the pathway by its p value, S = N/p, as we described previously (23). Subsequently the pathway candidate lists were sorted according to the scores.
Comparison of Pathway Status between Differently Treated Cells—
When experiments 1 and 2 were run for each of the three samples (control, low LET IR, and high LET IR) 2 × 3 analysis results were obtained: low LET IR experiment 1 (L1), low LET IR experiment 2 (L2), high LET IR experiment 1 (H1), high LET IR experiment 2 (H2), control experiment 1 (C1), and control experiment 2 (C2) (Fig. 2). The pathway score S was compared between different samples as indicated in Fig. 2. Besides the relative prominence of a pathway in a certain sample (score value S), the PSE also gives a measure of confidence in pathway activation for one sample compared with another through the relative degree of activation (ΔS value). The importance of a specific ΔS value can only be understood by comparing it with ΔS values of other pathway candidates within the same pairwise comparison. Thus for each comparison, a ΔS distribution was created and analyzed as described below.
Fig. 2.

Schematic overview of experiments. NSCLC cells (U-1810) were either left untreated or irradiated with low or high LET IR. At 4 h post-IR proteins were isolated and digested into peptides. Equal amounts of protein from each sample were divided into two experiments (experiment 1, control (C1), low LET 4-h IR (L1), and high LET 4-h IR (H1); and experiment 2, control (C2), low LET 4-h IR (L2), and high LET 4-h IR (H2)) and thereafter analyzed by nanoflow LC-FT-ICR-MS/MS using a 720-min (experiment 1) or 360-min solvent gradient (experiment 2) in the LC run. Protein identities were obtained by Mascot, quantified by label-free quantification, and subsequently uploaded in the ExPlain tool. Determination of pathway status in each sample was performed using PSE, and pathway status for each sample was compared between the samples both within one experiment and between the two experiments. Pathways predicted as deactivated or activated were thereafter validated as described under “Experimental Procedures.” WB, Western blotting; IF, immunofluorescence.
Statistical Methods—
The PSE used in this work functions in a way similar to that routinely used in transcriptomics for determination of signaling pathways from gene expression levels. We have added a mechanism of taking into account the protein abundances and a scoring system that evaluates relative activation of each pathway candidate. The important feature of the PSE is that it takes into account the abundance of all detected proteins at once without separating them into groups of up- and down-regulated or unchanged species. Moreover the PSE considers all known signaling pathways as potentially activated and identifies the pathway or pathways that are most activated. The consequence of these features is that it is not always possible to determine precisely which protein or group of proteins triggered the outcome. But in our view, such determination is not required for testing the PSE: validation has to be of the prediction (i.e. of the activated pathway) and not of the underlying data (input protein expression levels).
Between 241 and 283 pathway candidates and their score values S were found for each sample. The score difference ΔS between two samples was calculated for each pathway candidate for all 12 comparative analyses (Fig. 2). The ΔS formed a bell-shaped distribution (not necessarily Gaussian) centered at zero. For each ΔS distribution, its dispersion σ was calculated. Pathways that had a ΔS larger than 3σ or smaller than −3σ (outliers) were considered activated or deactivated, respectively. Because the properties of the ΔS distributions were unclear, p values for the outliers could not be directly calculated from a single ΔS distribution. Instead the ranks of the pathways in the ΔS sorted lists in the four pairwise comparisons were taken into consideration. The p value of the combined measurement comprising four comparison pairs was calculated as follows: p = r1·r2·r3·r4/N4 where r1, r2, r3, and r4 are the pathway ranks in the four ΔS lists sorted from high to low ΔS values, each containing N pathway candidates. N was set as the median value between 241 and 283, i.e. 261. For example, when comparing high LET IR with low LET IR cells, the JNK pathway was found on the positions 1, 3, 1, and 1 of 261 pathway candidates, which corresponds to the p value 6.4·1010. Such a straightforward calculation, however, may underestimate the p values because it assumes the ΔS lists to be completely random and the four comparisons to be completely independent. To compensate for the eventual non-random character of the ΔS lists and the presence of statistical correlation between the four pairwise comparisons, the pathways were randomized in the four comparisons using a random number generator, and the p* value distribution of the randomized data was calculated. None of the 283 p* values was below 1·10−4, which then was used as a conservative normalizing factor for the p values determined as described above. The normalized p value of the JNK pathway is 6·10−6. The p values reported for other pathways are the normalized p values. All statistical tests were two-sided.
Analysis of Apoptotic Morphology—
Apoptotic morphology was quantified as the number of cells with fragmented nuclei. Thus, control and low or high LET-irradiated cells in the presence or absence of the JNK inhibitor SP600125 or p38 inhibitor SB203850 were harvested with cell dissociation solution. After washing in PBS, cells were fixed for 10 min in 4% PFA solution. The nuclei of fixed cells were stained with mounting medium containing 4,6′diamidino-2-phenylindole dihydrochloride (DAPI) (Vector Laboratories). The presence of fragmented nuclei indicative of an apoptotic cell was examined and quantified using a fluorescence microscope (Zeiss Axioplan 2 imaging microscope). In each sample at least 100 cells were assessed, and apoptosis is presented as the percentage of cells with fragmented nuclei. The doses of IR and time points for harvest are indicated for each specific analysis.
Quantification of Caspase-3 Activity—
Caspase-3 activity was examined in intact cells using an antibody that recognize active caspase-3, i.e. DEVD, or by assessing the accumulation of caspase-cleaved cytokeratin-18 (CK18-Asp396-NE) in pooled cell lysates and culture medium using the M30-Apopotosense™ ELISA (Peviva). Antibody-based caspase-3 activity was quantified in 0.25% PFA solution-fixed cells at indicated times postirradiation by incubating the cells for 1 h with a FITC-conjugated caspase-3 antibody (BD Pharmingen; dilution, 1:20) in the presence of digitonin (100 μg/ml). After washing in PBS the corresponding caspase-3-associated fluorescence in each sample was analyzed on the FL-1 channel using a FACS-Calibur flow cytometer (BD Biosciences). Fractions of 10,000 cells/sample were analyzed, and the amounts of caspase-3-positive cells were quantified. For assessment of caspase-cleaved cytokeratin-18 (CK18-Asp396-NE), cells were seeded at a density of 10,000 cells/well in a 96-well microtiter plate and the next day irradiated in the presence or absence of JNK or p38 inhibitor. At 48 h post-IR, the amount of cleaved cytokeratin-18 in cell culture medium and intact cells was measured according to the manufacturer's instructions.
