

Abstract
Increased basal and loss of glucose-stimulated insulin secretion (GSIS) are hallmarks of β-cell dysfunction associated with type 2 diabetes. It has been proposed that elevated glucose promotes insulin secretory defects by activating sterol regulatory element binding protein (SREBP)-1c, lipogenic gene expression, and neutral lipid storage. Activation of liver X receptors (LXRs) also activates SREBP-1c and increases lipogenic gene expression and neutral lipid storage but increases basal and GSIS. This study was designed to characterize the changes in de novo fatty acid and triacylglyceride (TAG) synthesis in LXR-activated β-cells and determine how these changes contribute to elevated basal and GSIS. Treatment of INS-1 β-cells with LXR agonist T0901317 and elevated glucose led to markedly increased nuclear localization of SREBP-1, lipogenic gene expression, de novo synthesis of monounsaturated fatty acids and TAG, and basal and GSIS. LXR-activated cells had increased fatty acid oxidation and expression of genes involved in mitochondrial β-oxidation, particularly carnitine palmitoyltransferase-1. Increased basal insulin release from LXR-activated cells coincided with rapid turnover of newly synthesized TAG and required acyl-coenzyme A synthesis and mitochondrial β-oxidation. GSIS from LXR-activated INS-1 cells required influx of extracellular calcium and lipolysis, suggesting production of lipid-signaling molecules from TAG. Inhibition of diacylglyceride (DAG)-binding proteins, but not classic isoforms of protein kinase C, attenuated GSIS from LXR-activated INS-1 cells. In conclusion, LXR activation in β-cells exposed to elevated glucose concentrations increases de novo TAG synthesis; subsequent lipolysis produces free fatty acids and DAG, which are oxidized to increase basal insulin release and activate DAG-binding proteins to enhance GSIS, respectively.
LXR activation of INS-1 β-cells exposed to elevated glucose increases triacylglyceride (TAG) synthesis; subsequent TAG turnover can lead to the production of lipid signaling molecules resulting in elevated insulin release.
Type 2 diabetes mellitus occurs when pancreatic β-cells fail to secrete sufficient amounts of insulin necessary to overcome insulin resistance at peripheral tissues and maintain glucose homeostasis. Loss of β-cell function in type 2 diabetes has been suggested to occur when β-cells are chronically exposed to elevated circulating glucose and free fatty acids (FFAs), a state defined as glucolipotoxicity (1,2). Sterol regulatory element binding protein (SREBP)-1c, a basic helix loop helix transcription factor, plays a major role in inducing lipogenic gene expression in liver and adipose tissue (3,4,5) and thereby partitions glucose toward synthesis of lipid. The ability of elevated glucose to increase the nuclear form of SREBP-1c in β-cells (6,7,8) has been proposed to serve as a possible mechanism for glucolipotoxicity and explain the predominant role of elevated glucose in β-cell dysfunction (7). Consistent with this hypothesis, expression of a constitutively active nuclear form of SREBP-1c in islets or β-cell lines increased lipogenic gene expression, triacylglyceride (TAG) synthesis and storage, and suppressed glucose-stimulated insulin secretion (GSIS) (6,7,9,10,11). Islet failure in Zucker diabetic fatty (ZDF) rats is also associated with increased expression of SREBP-1c and TAG accumulation (12,13,14).
Although activation of SREBP-1c is an attractive mechanism to explain glucolipotoxicity, recent reports suggest that the role of SREPB-1c in β-cell function is more complex. First, SREBP-1c activation and lipid synthesis are required for adaptive changes leading to hypersecretion of insulin from mouse islets exposed to elevated glucose concentrations (15). Second, inactivation of SREBP-1c in ZDF rat islets failed to restore GSIS suggesting that increased SREBP-1c and intracellular TAG are not the principal cause of β-cell secretory dysfunction (16). Third, activation of liver X receptors (LXR) in islets and β-cell lines increased SREBP-1c, lipogenic gene expression, neutral lipid storage, and basal and GSIS (17,18).
LXRα (NR1H3) and LXRß (NR1H2) are nuclear receptors involved in transcriptional control of genes involved in cholesterol, fatty acid, and glucose metabolism (19). LXRα is primarily expressed in liver, kidney, intestine, and macrophages, whereas LXRß is ubiquitously expressed (20,21). LXR is activated by oxysterols (22) and glucose (23). In macrophages, LXR regulates reverse cholesterol transport through increased expression of genes encoding ATP-binding cassette (ABC) cholesterol transporters ABCA1 (24) and ABCG1 (25). In liver, LXR controls transcription of genes involved in conversion of cholesterol into bile acids (22,26) and excretion of biliary cholesterol (27). LXR also directly and indirectly, through increased SREBP-1c expression, activates lipogenic gene transcription including ACC [acetyl coenzyme A (CoA) carboxylase], fatty acid synthase (FAS), and stearoyl CoA desaturase (SCD)-1 and SCD2 (28,29,30,31).
LXRα and LXRβ are expressed in rodent and human pancreatic islets and β-cell lines (17,32). Islets from LXRβ knockout mice accumulated lipid, have reduced expression of cholesterol transporters, and reduced GSIS (18,33). Treatment of islets and β-cell lines with the LXR agonist T0901317 increased expression of lipogenic genes and lipid accumulation (17,34). Importantly, LXR agonists elevated basal and GSIS from islets and β-cell lines through a mechanism dependent on increased SREBP-1c and pyruvate carboxylase and ACCα activity (17,18). These findings suggest that LXR activation promotes insulin release by stimulating anaplerotic and cataplerotic pathways, possibly to supply malonyl-CoA for de novo fatty acid and lipid synthesis. The principal aims of the study presented herein were to characterize the changes in de novo fatty acid (FA) and TAG synthesis in LXR-activated β-cells and determine how these changes in lipid metabolism may contribute to elevated basal and GSIS.
Materials and Methods
Materials
T0901317 (Cayman Chemical, Ann Arbor, MI). [1-14C]palmitic acid and d-[5-3H]glucose were from PerkinElmer Life Sciences (Boston, MA). [2-14C]acetic acid and d-[U-14C]glucose were from ICN Pharmaceuticals, Inc. (Costa Mesa, CA). FA-free BSA was from Roche Applied Science (Indianapolis, IN).
INS-1 cell culture
INS-1 cells were routinely cultured in INS-1 media (RPMI 1640 media containing 11.1 mm glucose, 10% fetal bovine serum, 1 mm pyruvate, 10 mm HEPES, 50 μm 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin) as previously described (35). For T0901317 experiments, cells were seeded at a density of 0.2 × 106 cells/cm2 and cultured for 48 h in INS-1 media. Cells were then cultured for 48 h in INS-1 media containing 4 or 16.7 mm glucose and vehicle or 10 μm T0901317.
Human islets
Human islets were obtained from the Juvenile Diabetes Research Foundation Human Islet Distribution Program at the University of Minnesota (Minneapolis, MN), University of Miami (Miami, FL), and Northwest Tissue Center (Seattle, WA). Islets were maintained in N2 medium or RPMI 1640 plus 10% fetal bovine serum containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.5 μg/ml Fungizone. For gene expression experiments, islets were cultured for 3 d in N2 medium in the absence or presence of 10 μm T0901317. For lipid synthesis experiments, islets were cultured for 36 h in RPMI 1640 medium containing 11.1 or 22.1 mm glucose plus 5 μm T0901317 (see below) and then incubated for 12 h under the same conditions in the presence of 1 μCi [2-14C]acetic acid.
Western blot analysis
Microsomes and nuclear extracts were isolated as previously described (36). Proteins (50–100 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes. SREBP-1 immunoreactivity was detected using anti-SREBP-1 monoclonal antibodies (IgG-2A4; Santa Cruz Biotechnology. Santa Cruz, CA) and visualized using SuperSignal West Pico chemiluminescence kit (Pierce, Rockford, IL).
RNA analysis
Total RNA was obtained using TRIZOL (Invitrogen, Carlsbad, CA). For INS-1 cells, cDNA were synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Real-time quantitative PCR (qPCR) was performed using Platinum SYBR Green qPCR SuperMix-UGD (Invitrogen), gene-specific primers (supplemental Table 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://endo.endojournals.org) and a Mx3000P qPCR system (Stratagene, La Jolla, CA). Levels of mRNA in human islets were determined by real-time qPCR using Light Upon Extension fluorogenic primers (Invitrogen) (supplemental Table 1) as previously described (37). Relative amounts of mRNA were calculated using the comparative cycle threshold method. Results for human islets and INS-1 cells were normalized to the abundance of β-actin and cyclophilin mRNA, respectively.
Insulin secretion studies
INS-1 cells were preincubated twice for 30 min at 37 C in Krebs Ringer bicarbonate buffer (KRBB) (35) containing 2 mm glucose and subsequently incubated for 60 min in KRBB containing either 2 or 20 mm glucose. For studies using etomoxir (100 μm), orlistat (50 μm), or calphostin C (1 μm), agents were present throughout the incubation period in KRBB. Triacsin C (10 μm) was added 5 h before secretion studies. Verapamil (100 μm) was present only during the final 1 h incubation in KRBB. Insulin released into the media and insulin content from acid-ethanol-extracted cells were determined by RIA (Linco, St. Louis, MO). Total cell protein was determined by Lowry assay.
Glucose use studies
Glucose use was measured using a modification of the method of Zawalich and Matschinsky (38) as previously described (35).
De novo lipid synthesis and FA profile
INS-1 cells were cultured for 48 h in INS-1 media containing 4 or 16.7 mm glucose and vehicle or 10 μm T0901317. During the last 12 h, cells were cultured in INS-1 media containing 4 or 16.7 mm glucose, vehicle or 5 μm T0901317, and 1 μCi [2-14C]acetic acid (51 mCi/mmol). Cells were then extracted and lipids separated by thin-layer chromatography (TLC) as previously described (39). Quantification of 14C-labeled lipids was determined on a PhosphoImager 820 (Molecular Dynamics, Sunnyvale, CA).
Analysis of 14C-labeled FA profile was performed as previously described (40). Total lipid extracts from the [2-14C]acetic acid labeling studies were saponified, neutralized, extracted in diethyl ether, dried, and resuspended in methanol and 0.1 mm butylated hydroxytoluene. FAs were then fractionated by reverse-phase HPLC using a J’sphere ODS-H80 column (YMC-Waters, Milford, MA) and quantified by flow-through scintillation counting. Saturated and unsaturated FAs were quantified by evaporative light scatter, and unsaturated FAs were quantified by UV absorption at 192 nm.
For total FA synthesis from glucose during the insulin secretion study, cells were incubated for 1 h with either 2 or 20 mm glucose plus 4 or 40 μCi of [U-14C]glucose (260 mCi/mmol), respectively. Cells were extracted, extracts saponified, and 14C-labeled FA quantified as described above.
Palmitate oxidation
INS-1 cells were then incubated for 1 h in KRBB containing 2 mm glucose, after which cells were incubated for 1 h in KRBB containing 50 μm palmitate, 2 μCi/ml [1-14C]palmitic acid (56 mCi/mmol), and 2 or 20 mm glucose. Palmitate oxidation was determined by measuring [14C]CO2 released into the medium using the method of Parkera et al. (41).
Statistical analyses
All the data shown represent three to six independent experiments performed in duplicate. Experiments with more than two groups were analyzed using a one-way ANOVA followed by Bonferroni’s multiple comparison test. Experiments comparing two groups were analyzed using Student’s t test. P < 0.05 was considered significant.
Results
Effect of glucose and LXR activation on SREBP-1 and lipogenic gene expression
Culturing INS-1 cells in 16.7 mm glucose for 48 h increased protein levels of both the microsomal precursor (125 kDa) and nuclear active (65 kDa) forms of SREBP-1 (Fig. 1A) (36). Activation of LXR by the addition of T0901317 further increased microsomal precursor and nuclear forms of SREBP-1 for each glucose concentration tested. These results show that elevated glucose and direct activation of LXR by T0901317 can independently and synergistically activate SREBP-1 synthesis, processing, and nuclear localization in β-cells. Because LXR regulates only SREBP-1c and not SREBP-1a (29), these changes in SREBP-1 likely reflect increased SREBP-1c nuclear abundance.