ELISA-based Measurement of JNK and p38 Activation—
Activation of JNK and p38 was analyzed in fixed control or irradiated cells using FACE™ JNK ELISA and FACE-38 ELISA (both from Active Motif). Antibodies recognizing dually phosphorylated JNK (Thr183/Tyr185) and p38 (Thr180/Tyr182) or total JNK and p38 were used according to the manufacturer's instructions. Briefly cells were seeded at a density of 30,000/well in a 96-well plate and irradiated the next day in the adherent state with accelerated nitrogen ions as described above. At indicated time points postirradiation, the cells were rinsed in PBS and fixed in 4% PFA solution for 20 min at room temperature. Labeling with antibodies was done according to the manufacturer's protocol, and the resulting phosphoantibody signal was calculated after correction for number of cells and total JNK or p38 levels in each sample. The results are shown as -fold increase compared with control and are the mean of three independent experiments.
Immunofluorescence Analysis—
Cells were cytospun onto slides, directly fixed in 4% PFA solution for 10 min, and thereafter immersed in ice-cold acetone for 10 s. The cells were permeabilized using 0.5% Triton X-100 (10 min), blocked in 3% BSA buffer (10 min), and thereafter probed with phospho-JNK antibody (Cell Signaling Technology Inc.) diluted in BSA-containing buffer for 4 h at room temperature and subsequently washed in PBS. Finally slides were incubated for 1 h in darkness with FITC-conjugated secondary antibody, washed again, and mounted in DAPI-containing mounting medium before being analyzed by fluorescence microscopy (Zeiss Axioplan 2 imaging microscope). The images were acquired with Axiovision software and further processed in Adobe Photoshop CS3.
Western Blot Analysis—
Western blots were run using NuPAGE bis-Tris 3–12% gradient gels from Invitrogen according to the manufacturer's instructions. Equal amounts of protein (30 μg) were loaded, and gels were run in MES buffer for 35 min at 200 V. After blotting onto nitrocellulose membranes for 1 h at 30 V, the membranes were blocked in 5% BSA in TBS with Tween 20 for 60 min. Next the membranes were incubated with primary antibodies overnight at 4 °C. Primary antibodies used were JNK1 and JNK2 (phospho-Thr183 and -Tyr185) (Abcam), total JNK1 and JNK2 (Santa Cruz Biotechnology, Inc.), phospho-SEK1/mitogen-activated protein kinase kinase (MKK) 4 (Cell Signaling Technology Inc.), total SEK1/MKK4 (Cell Signaling Technology Inc.), 14-3-3σ (Abcam), and glyceraldehyde-3-phosphate dehydrogenase (Trevigen). After washing the membranes in TBS with Tween 20 for 30 min, secondary anti-mouse or anti-rabbit antibodies were added for 60 min at room temperature. Finally after washing, the membranes were developed on films, and band intensities were quantified using the Quantity One 4.6 software (Bio-Rad).
RESULTS
High LET Elicits Apoptosis in Low LET-resistant NSCLC Cells—
In this study we aimed to reveal signal transduction networks of importance for response to high LET IR in NSCLC cells. For this purpose, an NSCLC cell line, U-1810, considered resistant to low LET IR relative to other lung cancer cell lines was used (3, 6, 7). In response to high LET but not low LET IR, these cells showed a time-dependent increase in apoptotic morphology (Fig. 1A). Moreover a time-dependent increase in activation of caspase-3, one of the major executor caspases in apoptotic signaling, was observed after high LET IR (Fig. 1B). In accordance with our previous results (6), little or no increase in caspase-3 activity was observed in response to low LET IR in these NSCLC cells (Fig. 1B).
Fig. 1.

High LET but not low LET IR induces a time-dependent increase in apoptotic morphology and caspase-3 activity of U-1810 cells. U-1810 cells were irradiated with either 8 Gy of low LET γ-photons or 4 Gy of high LET accelerated ions. At the indicated time points (hours) after irradiation, the amount of cells with apoptotic morphology of the nuclei (A) or caspase-3 activation (B) was examined. A, fragmented nuclei indicative of an apoptotic cell were examined by counting DAPI-stained nuclei in duplicate samples for each treatment (100 cells examined). The results are presented as percentage of cells with apoptotic morphology. Data presented are the mean values (±S.D.) of two independent experiments. White bars, low LET IR; black bars, high LET IR. B, caspase-3 activity was analyzed using a FITC-conjugated antibody recognizing active caspase-3. Left, histogram showing caspase-3-associated immunofluorescence. Filled gray, untreated cells; black line, 44 h post-low or -high LET IR. Right, quantification of caspase-3-associated immunofluorescence after irradiation. Data presented are the mean values (±S.D.) of two independent experiments. White bars, low LET IR; black bars, high LET IR. Error bars refer to ±S.D.
Determination of Pathway Status in NSCLC Cells in Response to Different IR Qualities—
Proapoptotic mitochondria-mediated signaling occurs in NSCLC U-1810 cells between 12 and 16 h after DNA damage (6). To study proapoptotic signaling upstream of mitochondria, the MS analysis was therefore focused on an earlier time point, i.e. at 4 h post-IR, with the proteome being analyzed by LC-FT-ICR-MS/MS. In a few rare cases, proteins were identified and quantified with only one peptide. However, in those cases, the peptide always was identified both with electron capture dissociation and collision-associated dissociation fragmentation. The peptides used for identification and quantification of each protein and their corresponding identified sequences are presented for each run in supplemental Tables S1–S6. About 1,500 peptides and 650 proteins were identified from each sample. Quantitative data were based on at least two summed peptide intensities as described under “Experimental Procedures.” Lists of identified proteins and corresponding abundances are available in supplemental Table S1. Pathway analysis and determination of pathway status were carried out using PSE as described under “Experimental Procedures” and in Ref. 23 and was based on the proteomics data. Biological reproducibility was addressed by comparing high LET-irradiated cells not only with control but also with low LET-irradiated cells. Analytical reproducibility was addressed by running two technical replicates of each sample. A schematic overview of the study design is given in Fig. 2. One of the four lists showing (de)activated pathways in response to high LET IR compared with low LET IR is given in Table I. When analyzing the list of regulated cellular pathways, we noted that several of these have previously been attributed to apoptotic signaling, e.g. the JNK pathway, the Fas pathway, the p38 pathway, and the caspase network. Maps of these pathways as defined by the ExPlain 1.3 tool are provided in supplemental Figs. S2–S5.
Table I.