Figure 1.

Glucose and LXR activation increase SREBP-1 expression and nuclear localization and lipogenic gene expression. INS-1 cells were cultured for 48 h in media containing 4 or 16.7 mm glucose ± 10 μm T0901317. A, Microsomes and nuclear extracts were fractionated by SDS-PAGE and SREBP-1 immunoreactivity was detected by Western analysis. Results shown are representative of four independent experiments. B, Total RNA was isolated and analyzed for SREBP-1, ACC, FAS, SCD1, SCD2, ABCA1, and ABCG1 mRNA expression by real-time RT-PCR. Control genes cyclophilin and ribosomal protein L32 were unaffected by T0901317 (TO) or glucose (data not shown). Data are relative to cyclophilin and normalized to cells cultured in 4 mm glucose (mean ± sem, n = 4).
Activation of LXR in a number of tissues results in direct and indirect induction of genes involved in lipogenesis and cholesterol efflux. Culturing INS-1 cells in 16.7 mm glucose for 48 h led to a modest increase (1.4- to 2-fold) in mRNA levels of SREBP-1, FAS, SCD1, and SCD2 and a large increase in ACCα (6.4-fold) (Fig. 1B). LXR activation with T0901317 markedly increased the expression of SREBP-1, FAS, SCD1, SCD2, ABCA1, and ABCG1 irrespective of the glucose concentration. T0901317 also led to a 2.3-fold increase in ACCα gene expression in cell culture in low glucose but did not further increase ACCα expression in cells cultured in elevated glucose. Treatment of human islets with T0901317 (10 μm) for 72 h also led to a 6- to 13-fold increase in expression of SREBP-1, FAS, and ACCα mRNA (supplemental Fig. 1).
Effect of glucose and LXR activation on lipogenesis
To assess de novo lipogenesis, INS-1 cells were incubated for 36 h in 4 or 16.7 mm glucose ± T0901317 and then incubated for 12 h under the same conditions in the presence of [2-14C]acetic acid. Total lipids were analyzed by TLC to measure 14C incorporation into polar lipids (phospho- and sphingolipids), FFAs, TAG, cholesterol, and cholesterol esters (39). Figure 2A illustrates the fractional distribution of 14C in neutral, polar, and nonesterified lipid fractions. The percent of 14C in the polar lipid fraction ranged from 88 to 71% and was sensitive to both glucose and T0901317. For cells incubated in 4 mm glucose, 83.1 ± 1.5% (n = 4) of 14C-labeled lipid were polar lipids, and the fractional distribution was not altered by T0901317 (82.2 ± 1.4%). Treatment of cells with 16.7 mm glucose shifted the fractional 14C distribution from polar lipids (77.6 ± 2.0%) to neutral lipids, and this was further shifted by T0901317 (71.1 ± 1.3%).
Figure 2.

Elevated glucose and T0901317 increase de novo lipid synthesis in INS-1 cells. INS-1 cells were cultured for 48 h in 4 or 16.7 mm glucose ± 10 μm T0901317. Cells were then incubated overnight under the same conditions in the presence of [2-14C]acetic acid. A, Total lipids were extracted from the cells and separated on silica TLC plates with hexane-ether-acetic acid (90:30:1). Plates were dried, and radioactivity was detected and quantified on a PhosphoImager (Molecular Dynamics). Data are reported as percentage of total labeled lipid. Percent of labeled polar lipid is reported as a numeric value at the top of the graph. B, Total lipids were saponified and incorporation of 14C into fatty acids was determined by reverse-phase HPLC. Data are reported as percentage of total labeled fatty acid. Values are the mean ± sem for four independent experiments.
In cells cultured in 4 mm glucose, the fractional distribution of 14C assimilation into cholesterol, cholesterol esters (CEs), FFAs, and TAG was 7.7 ± 0.6, 0.6 ± 0.1, 1.1 ± 0.2, and 1.7 ± 0.3%, respectively, and this was unaltered by treatment with T0901317 (Fig. 2A). Culturing cells in 16.7 mm glucose led to a 5.4- and 2.5-fold increase in 14C-labeled TAG (9.1 ± 1.6%, P < 0.01) and CE (1.4 ± 0.3%, P < 0.03), respectively. Addition of T0901317 led to an additional 2-fold increase in 14C-labeled TAG (16.7 ± 1.9%, P < 0.01) and an approximately 50% reduction in 14C-labeled cholesterol (4.0 ± 0.4, P < 0.03) and CE (0.7 ± 0.1, P < 0.04), respectively. Similar changes in fractional distribution of 14C-labeled polar lipids and TAG were observed in experiments using d-[U-14C]glucose (data not shown). Changes in 14C-labeled TAG were also proportional to changes in TAG mass (data not shown). Treatment of human islets with T0901317 also increased 14C-labeled TAG and reduced 14C-labeled cholesterol (supplemental Fig. 2A). Overall, these results demonstrate that activation of lipogenic genes in β-cells by elevated glucose and T0901317 markedly increase de novo lipid synthesis and partitioning of carbon from acetic acid or glucose into complex neutral lipids, particularly TAG.
Effect of LXR activation on fatty acid profile
Total lipids extracted from [2-14C]acetic acid-labeled INS-1 cells were saponified, and the FA profile was determined by reverse-phase HPLC. When INS-1 cells were incubated in 4 mm glucose, 4.5, 53.1, 23.6, 5.7, 5.3, and 7.8% of 14C-labeled FAs were myristic acid (14:0), palmitic acid (16:0), stearic acid (18:0), palmitoleic acid (16:1, n-7), vaccenic (18:1, n-7), and oleic acid (18:1, n-9), respectively (Fig. 2B). Incubation of cells in 16.7 mm glucose only modestly affected the fractional distribution of 14C-labeled FAs, and this was reflected by a small increase in 14C-labeled palmitoleic acid (16:1, n-7). In contrast, treatment of cells with T0901317 led to a large change in the fractional distribution of 14C-labeled FAs such that 14C-labeled palmitoleic (16:1, n-7), vaccenic (18;1, n-7), and oleic (18:1, n-9) acids increased by approximately 2-fold. T0901317 also led to an approximately 25% reduction in 14C-labeled palmitic (16:0) and stearic (18:0) acid. The T0901317-induced change in 14C-labeled FAs occurred with a commensurate increase in monounsaturated fatty acid (MUFA) pool size and decrease in saturated FA pool size (data not shown). Treatment of isolated human islets with T0901317 also changed the fractional distribution of 14C-labeled FAs with an increase in palmitoleic acid (16:1, n-7) synthesis and a reduction in stearic acid (18:0) production (supplemental Fig. 2B). These results show that activation of LXR in β-cells increases de novo synthesis of MUFA.
Impact of LXR activation on basal and glucose-stimulated insulin secretion
INS-1 cells were cultured for 48 h in 4 or 16.7 mm glucose ± 10 μm T0901317, after which insulin release was measured in response to a 1 h challenge with either 2 or 20 mm glucose. Acute treatment of INS-1 cells, previously cultured in 4 mm glucose ± T0901317, with 20 mm glucose led to an approximately 2-fold increase in GSIS (Fig. 3A). In comparison, INS-1 cells cultured in 16.7 mm glucose for 48 h had reduced basal insulin release (2 mm glucose) and GSIS. In cells cultured in 16.7 mm glucose plus T0901317, insulin release was increased when cells were stimulated with 2 or 20 mm glucose, and there appeared to be a partial recovery of GSIS. Although long-term exposure of INS-1 cells to elevated glucose reduces insulin expression (42), increased insulin release from cells cultured in 16.7 mm glucose plus T0901317 was not due to increased cellular insulin content (Fig. 3B). Moreover, treatment of cells with T0901317 did not significantly alter glucose utilization at either 2 or 16.7 mm glucose (Fig. 3C). Similar findings were also observed for glucose oxidation (data not shown). These data indicate that LXR-activated INS-1 cells cultured under an elevated glucose load have increased basal and stimulated insulin release, and this is independent of increased insulin content or changes in acute glucose metabolism.
Figure 3.

INS-1 cells cultured in elevated glucose and T0901317 have increased insulin release. INS-1 cells were cultured for 48 h in 4 and 16.7 mm glucose ± 10 μm T0901317. Cells were washed and preincubated at 37 C in KRBB containing 2 mm glucose for 60 min. A, Acute insulin release was then determined by incubating cells for 60 min at 37 C in KRBB containing 2 or 20 mm glucose. Data represent the mean ± sem for four independent experiments performed in triplicate. *, P < 0.03 when compared with cells cultured in 16.7 mm glucose. B, Intracellular insulin content from INS-1 cells that under went an acute glucose (2 mm) challenge as described above. Data represent the mean ± se of four independent experiments. *, P < 0.05 when compared with cells cultured in 4 mm glucose. C, Glucose utilization was measured by incubating cells for 30 min at 37 C in KRBB containing either 2 or 16.7 mm glucose and [5-3H]d-glucose. Conversion of [5-3H]glucose to [3H]H2O was determined. Data represent the mean ± se of five independent experiments. * and #, P < 0.04 or P < 0.001, respectively, when compared with cells cultured in 4 mm glucose.
Role of fatty acid oxidation in elevated insulin release
To investigate the role of FA oxidation in insulin release, secretion studies were performed in the presence of an inhibitor of long-chain acyl CoA synthetase, triacsin C, or an inhibitor of carnitine palmitoyltransferase (CPT)-1, etomoxir. Triacsin C prevents conversion of FFA to long-chain acyl CoA, thereby indirectly inhibiting β oxidation of FFA, whereas etomoxir directly inhibits the rate-limiting enzyme of β-oxidation. In INS-1 cells cultured for 48 h in 4 mm glucose ± T0901317 or 16.7 mm glucose, etomoxir (100 μm) had no effect on insulin release in response to a 1 h challenge with 2 or 20 mm glucose (Fig. 4A and supplemental Fig. 3). In contrast, the enhanced basal insulin release (2 mm glucose) from INS-1 cells cultured in 16.7 mm glucose plus T0901317 was reduced 25% (P < 0.001, n = 6) by etomoxir. Triacsin C (10 μm) tended to lower basal insulin release from cells cultured in 4 mm glucose ± T0901317 (supplemental Fig. 3). In LXR-activated cells cultured in 16.7 mm glucose, triacsin C reduced basal insulin release by 38% (P < 0.001, n = 6) (Fig. 4A). These data suggest that enhanced basal insulin release from LXR-activated cells cultured in 16.7 mm glucose requires conversion of FFA to LC-CoA and increased β-oxidation. Consistent with this possibility, [14C]palmitate oxidation under basal glucose conditions (2 mm) was elevated approximately 3-fold in INS-1 cells cultured for 48 h in T0901317 (Fig. 4B). Although palmitate oxidation was suppressed by elevated glucose (20 mm), [14C]palmitate oxidation remained elevated approximately 3.5-fold in LXR-activated INS-1 cells. Oxidation of [14C]palmitate in LXR-activated cells was also inhibited approximately 65% by triacsin C and approximately 90% by etomoxir. Increased FA oxidation in T0901317-cultured INS-1 cells correlated with increased mRNA levels of genes involved in mitochondrial β-oxidation (Fig. 4C) including CPT-1α, carnitine palmitoyl transferase-2, and long-chain acyl CoA dehydrogenase but not very long-chain acyl CoA dehydrogenase. These data strongly support the hypothesis that LXR activation increases basal insulin release from INS-1 cells by a mechanism involving increased β-oxidation of fatty acids.
Figure 4.