Exemplified data when comparing high LET IR cells with low LET IR cells
Here data from one of the four analyses comparing high LET IR with low LET IR are presented. The pathways are sorted according to their difference in score, i.e. ΔS between the two different radiation qualities. Thus, activation of a certain pathway is presented as a positive delta score for the corresponding pathway, and by analogy, deactivation of a specific pathway is given as a negative delta score. In this data set, 3σ corresponds to a ΔS of 4,656, meaning that every pathway with a ΔS larger than 4,656 or smaller than −4,656 is characterized as an outlier. The top three ranked (de)activated pathways are in bold. When the name of the pathway is reported this was the only pathway given the corresponding delta score. When the delta score approaches zero, as indicated, several different pathways can share the same delta score. The complete tables containing also the name of all these different pathways and all analyses are provided in supplemental Tables S1–S12. EGF, epidermal growth factor; LAT, linker for activation of T cells; PKC, protein kinase C; PRL, prolactin; MITF, microphthalmia transcription factor; ERK, extracellular signal-regulated kinase; TGF, transforming growth factor.
| ΔS | |
|---|---|
| Activated pathways | |
| JNK pathwaya | 24,897 |
| Fas pathway | 2,929 |
| EGF pathway | 2,625 |
| JNK1 → MKK4 | 1,302 |
| IL-8 pathway | 175 |
| Neurotensin pathway | 151 |
| Caspase network | 127 |
| LAT → PKCβ | 86 |
| Two different pathways | 77 |
| Three different pathways | 36 |
| Two different pathways | 27 |
| Wnt pathway | 25 |
| Seven different pathways | 5 |
| Deactivated pathways | |
| p38 pathway | −3,561 |
| RANKL pathway | −1,897 |
| Epo pathway | −1,713 |
| PRL pathway | −633 |
| BMP2 → p38α | −519 |
| TAK1 → ATF-2 | −287 |
| EGF → PAK1 | −185 |
| Apo2L pathway | −159 |
| RANKL → MITF | −130 |
| Epo → ERK1 | −88 |
| TGFβ pathway | −74 |
| ZAP-70 → c-Jun | −50 |
| Two different pathways | −40 |
| Five different pathways | −33 |
| Three different pathways | −25 |
| 25 different pathways | −14 |
| 197 different pathways | −4 |
The outlier.
One approach to identify significantly regulated pathways was to find the outliers of the ΔS distribution. To be characterized as an outlier the corresponding ΔS had to be larger than 3 standard deviations, 3σ, which in the exemplified data set (Table I and Fig. 3) corresponds to ΔS > 4,656. The spread of ΔS for the different pathways in Table I is presented graphically in Fig. 3. As can be observed, the JNK pathway is a clear outlier in this data set with ΔS = 24,897. The total 12 lists and corresponding graphic views of pathways regulated in response to the different treatments are given in supplemental Tables S7–S18. In these analyses, the stress pathway was ignored for the following reason as reported previously (23): the score value of the stress pathway is usually so large that its statistical fluctuation is comparable with the non-random ΔS value for other signaling pathways. Consequently it was difficult to interpret the reliability of the biological outcome from the analysis result of the stress pathway.
Fig. 3.

Spread in ΔS of regulated pathways in response to high LET IR versus low LET IR in U-1810 cells. Using the data from Table I, the numbers of differentially regulated pathways from high versus low LET-irradiated cells are plotted against their corresponding ΔS. Each dot in the graph represents a specific ΔS, and when ΔS approaches zero several pathways share the same ΔS, and therefore a dot could represent one or several pathways (see Table I). One procedure to identify significantly (de)activated pathways was to find the pathways that have a delta score that is so large that it is considered an outlier in the data set in point. Pathways are considered as outliers if the pathway has a delta score >|3σ|, which in the exemplified data set corresponds only to the JNK pathway (delta score, 24,897).
Criteria for Assigning Regulated Pathways—
Statistical outliers (|ΔS| > 3σ) represent significantly regulated pathways, whereas less significantly regulated pathways may appear with ΔS values within the −3σ to +3σ area (Fig. 3). These pathways can still be identified as (de)activated if they systematically appear among the top three pathways in the ΔS sorted candidate list. Therefore, we kept separate records for the outliers (Fig. 4, A–C, upper panel) and the top three pathways (Fig. 4, A–C, lower panel). These two types of pathway identification correspond to different degrees of analytical sensitivity with the top 3 method being the most sensitive. The most significantly (de)activated pathways were identified with both methods. A summary of the four analyses from each of the three comparisons, low LET-irradiated cells versus untreated cells, high LET-irradiated cells versus untreated cells, and high LET-irradiated cells versus low LET-irradiated cells, are presented in Fig. 4.
Fig. 4.

Summary of the four analyses for each pairwise comparison, showing significant outliers and top three ranked deactivated/activated pathways. Using PSE, pathways could be identified as significantly (*) (de)activated by fulfilling one of two requirements or both of them. The most apparent identification of a (de)activated pathway is when the pathway is classified as an outlier in at least three of the four analyses (upper panel). A pathway that was not classified as an outlier could still be identified as significantly (de)activated by a more sensitive method, i.e. if it was ranked as top 3 according to ΔS in at least three of the four analyses (lower panel). The most significant (de)activated pathways are found in both upper and lower panels. The requirements for a significant (de)activated pathway were fulfilled for the JNK pathway (p = 2·10−6), the Fas pathway (p = 3·10−5), and the p38 pathway (p = 0.001) when comparing high LET-irradiated versus control cells (A) and for the JNK pathway (p = 6·10−6) and the Fas pathway (p = 3·10−5) when comparing high LET- versus low LET-irradiated cells (C). When comparing low LET-irradiated versus control cells no significantly (de)activated pathways were identified (B). Nr., number; LPS, lipopolysaccharide; IFN, interferon; EGF, epidermal growth factor; PRL, prolactin.
In Silico Analysis Suggested the JNK Pathway as Activated after High LET IR—
In the case of high LET IR versus control the JNK pathway was a positive outlier in four of four analyses (p = 2·10−6) (Fig. 4, A, upper panel). This corresponds to the highest possible degree of activation. In contrast, in the case of low LET IR versus control, no pathway was an outlier more than twice, even in the sensitive top 3 detection (Fig. 4B). This indicates that low LET IR does not produce a serious disturbance in the pathway domain in U-1810 cells at the 4-h time point examined. Importantly when comparing high LET- versus low LET-irradiated NSCLC U-1810 cells, the JNK pathway was again a positive outlier (p = 6·10−6) in four of four analyses (Fig. 4C). Also the Fas pathway was significantly activated both when comparing high LET IR versus control (p = 3·10−5) and when comparing high LET- versus low LET-irradiated cells (p = 3·10−5), classified as an outlier or appearing as a top 3 ranked pathway in three of the four pairwise analyses (Fig. 4C). In addition, the p38 pathway was significantly deactivated (p = 0.001) when comparing high LET IR versus control cells (Fig. 4A). The interferon γ/STAT1α pathway, the RANKL pathway, and the Epo pathway were also predicted to be significantly deactivated (Fig. 4, A and C) but were not considered further in this study. Pathways that appeared as outliers or as the top three less than three times but more than zero are also presented in Fig. 4, but they were not considered significantly regulated.