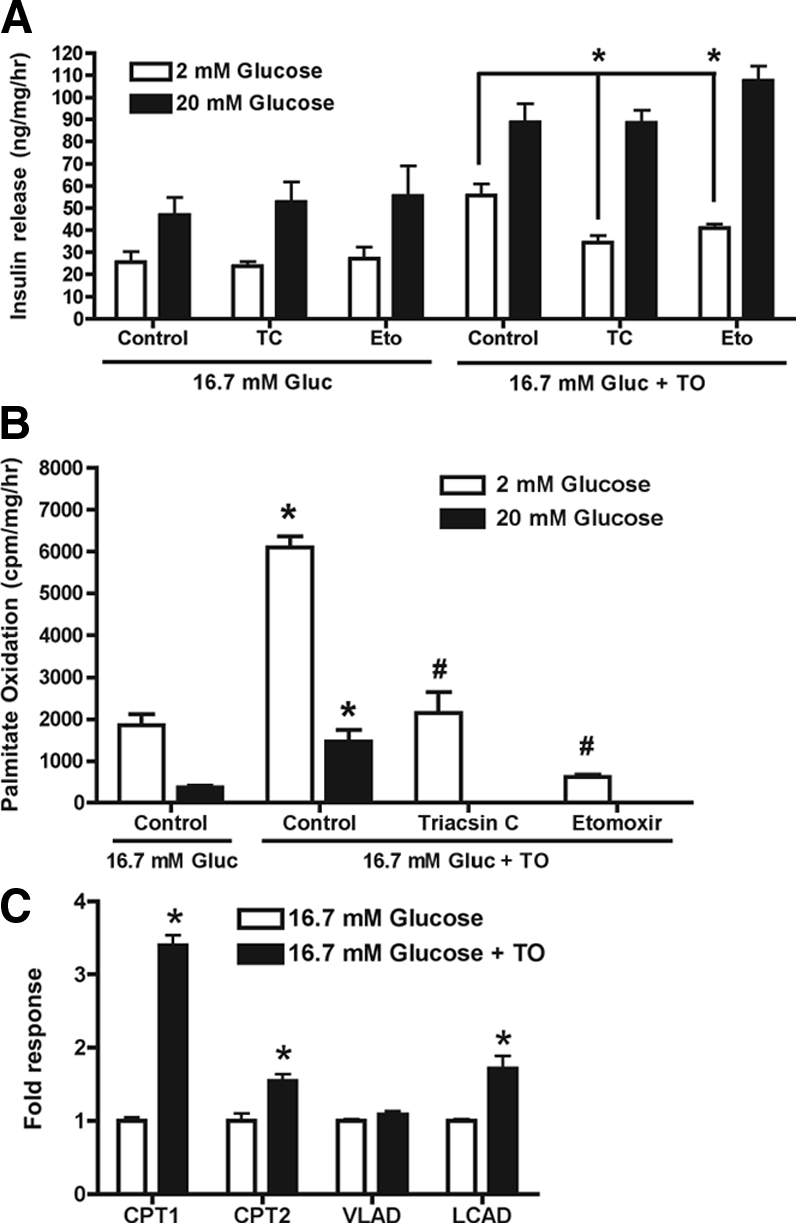
Elevated basal insulin release from LXR-activated INS-1 cells involves increased FA oxidation. INS-1 cells were incubated for 48 h in 16.7 mm glucose (Gluc) ± T0901317 (TO; 10 μm). A, Insulin release (60 min) in response to 2 or 20 mm glucose was then assessed in the presence or absence of etomoxir (Eto; 100 μm) or triacsin C (TC; 10 μm). Values are mean ± sem for six independent experiments. *, P < 0.001 when etomoxir- or triacsin C-treated cells are compared with control cells. B, FA oxidation was determined by measuring [14C]CO2 production from cells incubated for 1 h in 2 mm glucose followed by a 1-h incubation with palmitic acid (50 μm), [14C]palmitic acid, and 2 or 20 mm glucose. Data are mean ± sem for three independent experiments. *, P < 0.01 when cells cultured in 16.7 mm glucose plus T0901317 are compared with cells cultured in 16.7 mm glucose; #, P < 0.001 when triacsin C- or etomoxir-treated cells are compared with cells cultured in 16.7 mm glucose plus T0901317. C, Total RNA was extracted and mRNA levels for CPT-1α (CPT1), CPT-2, very long-chain acyl CoA dehydrogenase (VLAD), and long-chain acyl CoA dehydrogenase (LCAD) were determined by real time RT-PCR. Data are mean ± sem for three independent experiments. *, P < 0.02 when cells cultured in 16.7 mm glucose are compared with cells cultured in 16.7 mm glucose plus T0901317.
Role of TAG turnover in elevated insulin secretion
The relationship between enhanced insulin release and the turnover of TAG was examined using the general lipase inhibitor orlistat. Treatment of INS-1 cells cultured in 4 mm glucose with orlistat (5–50 μm) increased basal insulin secretion (supplemental Fig. 3). The mechanism accounting for the elevated basal insulin release is unknown but may be related to metabolic stress associated with low glucose and the inability to turnover lipid pools. Consistent with this, orlistat (50 μm) had no effect on basal insulin release from INS-1 cells initially cultured in 16.7 mm glucose ± T0901317 (Fig. 5A). Orlistat, however, completely blocked insulin release in response to 20 mm glucose. These results suggested that enhanced GSIS from LXR-activated INS-1 cells cultured in 16.7 mm glucose was associated with turnover of TAG. To examine TAG turnover, INS-1 cells were cultured in 16.7 mm glucose and T0901317 for 48 h and during the last 6 h were labeled with [2-14C]acetic acid. After labeling, cells were subjected to conditions mimicking an acute insulin release assay in the presence of orlistat and turnover of labeled lipid was measured. Incubation of cells for 2 h in 2 mm glucose led to an approximately 65% decrease in TAG and diacylglyceride (DAG) labeling and an approximately 50% decrease in FFA labeling (Fig. 5B). Treatment of cells for 1 h in 2 mm glucose followed by 1 h in 20 mm glucose tended to slow the turnover of TAG and DAG. Orlistat completely blocked the turnover TAG and led to a precipitous fall in labeled FFA, possibly due to FA oxidation of cellular FFA. Orlistat has also been shown to inhibit FAS (43), suggesting that orlistat may block GSIS from LXR-activated INS-1 cells by inhibiting de novo lipid synthesis. To test this possibility, INS-1 cells cultured for 48 h in 16.7 mm glucose ± T0901317 were subjected to an acute insulin release assay in the presence of 2 or 20 mm glucose containing [U-14C]glucose and incorporation of 14C into FA from methanol-soluble lipids was determined. Incubation of control and LXR-activated cells for 1 h with 20 mm glucose markedly increased 14C-labeled FA, and this was further increased by orlistat (Fig. 5C), suggesting orlistat’s action on GSIS is independent of inhibition of de novo lipogenesis. These data suggest that a byproduct of TAG turnover such as DAG or FFA may participate in enhanced GSIS from LXR-activated INS-1 cells.
Figure 5.

TAG turnover is required for enhanced glucose-stimulated insulin release from LXR-activated INS-1 cells. A, Impact of orlistat on insulin release. INS-1 cells were incubated for 48 h in 16.7 mm glucose (Gluc) ± T0901317 (TO; 10 μm). Insulin release (60 min) in response to 2 or 20 mm glucose was then assessed in the presence or absence of orlistat (50 μm). Values are mean ± sem for six independent experiments. *, P < 0.001 when orlistat-treated cells are compared with control cells. B, Impact of orlistat on turnover of de novo-derived TAG, DAG, and FFA. INS-1 cells were cultured for 48 h in 16.7 mm glucose ± T0901317 (10 μm). During the last 6 h, cells were incubated with [2-14C]acetic acid (t = 0), after which cells were subjected to conditions for an acute insulin release study: 1 h incubation in 2 mm glucose followed by a 1-h incubation in 2 or 20 mm glucose. Total lipids were extracted and analyzed as described in Fig. 3A. Values are mean ± sem for three independent experiments. Data are presented relative to 14C-labeling at t = 0. PhosphoImager intensity values at t = 0 for TAG are 482,621 ± 59,573, for DAG are 39,461 ± 4,451 and for FFA are 37,267 ± 3,691. C, Impact of orlistat (50 μm) on de novo FA synthesis from glucose. INS-1 cells were incubated for 48 h in 16.7 mm glucose ± T0901317, and subjected to a insulin release assay with 2 or 20 mm glucose containing 4 or 40 μCi of [U-14C]glucose, respectively. 14C-labeled FAs were quantified as described in Materials and Methods. Values are mean ± sem of three independent experiments. *, P < 0.001 orlistat-treated cells are compared with control cells.
To investigate a potential role for DAG, acute insulin release studies were performed in the presence of calphostin C, an inhibitor of protein kinase C (PKC) that competitively interferes with DAG and phorbol ester binding. GSIS from LXR-activated INS-1 cells was attenuated by calphostin C (1 μm) (Fig. 6) but not other PKC inhibitors, GÖ6976 or GÖ6983 (data not shown). Because calphostin C and orlistat affected only GSIS, the role for influx of calcium through the L-type voltage-gated calcium channel (L-VGCC) was tested. The L-VGCC inhibitor verapamil (100 μm) completely inhibited GSIS from LXR-activated INS-1 cells without affecting basal insulin release.
Figure 6.

Enhanced glucose-stimulated insulin release from LXR-activated INS-1 cells is attenuated by verapamil (Verap) or calphostin C (Cal C). Insulin release (60 min) in response to 2 or 20 mm glucose was then assessed in the presence or absence of verapamil (100 μm) or calphostin C (1 μm). Values are mean ± sem for six independent experiments. Gluc, Glucose; TO, T0901317. *, P < 0.01 when verapamil- or calphostin C-treated cells are compared with control cells.
Discussion
Culturing INS-1 cells in elevated glucose led to increased nuclear SREBP-1c, lipogenic gene expression, TAG synthesis, and loss of GSIS. These findings are consistent with reports that SREBP-1c activation, either by elevated glucose or overexpression, in β-cell lines or islets increased lipogenic gene expression and TAG synthesis and decreased GSIS (6,7,8,9,10,11,44,45). Compared with INS-1 cells cultured in elevated glucose, LXR-activated INS-1 cells had significantly elevated microsomal and nuclear forms of SREBP-1c and lipogenic gene expression, particularly SREBP-1, FAS, SCD1, and SCD2. These findings are consistent with the role of LXR in regulating SREBP-1 gene transcription and LXR and SREBP-1c in regulating FAS, SCD1, and SCD2 gene transcription (46,47). Of the lipogenic genes examined, only ACCα mRNA levels were more strongly induced by elevated glucose than LXR activation. Glucose has been reported to bind and activate LXR (23), but this does not appear to play a prominent role in INS-1 cells because elevated glucose did not induce expression of LXR target genes including ABCA1 and ABCG1. As expected, increased lipogenic gene expression in LXR-activated INS-1 cells cultured in elevated glucose markedly increased de novo neutral lipid synthesis. Because LXR activation affected only lipid synthesis and insulin secretion in cells cultured in elevated glucose suggests that the two events are linked. This is likely an adaptive effect because it has recently been shown that metabolic flux through lipogenic pathways is not required for normal GSIS (48,49). Hypersecretion of insulin, however, has been shown to involve the induction of SREBP-1c and enhanced lipid synthesis in mouse islets cultured in elevated glucose (15).
FA Δ9 desaturases (SCD1 and SCD2) function as the rate-limiting step for MUFA synthesis and play an integral role in neutral lipid (TAG and CE) synthesis (47). In agreement with increased SCD1 and SCD2 mRNA levels, LXR-activated INS-1 cells exhibited a 2-fold increase in de novo derived MUFA (16:1, n-7; 18:1, n-7; 18:1, n-9) and a commensurate drop in synthesis of saturated FA (16:0, 18:0). The increase MUFA synthesis in LXR-activated INS-1 cells cultured in elevated glucose corresponded with increased MUFA mass (data not shown). Chronic exposure of islets and β-cell lines to oleic (18:1, n-9) or vaccenic (18:1, n-7) acid have been reported to increase basal insulin release (50,51,52), suggesting that increased MUFA synthesis in LXR-activated cells might be directly involved in basal insulin release. To test this possibility, small interfering RNA targeting SCD1 and SCD2 were introduced into LXR-activated INS-1 cells. SCD1/2 small interfering RNA effectively decreased SCD1 and SCD2 mRNA levels and MUFA synthesis but did not lower insulin release from LXR-activated INS-1 cells (supplemental Fig. 4). These data indicate that increased de novo MUFA synthesis is not an obligatory step for enhanced insulin release from LXR-activated INS-1 cells but likely facilitates neutral lipid synthesis.