In Vitro Validation of JNK Pathway Activation in U-1810 Cells after High LET IR—
We have previously reported that defective activation of JNK in NSCLC may contribute to impaired apoptotic response to low LET IR (6). Given this and the fact that predictions from the pathway analysis identified the JNK pathway as a key event in high LET IR-induced cellular signaling, we aimed to validate the activation and function of JNK in high LET IR-induced proapoptotic signaling. As a start we examined whether an increased phosphorylation of JNK, indicative of activation, is observed in response to high LET IR in these NSCLC U-1810 cells. At 4 h post-IR with high LET particles (absorbed dose, 4 Gy), an activation of JNK could be confirmed by immunofluorescence analysis (Fig. 5A, left). We also examined JNK activation over time (1, 4, and 16 h) in response to high LET IR using an ELISA-based assay, and a 2-fold increase in phosphorylated JNK was observed at 1 and 4 h compared with control cells (Fig. 5A, right). To compare the effect of low LET IR versus high LET IR another time course study (2, 4, and 24 h) was performed using classic Western blot analysis. With respect to JNK, we observed a clear activation of both JNK1 and JNK2 at 2 h in response to high but not low LET IR. However, the most prominent activation of JNK was detected at 24 h where a strong activation of JNK2 was observed in response to high but not low LET IR (Fig. 5B, left). Moreover the band intensities from the JNK Western blot were quantified. A graph showing variations of the total phosphorylation status (pJNK1 and pJNK2) of JNK (after normalization to loading control and to total JNK1 and JNK2 expression) over time in cells treated with low and high LET IR, respectively, is shown in Fig. 5B, right.
Fig. 5.

High LET IR activates JNK, which in turn is required for induction of apoptosis in U-1810 cells. A, left, U-1810 cells were irradiated with 4 Gy of high LET IR and after 4 h were stained with antibody specific for phosphorylated JNK1/2. The nucleus of the cells is visualized by blue fluorescence (DAPI), and phosphorylated JNK is visualized by green fluorescence (FITC). Right, activation of JNK was assessed at the indicated time points after high LET IR (4 Gy) using phosphospecific activity-related antibodies to JNK in a 96-well ELISA-based assay. -Fold increase in phospho-JNK-associated signal in irradiated cells compared with control was examined after correction for the number of cells and the level of total JNK in each sample. Data shown are the mean (±S.D.) of three independent experiments with duplicate samples. B, left, Western blots showing levels of total expression of JNK1 and JNK2 as well as phosphorylated JNK1 and phosphorylated JNK2 in response to low LET and high LET IR, respectively, at the indicated time points. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is used as loading control. Left, the graph shows quantified data from the Western blot in the right panel. Total phosphorylation of JNK (pJNK1 and pJNK2) and total protein expression of JNK (Tot JNK1 and Tot JNK2) were both normalized to loading control (glyceraldehyde-3-phosphate dehydrogenase). Next corrected total phosphorylated JNK was normalized to corrected total JNK protein expression. Untreated cells were used as control (C), and the curves illustrate the -fold change to control distributed over time of total phosphorylated JNK in response to low LET and high LET IR, respectively. C, Western blots showing levels of total expression of 14-3-3σ and MKK4 as well as phosphorylated MKK4 in response to low LET (8 Gy) and high LET IR (4 Gy), respectively, at the indicated time points. Glyceraldehyde-3-phosphate dehydrogenase is used as loading control. D, U-1810 cells were irradiated with high LET IR (4 Gy) in the presence or absence of SP600125 (10 μm). At 44 h post-treatment, apoptosis was assessed either as inhibition of apoptotic morphology of the cell nuclei (left) or as inhibition of caspase-3 (Casp.3) activity assessed as cleavage of cytokeratin 18 (M30 signal) (right). The data shown are the mean (±S.D.) of three independent experiments with duplicate samples. Error bars refer to ±S.D.
To further validate the activation of the JNK pathway, we also checked activation of MKK4 and MKK7, which are upstream of JNK itself in the JNK pathway. Thus, MKK4 and MKK7 were suggested from the pathway analysis as potential important players of this pathway (supplemental Fig. S3). Interestingly our data showed no changes in activation of MKK7 (data not shown), but in contrast, MKK4 was strongly activated at 4 h in response to high but not to low LET IR (Fig. 5C).
In addition, we validated activation of the ζ isoform and the expression turnover of the σ isoform of the scaffold protein 14-3-3; they also were suggested as candidate players in the JNK pathway (supplemental Fig. S3). However, with respect to 14-3-3ζ, no change in activation (phosphorylation) was detected (data not shown).
With regard to 14-3-3σ, its expression level is reported to be elevated in tumors, and this isoform is thereby suggested as a tumor marker. Because 14-3-3σ is reported to be overexpressed specifically in NSCLC it is a potential drug target for this form of cancer in particular (27). The σ isoform of 14-3-3 is not reported to be phosphorylated in the literature; thus, in this case the validation was focused on the expression turnover. As expected, we found that 14-3-3σ was strongly expressed in our NSCLC cell model system; however, at the 4-h time point postirradiation with high LET IR it was clear that the protein level of 14-3-3σ was significantly decreased (Fig. 5C).
Inhibition of JNK Blocks High LET IR-induced Apoptotic Signaling in U-1810 Cells—
To examine the importance of JNK-mediated signaling for proapoptotic response to high LET IR, U-1810 cells were pretreated with the JNK inhibitor SP600125 prior to IR. The JNK inhibitor was able to efficiently block apoptosis induced by high LET IR (Fig. 5D). Thus the amount of apoptotic cells when combining high LET IR and SP600125 was almost the same as that seen for SP600125 alone (Fig. 5D, left). Importantly SP600125 was also found to block high LET IR-induced caspase-3 activity (Fig. 5D, right). In conclusion, we for the first time show that apoptotic signaling in response to high LET IR involves JNK activation.
High LET IR Deactivate p38 in U-1810 Cells—
Given the reported dissimilar roles of p38 with respect to DNA damage-induced apoptotic signaling and the fact that PSE pathway analyses indicated that the p38 pathway was deactivated when comparing high LET IR versus control cells, we also analyzed the function of p38 in response to high LET IR. In agreement with the PSE prediction, analysis of p38 activity at 4 and 16 h post-high LET IR revealed a reduction in activity to approximately half (Fig. 6A). Accordingly blocking p38 activity by SB203850 in U-1810 cells prior to IR neither inhibited high LET IR-induced apoptotic propensity (Fig. 6B) nor affected high LET IR-induced caspase-3 activity (Fig. 6C). Rather SB203850 somewhat increased high LET IR-induced apoptotic morphology (Fig. 6B). These results indicate that JNK and p38 have opposite roles in high LET IR-induced apoptotic signaling in U-1810 cells.
Fig. 6.

Activation of p38 is dispensable for induction of apoptosis after high LET IR in U-1810 cells. Activation of p38 was assessed at the indicated time points after high LET IR (4 Gy) using phosphospecific antibodies to p38 in a 96-well ELISA-based assay. -Fold increase in phospho-p38-associated fluorescence in irradiated cells compared with control after correction for the number of cells and amount of total p38 is presented (A). U-1810 cells were irradiated with accelerated nitrogen ions in the presence or absence of SB203850 (10 μm). At 44 h post-treatment, apoptosis was assessed either as inhibition of apoptotic morphology of the cell nuclei (B) or as inhibition of caspase-3 activity assessed as cleavage of cytokeratin 18 (M30 signal) (C). The data shown are the mean (±S.D.) of three independent experiments with duplicate samples. Error bar refers to ± S.D.
DISCUSSION
Accelerated ions conferring high LET IR are known to have a higher relative biological effect compared with conventional IR, i.e. low LET. In NSCLC cases that showed resistance to conventional low LET radiotherapy, high LET IR (accelerated ions) have demonstrated promising clinical results (8, 10). In this setting, the identification of specific biomarkers would be of high clinical impact. In fact, the selection of patients for therapy with high LET IR could be based on the evaluation of biomarkers of resistance to conventional radiotherapy and sensitivity to high LET IR. In addition monitoring the expression of IR-induced downstream effectors could be of importance for early prediction of patients’ response to treatment. Furthermore key molecules could be targeted to sensitize NSCLC cells to conventional low LET IR. In the search for such biomarkers, identification of signaling pathways operative after IR with high LET as opposed to low LET IR is highly relevant.
In this study an NSCLC cell line sensitive to high and resistant to low LET IR was used. We have previously found defective mitochondrial activation in response to low LET IR in this cell line, also reflected by reduced caspase-3 activation (6). As shown in Fig. 1, it is clear that high LET IR more efficiently induces both caspase-3 activation and apoptotic morphology in these cells compared with low LET IR. This indicates that our model system suits the purpose of using it for revealing signaling pathways of importance for high LET IR-induced apoptosis. For analyses of signal transduction events triggered by high and low LET IR we used PSE, a novel search engine, the basis of which is an unsupervised algorithm that compares extensive sets of proteomics data to predict signaling status in cells. When using this strategy to compare high and low LET-irradiated U-1810 cells, several signal transduction pathways were identified as differentially regulated (Table I, Fig. 3, and supplemental Tables S7–S18). After repeatedly analyzing the pathway status in response to high LET IR in this cell system, JNK- and Fas-mediated signal transduction pathways were consistently identified as the most striking alterations, a finding that was not seen in response to low LET IR (Fig. 4, A–C). Moreover the p38 pathway was significantly deactivated when comparing high LET IR with control cells (Fig. 4A).
Undoubtedly there are large advantages and potential in using global and unsupervised methods to get a fast and holistic understanding of critical signaling events. However, it is important to be aware that the PSE predictions rely on the information stored in the database. If the database is incomplete, there is a risk of generating invalid hypotheses. This emphasizes the importance of validation of PSE predictions and encourages that both traditional hypothesis-driven research and the hypothesis-generating approach described here should be used in parallel to accomplish fast and reliable medical development.
The JNK pathway is one of the most well characterized pathways within cells. In addition, databases that work with public domain annotated data will more often report highly annotated pathways simply because more information about them is available. Hence the chance that the JNK pathway will be reported as affected with PSE-mediated pathway analysis could be considered high. Importantly in our comparison of the pathways differentially regulated between low LET-irradiated U-1810 cells and untreated, PSE did not report the JNK pathway as activated in the low LET-irradiated sample (Fig. 4B). This indicates that the JNK pathway was not randomly identified. Moreover the robustness of the predictions is highlighted by the fact that the pathway analysis consistently generated similar results although different solvent gradients were used for the LC runs and despite the fact that there was a 3-month time lag between the two experiments (Fig. 4).
It should be stressed that the PSE analysis is based on pathway information assembled from thousands of published sources. None of these sources has reported JNK activation in response to high LET IR, which was an original finding of this work. Moreover PSE was ignorant of the experimental specifics. Therefore, the JNK activation prediction was based on objective proteomics data and not on a priori knowledge of the processes considered. At no point was the PSE aware of the plausible involvement of apoptosis, cancer treatment, DNA damage, or any other information that could have triggered the output. This indicates that it is possible to use large sets of global proteomics data in combination with previous knowledge stored in the database to create a plausible suggestion of the pathway status in cells even in the absence of other a priori information. However, as always, in silico-based findings need to be validated. In accordance, because PSE is mostly a hypothesis-generating tool, it was crucial that we could validate its predictions and confirm that high LET IR activated signaling through the JNK pathway in U-1810 cells (Fig. 5).
The presented data strongly indicate a role of JNK signaling for efficient induction of apoptosis after high LET IR because pharmacological inhibition of JNK inhibited DNA fragmentation as well as caspase-3 activation in this cell system (Fig. 5D). Because we have shown that the sensitivity of NSCLC cells to low LET IR is linked to whether or not the JNK pathway is activated (6), the fact that we found activation of the JNK pathway by high LET IR is of considerable interest. This suggests that in response to high LET IR, NSCLC cells may circumvent the resistance to apoptotic cell death seen in the case of low LET IR by being capable of activating the JNK pathway in a proper way.
It is interesting to consider which upstream signaling events might be implicated in JNK activation after high LET IR. As we did see an activation of MKK4 (Fig. 5C) but not MKK7 (data not shown) at 4 h post-high LET IR our data indicate that MKK4 might be involved in the activation of JNK at 24 h but not be responsible for the observed early JNK activation i.e. at 2 h (Fig. 5B). Further upstream of MKK4 activation one may speculate that the non-receptor tyrosine kinase c-Abl is involved as it has been shown to activate MKK4 and subsequently JNK via MEKK1-mediated signaling after low LET IR as well as other DNA-damaging treatments (28–31). It was recently shown that an early initial JNK activation is required to trigger release of c-Abl from the adaptor protein 14-3-3 and localization of c-Abl within the cell nuclei and subsequently allow c-Abl and late JNK activation (32–34). Given the fact that we did see a reduction in 14-3-3σ in U-1810 cells after high LET IR (Fig. 5C) and because 14-3-3 proteins are known to form cytoplasmic complexes with c-Abl, one might hypothesize that high LET but not low LET IR is capable of causing redistribution of c-Abl to the nucleus and that c-Abl is responsible for the observed late activation of JNK in this system. Therefore, further analyses of c-Abl in response to high LET IR would indeed be an interesting future research approach.