Enhanced basal insulin release from LXR-activated INS-1 cells was attenuated by triacsin C and etomoxir, indicating a role for increased acyl-CoA formation and FA oxidation. Under our experimental paradigm for insulin release studies, INS-1 cells are first preincubated for 1 h in 2 mm glucose followed by incubation for 1 h in either 2 or 20 mm glucose. During this time frame, newly synthesized TAG is rapidly turned over (Fig. 5) and likely serves as the source of the FFA for acyl-CoA formation and oxidation. INS-1 cells cultured in elevated glucose also have increased de novo synthesized TAG but do not have elevated basal insulin release. This suggests that INS-1 cells cultured in elevated glucose either do not synthesize sufficient quantities of TAG to sustain increased basal insulin release or that LXR activation stimulates additional pathways associated with lipid metabolism. Consistent with the latter, LXR activation was shown here to increase FA oxidation, and this correlated with increased expression of genes involved in mitochondrial β-oxidation, particularly CPT-1α. Recently Colin et al. (53) showed that activation of LXR with synthetic agonists induced peroxisomal proliferator-activated receptor-α and subsequently its target CPT-1 in the intestine but not the liver. This suggests that LXR agonists may also induce CPT-1 through peroxisomal proliferator-activated receptor-α in β-cells. Alternatively, LXR activation of INS-1 cells may increase de novo-synthesized FA to levels sufficient to induce CPT-1α. This possibility is supported by the observation that long-term exposure of INS-1 cells to long-chain FA increases CPT-1 gene expression and FA oxidation (51,54). Our findings also raise the possibility that LXR activation can protect the β-cell from glucose toxicity by shuttling glucose toward FA, which can be oxidized immediately or after release from TAG.
Lipolysis of intracellular TAG and the subsequent generation of lipid signaling molecules including FFA, acyl-CoA, and DAG have been proposed to mediate GSIS (reviewed in Ref. 55). TAG turnover produces a FFA and a predominantly less biologically active sn2,3-DAG species, which can be further broken down to monoacylglyceride, glycerol, and FFA (56,57,58). These latter products, along with de novo-derived FA, can be reincorporated into biologically active sn1,2-DAG species through a glycerolipid/FFA cycle (59). Based on this, we hypothesize that enhanced GSIS from LXR-activated INS-1 cells results from elevated lipolysis and formation of lipid products that can directly serve as signaling molecules (e.g. FFA) or used as substrates for the glycerolipid/FFA cycle to generate sn1,2-DAG. Consistent with this hypothesis, the general lipase inhibitor orlistat blocked turnover of de novo-derived TAG and GSIS in LXR-activated INS-1 cells but did not block de novo synthesis of FA (Fig. 5). Mulder et al. (60) also proposed that orlistat attenuates GSIS by blocking the formation of an acylglyceride-coupling factor. If the glycerolipid/FFA cycle is involved in enhanced GSIS from LXR-activated INS-1 cells, one would predict that inhibition of acyl-CoA formation with triacsin C would have also blocked GSIS, which did not occur (Fig. 4). This might be due to the inability of triacsin C to inhibit all acyl CoA synthetase isoforms (61) and that triacsin C is more efficacious at inhibiting FA oxidation than lipid synthesis in β-cells (62). It remains a possibility that enhanced GSIS from LXR-activated INS-1 cells might also involve turnover of phospholipids and direct production of sn1,2-DAG (63). Polar lipid turnover, however, was much slower than TAG turnover in INS-1 cells and not effectively blocked by orlistat (data not shown).
DAG generated from the glycerol/FFA cycle might serve as the coupling factor to enhance GSIS from LXR-activated INS-1 cells. Classic (α, βI, βII, γ) and novel (δ, ε, η, θ) isoforms of PKC are activated by DAG in Ca2+-dependent and -independent manners, respectively. Pharmacological inhibition of many of these PKC isoforms with GÖ6976 (inhibits PKCα, -ßI) and GÖ6983 (inhibits PKCα, -β, -γ, -δ, -ζ), however, did not attenuate GSIS from LXR-activated INS-1 cells (data not shown). Calphostin C, which competitively blocks DAG-binding sites on classic and novel PKC isoforms, protein kinase D (PKCμ), and DAG-binding proteins, significantly attenuated GSIS from LXR-activated INS-1 cells (Fig. 6). Blockade of the influx of extracellular Ca2+ with the L-VGCC inhibitor also completely abrogated GSIS from LXR-activated INS-1 cells. Taken as a whole, these data suggest that enhanced GSIS from LXR-activated INS-1 cells does not involve classic or novel PKC isoforms but involves activation of a DAG-binding protein that is calcium dependent or mediates biochemical events upstream from the influx of calcium. There are a number of families of DAG-binding proteins that could be involved including protein kinase D (PKCμ), chimaerins, Ras guanyl nucleotide-releasing proteins, mammalian homolog of the caenorhabditis elegans UNC13 proteins (MUNC13s), or DAG kinases (reviewed in Ref. 64). Further experimentation is necessary to determine the exact DAG-binding protein(s) involved. Straub et al. (65) proposed a similar mechanism to explain how FA depletion of rat islets caused large increases in GSIS. In their model, FA depletion is proposed to cause lipid remodeling or increase breakdown of intracellular TAG, which increases DAG production, activates a DAG-binding protein, and augments GSIS.
Enhanced insulin release from LXR-activated β-cells has been reported to be associated with increased mRNA levels of pancreatic duodenal homeobox-1, insulin, and glucose transporter 2 (18), suggesting a role for LXR or SREBP-1c in augmenting β-cell phenotype and glucose sensing. SREBP-1 is also required for elevated glucose to increase mRNA levels of pancreatic duodenal homeobox-1 and genes involved in glucose sensing including glucose transporter 2 and glucokinase (15). Similar changes in gene expression may play a role in enhanced insulin release from LXR-activated INS-1 cells. Nevertheless, this seems unlikely because glucose utilization and insulin content were not significantly increased in LXR-activated INS-1 cells.
In conclusion, our study shows that LXR activation of INS-1 β-cells exposed to elevated glucose increases TAG synthesis, and subsequent TAG turnover can lead to the production of lipid signaling molecules resulting in elevated insulin release. Similar mechanisms may account for the ability of SREBP-1c to establish hypersecretion of insulin in some models of hyperglycemia.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Daniela Botolin and Ms. Katrina D. Linning for their expert technical assistance.