The PSE analysis also suggested that the Fas pathway is activated in response to high but not low LET IR (Fig. 4C). Given that we and others have previously shown that a Fas-caspase-8-mediated pathway is not of importance for low LET IR (3, 35, 36), one may also speculate that a non-mitochondria-mediated pathway involving Fas is responsible for the higher efficiency of high LET IR-induced apoptotic signaling. Interestingly the JNK and Fas pathways have proven connections and work in concert. Thus, JNK activates c-Jun through phosphorylation, which in turn acts as a transcriptional regulator of Fas and subsequent activation of the Fas pathway (37, 38). Yet another route to increased Fas activity is mediated via inhibition of the phosphatidylinositol 3-kinase/Akt pathway, which subsequently can activate the FKHR-L1/FOXO3A transcription factor and trigger expression of Fas (39). Given that we previously showed that low LET IR efficiency can be increased by blocking phosphatidylinositol 3-kinase/Akt signaling either indirectly via IGF-1R (40) or protein kinase C blockade or directly via LY-294002 (40–42), one may speculate that high LET IR in contrast to low LET IR may cause inhibition of the phosphatidylinositol 3-kinase/Akt pathway in U-1810 cells and in this way regulate the function of Fas signaling.
The SAPK p38 has at least in part been associated with apoptotic signaling after DNA damage (6, 18, 43, 44). However, we could not in response to high LET IR predict any increased activation of the p38 pathway when determining the pathway status (Fig. 4). In accordance, no activation of p38 could be detected in vitro after high LET IR (Fig. 6A). In addition, when inhibiting p38 pharmacologically before high LET IR, decreased apoptosis was not observed (Fig. 6, B and C). This suggests that induction of p38 is not an absolute requirement for high LET IR-induced apoptotic signaling. We have previously shown that p38, rather than being a proapoptotic kinase in response to low LET IR, is involved in an IGF-1R-mediated prosurvival signal in this NSCLC cell line (40). Thus one may speculate that the observed down-regulation of p38 after high LET IR (Fig. 4A and Fig. 6A) reflects a blockade of an IGF-1 R-mediated survival signaling. In contrast, p38 was recently implicated in low LET IR-induced proapoptotic signaling involving Bak and Bax (20) pointing to its dual function in signaling in response to IR in NSCLC cells. A hypothetical illustration reflecting the differences in cellular response between low LET and high LET IR in U-1810 cells as discussed above is presented in Fig. 7.
Fig. 7.

A simplified hypothetical illustration of signaling in response to low and high LET IR in U-1810 cells. The NSCLC cell line U-1810 can resist apoptosis when treated with low LET IR. However, in response to high LET IR, these cells start a proapoptotic signaling through an early activation of JNK. Left, a hypothetical explanation is that high LET IR causes a decrease of 14-3-3 and possibly phosphorylation of c-Abl by JNK (I). As a consequence, c-Abl cannot form a complex with 14-3-3 (II) and is allowed to translocate to the nucleus (III) where it is activated by IR-induced DNA damage that in turn through activation of MKK4 mediates a late JNK activation (IV). This subsequently leads to activation of various proapoptotic signaling molecules and activation of mitochondria resulting in an apoptotic cell death. JNK may also phosphorylate the transcriptional factor c-Jun, which can increase expression of Fas ligand (V) and thereby activate non-mitochondria-mediated apoptotic signaling (VI). High LET IR also deactivates p38 that in turn inhibits the IGF-1R-mediated survival signaling in these NSCLC cells (VII). Right, in contrast, low LET IR can neither activate JNK-mediated apoptotic signaling (VIII) nor inhibit p38-mediated survival signaling via the IGF-1R/phosphatidylinositol 3-kinase (PI3K)/Akt pathway (IX). As a result, U-1810 cells can resist low LET IR-induced apoptotic cell death. Fas R, Fas receptor; cyt. c, cytochrome c. FOX, Forkhead box.
When considering approaches focused on finding biomarkers of IR sensitivity or resistance, one may suppose that one or a few molecules may be critical for radiosensitivity or, alternatively, that one or several protein networks together contribute to a radiosensitive phenotype. This also has an implication on development of therapeutic strategies. Depending on the ultimate result of such analyses one or several regulated pathways may have to be targeted. This promotes the concept of multipathway targeting in analogy with using either specific or multitargeted tyrosine kinase inhibitors in cancer therapy (45, 46). Similarly it will be important to understand whether activation of the JNK and Fas pathways will be the optimal way to sensitize tumors to low LET IR. The JNK pathway consists of a large network of molecules, and the critical parts of this network may be the known activators and substrates for JNK discussed above but might as well consist of molecules or networks of molecules that are not yet well characterized. The PSE may here, by coupling back to the large set of input molecules from the proteomics analysis, play a very important role. By revealing a selection of relations between proteins, PSE can enable the findings of key molecules and networks containing possible targets to circumvent resistance and thereby provide a rich resource for future follow-up studies. The information generated in these kinds of analyses will most probably be a requirement for the development of future cancer therapy.
Supplementary Material
Footnotes
Published, MCP Papers in Press, January 23, 2009, DOI 10.1074/mcp.M800274-MCP200
The abbreviations used are: IR, ionizing radiation; LET, linear energy transfer; NSCLC, non-small cell lung carcinoma; PSE, pathway search engine; SAPK, stress-activated protein kinase; JNK, c-Jun NH2-terminal kinase; Gy, gray(s); PFA, paraformaldehyde; DAPI, 4,6′diamidino-2-phenylindole dihydrochloride; bis-Tris, 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol; RANKL, receptor activator of nuclear factor κB ligand; Epo, erythropoietin; pJNK, phosphorylated JNK; MKK, mitogen-activated protein kinase kinase; IGF-1 R, insulin-like growth factor receptor.
This work was supported by Swedish Cancer Society Grant CAN 2006/1149 (to R. L.); Stockholm Cancer Society Grants 05:1201 (to R. L.), 06:1222 (to R. L.), 05:1213 (to R. L.), and 06:1462 (to K. V.); the Stockholm County Council, and the European Commission (6th framework program CHEMORES project and 7th framework program ApoSYS project).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Turesson, I., Carlsson, J., Brahme, A., Glimelius, B., Zackrisson, B., and Stenerlow, B. ( 2003) Biological response to radiation therapy. Acta Oncol. 42, 92–106 [DOI] [PubMed] [Google Scholar]
- 2.Schulz-Ertner, D., Jakel, O., and Schlegel, W. ( 2006) Radiation therapy with charged particles. Semin. Radiat. Oncol. 16, 249–259 [DOI] [PubMed] [Google Scholar]
- 3.Joseph, B., Ekedahl, J., Lewensohn, R., Marchetti, P., Formstecher, P., and Zhivotovsky, B. ( 2001) Defective caspase-3 relocalization in non-small cell lung carcinoma. Oncogene 20, 2877–2888 [DOI] [PubMed] [Google Scholar]
- 4.Joseph, B., Marchetti, P., Formstecher, P., Kroemer, G., Lewensohn, R., and Zhivotovsky, B. ( 2002) Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene 21, 65–77 [DOI] [PubMed] [Google Scholar]
- 5.Sirzen, F., Zhivotovsky, B., Nilsson, A., Bergh, J., and Lewensohn, R. ( 1998) Spontaneous and radiation-induced apoptosis in lung carcinoma cells with different intrinsic radiosensitivities. Anticancer Res. 18, 695–699 [PubMed] [Google Scholar]
- 6.Viktorsson, K., Ekedahl, J., Lindebro, M. C., Lewensohn, R., Zhivotovsky, B., Linder, S., and Shoshan, M. C. ( 2003) Defective stress kinase and Bak activation in response to ionizing radiation but not cisplatin in a non-small cell lung carcinoma cell line. Exp. Cell Res. 289, 256–264 [DOI] [PubMed] [Google Scholar]
- 7.Viktorsson, K., Lewensohn, R., and Zhivotovsky, B. ( 2005) Apoptotic pathways and therapy resistance in human malignancies. Adv. Cancer Res. 94, 143–196 [DOI] [PubMed] [Google Scholar]
- 8.Austin-Seymour, M., Griffin, T., Laramore, G., Maor, M., and Parker, R. ( 1989) High-LET radiation therapy of non-small cell lung cancer. Chest 96, 72S–73S [DOI] [PubMed] [Google Scholar]
- 9.Nakano, T., Suzuki, Y., Ohno, T., Kato, S., Suzuki, M., Morita, S., Sato, S., Oka, K., and Tsujii, H. ( 2006) Carbon beam therapy overcomes the radiation resistance of uterine cervical cancer originating from hypoxia. Clin. Cancer Res. 12, 2185–2190 [DOI] [PubMed] [Google Scholar]
- 10.Tsujii, H., Mizoe, J. E., Kamada, T., Baba, M., Kato, S., Kato, H., Tsuji, H., Yamada, S., Yasuda, S., Ohno, T., Yanagi, T., Hasegawa, A., Sugawara, T., Ezawa, H., Kandatsu, S., Yoshikawa, K., Kishimoto, R., and Miyamoto, T. ( 2004) Overview of clinical experiences on carbon ion radiotherapy at NIRS. Radiother. Oncol. 73, Suppl. 2, S41–S49 [DOI] [PubMed] [Google Scholar]
- 11.Stenerlow, B., Carlsson, J., Blomquist, E., and Erixon, K. ( 1994) Clonogenic cell survival and rejoining of DNA double-strand breaks: comparisons between three cell lines after photon or He ion irradiation. Int. J. Radiat. Biol. 65, 631–639 [DOI] [PubMed] [Google Scholar]
- 12.Goodhead, D. T. ( 1994) Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int. J. Radiat. Biol. 65, 7–17 [DOI] [PubMed] [Google Scholar]
- 13.Radulescu, I., Elmroth, K., and Stenerlow, B. ( 2004) Chromatin organization contributes to non-randomly distributed double-strand breaks after exposure to high-LET radiation. Radiat. Res. 161, 1–8 [DOI] [PubMed] [Google Scholar]
- 14.Davis, R. J. ( 2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 [DOI] [PubMed] [Google Scholar]
- 15.Dent, P., Yacoub, A., Fisher, P. B., Hagan, M. P., and Grant, S. ( 2003) MAPK pathways in radiation responses. Oncogene 22, 5885–5896 [DOI] [PubMed] [Google Scholar]
- 16.Sanchez-Perez, I., Murguia, J. R., and Perona, R. ( 1998) Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene 16, 533–540 [DOI] [PubMed] [Google Scholar]
- 17.Tournier, C., Hess, P., Yang, D. D., Xu, J., Turner, T. K., Nimnual, A., Bar-Sagi, D., Jones, S. N., Flavell, R. A., and Davis, R. J. ( 2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 18.Brozovic, A., Fritz, G., Christmann, M., Zisowsky, J., Jaehde, U., Osmak, M., and Kaina, B. ( 2004) Long-term activation of SAPK/JNK, p38 kinase and fas-L expression by cisplatin is attenuated in human carcinoma cells that acquired drug resistance. Int. J. Cancer 112, 974–985 [DOI] [PubMed] [Google Scholar]
- 19.Chen, Y. R., Wang, X., Templeton, D., Davis, R. J., and Tan, T. H. ( 1996) The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and γ radiation. Duration of JNK activation may determine cell death and proliferation. J. Biol. Chem. 271, 31929–31936 [DOI] [PubMed] [Google Scholar]
- 20.Choi, S. Y., Kim, M. J., Kang, C. M., Bae, S., Cho, C. K., Soh, J. W., Kim, J. H., Kang, S., Chung, H. Y., Lee, Y. S., and Lee, S. J. ( 2006) Activation of Bak and Bax through c-abl-protein kinase Cδ-p38 MAPK signaling in response to ionizing radiation in human non-small cell lung cancer cells. J. Biol. Chem. 281, 7049–7059 [DOI] [PubMed] [Google Scholar]
- 21.Buza, J. J., and Burgess, S. C. ( 2008) Different signaling pathways expressed by chicken naive CD4+ T cells, CD4+ lymphocytes activated with staphylococcal enterotoxin B, and those malignantly transformed by Marek's disease virus. J. Proteome Res. 7, 2380–2387 [DOI] [PubMed] [Google Scholar]
- 22.Ralhan, R., Desouza, L. V., Matta, A., Chandra Tripathi, S., Ghanny, S., Datta Gupta, S., Bahadur, S., and Siu, K. W. ( 2008) Discovery and verification of head-and-neck cancer biomarkers by differential protein expression analysis using iTRAQ labeling and multidimensional liquid chromatography and tandem mass spectrometry. Mol. Cell. Proteomics 7, 1162–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zubarev, R. A., Nielsen, M. L., Fung, E. M., Savitski, M. M., Kel-Margoulis, O., Wingender, E., and Kel, A. ( 2008) Identification of dominant signaling pathways from proteomics expression data. J. Proteomics 1, 89–96 [DOI] [PubMed] [Google Scholar]
- 24.Kel, A., Voss, N., Jauregui, R., Kel-Margoulis, O., and Wingender, E. ( 2006) Beyond microarrays: Finding key transcription factors controlling signal transduction pathways. BMC Bioinformatics 7, Suppl. 2, S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krull, M., Pistor, S., Voss, N., Kel, A., Reuter, I., Kronenberg, D., Michael, H., Schwarzer, K., Potapov, A., Choi, C., Kel-Margoulis, O., and Wingender, E. ( 2006) TRANSPATH: an information resource for storing and visualizing signaling pathways and their pathological aberrations. Nucleic Acids Res. 34, D546–D551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wingender, E. ( 2008) The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 9, 326–332 [DOI] [PubMed] [Google Scholar]
- 27.Qi, W., Liu, X., Qiao, D., and Martinez, J. D. ( 2005) Isoform-specific expression of 14-3-3 proteins in human lung cancer tissues. Int. J. Cancer 113, 359–363 [DOI] [PubMed] [Google Scholar]
- 28.Kharbanda, S., Pandey, P., Ren, R., Mayer, B., Zon, L., and Kufe, D. ( 1995) c-Abl activation regulates induction of the SEK1/stress-activated protein kinase pathway in the cellular response to 1-β-d-arabinofuranosylcytosine. J. Biol. Chem. 270, 30278–30281 [DOI] [PubMed] [Google Scholar]
- 29.Kharbanda, S., Pandey, P., Yamauchi, T., Kumar, S., Kaneki, M., Kumar, V., Bharti, A., Yuan, Z. M., Ghanem, L., Rana, A., Weichselbaum, R., Johnson, G., and Kufe, D. ( 2000) Activation of MEK kinase 1 by the c-Abl protein tyrosine kinase in response to DNA damage. Mol. Cell. Biol. 20, 4979–4989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kharbanda, S., Ren, R., Pandey, P., Shafman, T. D., Feller, S. M., Weichselbaum, R. R., and Kufe, D. W. ( 1995) Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature 376, 785–788 [DOI] [PubMed] [Google Scholar]
- 31.Kharbanda, S., Yuan, Z. M., Weichselbaum, R., and Kufe, D. ( 1998) Determination of cell fate by c-Abl activation in the response to DNA damage. Oncogene 17, 3309–3318 [DOI] [PubMed] [Google Scholar]
- 32.Yoshida, K. ( 2007) Regulation for nuclear targeting of the Abl tyrosine kinase in response to DNA damage. Adv. Exp. Med. Biol. 604, 155–165 [DOI] [PubMed] [Google Scholar]
- 33.Yoshida, K., and Miki, Y. ( 2005) Enabling death by the Abl tyrosine kinase: mechanisms for nuclear shuttling of c-Abl in response to DNA damage. Cell Cycle 4, 777–779 [DOI] [PubMed] [Google Scholar]
- 34.Yoshida, K., Yamaguchi, T., Natsume, T., Kufe, D., and Miki, Y. ( 2005) JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell 7, 278–285 [DOI] [PubMed] [Google Scholar]
- 35.Belka, C., Heinrich, V., Marini, P., Faltin, H., Schulze-Osthoff, K., Bamberg, M., and Budach, W. ( 1999) Ionizing radiation and the activation of caspase-8 in highly apoptosis-sensitive lymphoma cells. Int. J. Radiat. Biol. 75, 1257–1264 [DOI] [PubMed] [Google Scholar]
- 36.Belka, C., Marini, P., Lepple-Wienhues, A., Budach, W., Jekle, A., Los, M., Lang, F., Schulze-Osthoff, K., Gulbins, E., and Bamberg, M. ( 1999) The tyrosine kinase lck is required for CD95-independent caspase-8 activation and apoptosis in response to ionizing radiation. Oncogene 18, 4983–4992 [DOI] [PubMed] [Google Scholar]
- 37.Kuwabara, M., Takahashi, K., and Inanami, O. ( 2003) Induction of apoptosis through the activation of SAPK/JNK followed by the expression of death receptor Fas in X-irradiated cells. J Radiat. Res. (Tokyo) 44, 203–209 [DOI] [PubMed] [Google Scholar]
- 38.Li, X. R., Chong, A. S., Wu, J., Roebuck, K. A., Kumar, A., Parrillo, J. E., Rapp, U. R., Kimberly, R. P., Williams, J. W., and Xu, X. ( 1999) Transcriptional regulation of Fas gene expression by GA-binding protein and AP-1 in T cell antigen receptor·CD3 complex-stimulated T cells. J. Biol. Chem. 274, 35203–35210 [DOI] [PubMed] [Google Scholar]
- 39.Jia, L., Yu, W., Wang, P., Li, J., Sanders, B. G., and Kline, K. ( 2008) Critical roles for JNK, c-Jun, and Fas/FasL signaling in vitamin E analog-induced apoptosis in human prostate cancer cells. Prostate 68, 427–441 [DOI] [PubMed] [Google Scholar]
- 40.Cosaceanu, D., Budiu, R. A., Carapancea, M., Castro, J., Lewensohn, R., and Dricu, A. ( 2007) Ionizing radiation activates IGF-1R triggering a cytoprotective signaling by interfering with Ku-DNA binding and by modulating Ku86 expression via a p38 kinase-dependent mechanism. Oncogene 26, 2423–2434 [DOI] [PubMed] [Google Scholar]
- 41.Hemstrom, T. H., Joseph, B., Schulte, G., Lewensohn, R., and Zhivotovsky, B. ( 2005) PKC 412 sensitizes U1810 non-small cell lung cancer cells to DNA damage. Exp. Cell Res. 305, 200–213 [DOI] [PubMed] [Google Scholar]
- 42.Hemstrom, T. H., Sandstrom, M., and Zhivotovsky, B. ( 2006) Inhibitors of the PI3-kinase/Akt pathway induce mitotic catastrophe in non-small cell lung cancer cells. Int. J. Cancer 119, 1028–1038 [DOI] [PubMed] [Google Scholar]
- 43.Benhar, M., Dalyot, I., Engelberg, D., and Levitzki, A. ( 2001) Enhanced ROS production in oncogenically transformed cells potentiates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase activation and sensitization to genotoxic stress. Mol. Cell. Biol. 21, 6913–6926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chattopadhyay, S., Machado-Pinilla, R., Manguan-Garcia, C., Belda-Iniesta, C., Moratilla, C., Cejas, P., Fresno-Vara, J. A., de Castro-Carpeno, J., Casado, E., Nistal, M., Gonzalez-Baron, M., and Perona, R. ( 2006) MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 25, 3335–3345 [DOI] [PubMed] [Google Scholar]
- 45.Marinov, M., Fischer, B., and Arcaro, A. ( 2007) Targeting mTOR signaling in lung cancer. Crit. Rev. Oncol. Hematol. 63, 172–182 [DOI] [PubMed] [Google Scholar]
- 46.Gollob, J. A., Wilhelm, S., Carter, C., and Kelley, S. L. ( 2006) Role of Raf kinase in cancer: therapeutic potential of targeting the Raf/MEK/ERK signal transduction pathway. Semin. Oncol. 33, 392–406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.