Footnotes
This work was supported by funds from the Michigan Life Science Corridor (GR352); the American Diabetes Association (7-06-RA-103, to L.K.O.); the National Institutes of Health (DK43220); the National Research Initiative of the U.S. Department of Agriculture Cooperative State Research, Education and Extension Service (2003-35200-13400); and the Michigan Agriculture Experiment Station (to D.B.J.).
Disclosure Summary: C.D.G., D.B.J., and L.K.O. have nothing to disclose.
First Published Online February 19, 2009
Abbreviations: ABC, ATP binding cassette; ACC, acetyl CoA carboxylase; CE, cholesterol ester; CoA, coenzyme A; CPT, carnitine palmitoyl transferase; DAG, diacylglyceride; FA, fatty acid; FAS, fatty acid synthase; FFA, free fatty acid; GSIS, glucose-stimulated insulin secretion; KRBB, Kreb’s Ringer bicarbonate buffer; L-VGCC, L-type voltage-gated calcium channel; LXR, liver X receptor; MUFA, monounsaturated fatty acid; PKC, protein kinase C; qPCR, quantitative PCR; SCD, stearoyl CoA desaturase; SREBP, sterol response element binding protein; TAG, triacylglyceride.
References
- Prentki M, Joly E, El-Assaad W, Roduit R 2002 Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in β-cell adaptation and failure in the etiology of diabetes. Diabetes 51(Suppl 3):S405–S413 [DOI] [PubMed] [Google Scholar]
- Poitout V, Robertson RP 2008 Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev 29:351–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Guichard C, Ferré P, Foufelle F 1999 Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci USA 96:12737–12742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Lièpvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferré P, Foufelle F 1999 ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Mol Cell Biol 19:3760–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM 1998 Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 101:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreolas C, da Silva Xavier G, Diraison F, Zhao C, Varadi A, Lopez-Casillas F, Ferré P, Foufelle F, Rutter GA 2002 Stimulation of acetyl-CoA carboxylase gene expression by glucose requires insulin release and sterol regulatory element binding protein 1c in pancreatic MIN6 β-cells. Diabetes 51:2536–2545 [DOI] [PubMed] [Google Scholar]
- Wang H, Maechler P, Antinozzi PA, Herrero L, Hagenfeldt-Johansson KA, Bjorklund A, Wollheim CB 2003 The transcription factor SREBP-1c is instrumental in the development of β-cell dysfunction. J Biol Chem 278:16622–16629 [DOI] [PubMed] [Google Scholar]
- Sandberg MB, Fridriksson J, Madsen L, Rishi V, Vinson C, Holmsen H, Berge RK, Mandrup S 2005 Glucose-induced lipogenesis in pancreatic β-cells is dependent on SREBP-1. Mol Cell Endocrinol 240:94–106 [DOI] [PubMed] [Google Scholar]
- Diraison F, Parton L, Ferré P, Foufelle F, Briscoe CP, Leclerc I, Rutter GA 2004 Over-expression of sterol-regulatory-element-binding protein-1c (SREBP1c) in rat pancreatic islets induces lipogenesis and decreases glucose-stimulated insulin release: modulation by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR). Biochem J 378:769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Eto K, Okazaki Y, Yamashita S, Yamauchi T, Sekine N, Nagai R, Noda M, Kadowaki T 2004 Role of uncoupling protein-2 up-regulation and triglyceride accumulation in impaired glucose-stimulated insulin secretion in a β-cell lipotoxicity model overexpressing sterol regulatory element-binding protein-1c. Endocrinology 145:3566–3577 [DOI] [PubMed] [Google Scholar]
- Takahashi A, Motomura K, Kato T, Yoshikawa T, Nakagawa Y, Yahagi N, Sone H, Suzuki H, Toyoshima H, Yamada N, Shimano H 2005 Transgenic mice overexpressing nuclear SREBP-1c in pancreatic β-cells. Diabetes 54:492–499 [DOI] [PubMed] [Google Scholar]
- Kakuma T, Lee Y, Higa M, Wang Z, Pan W, Shimomura I, Unger RH 2000 Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc Natl Acad Sci USA 97:8536–8541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Hirose H, Zhou YT, Esser V, McGarry JD, Unger RH 1997 Increased lipogenic capacity of the islets of obese rats: a role in the pathogenesis of NIDDM. Diabetes 46:408–413 [DOI] [PubMed] [Google Scholar]
- Zhou YT, Shimabukuro M, Lee Y, Koyama K, Higa M, Ferguson T, Unger RH 1998 Enhanced de novo lipogenesis in the leptin-unresponsive pancreatic islets of prediabetic Zucker diabetic fatty rats: role in the pathogenesis of lipotoxic diabetes. Diabetes 47:1904–1908 [DOI] [PubMed] [Google Scholar]
- Diraison F, Ravier MA, Richards SK, Smith RM, Shimano H, Rutter GA 2008 SREBP1 is required for the induction by glucose of pancreatic β-cell genes involved in glucose sensing. J Lipid Res 49:814–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, McMillen PJ, Shen Y, Docherty E, Sharpe E, Diraison F, Briscoe CP, Rutter GA 2006 Limited role for SREBP-1c in defective glucose-induced insulin secretion from Zucker diabetic fatty rat islets: a functional and gene profiling analysis. Am J Physiol Endocrinol Metab 291:E982–E994 [DOI] [PubMed] [Google Scholar]
- Efanov AM, Sewing S, Bokvist K, Gromada J 2004 Liver X receptor activation stimulates insulin secretion via modulation of glucose and lipid metabolism in pancreatic β-cells. Diabetes 53(Suppl 3):S75–S78 [DOI] [PubMed] [Google Scholar]
- Zitzer H, Wente W, Brenner MB, Sewing S, Buschard K, Gromada J, Efanov AM 2006 Sterol regulatory element-binding protein 1 mediates liver X receptor-β-induced increases in insulin secretion and insulin messenger ribonucleic acid levels. Endocrinology 147:3898–3905 [DOI] [PubMed] [Google Scholar]
- Mohan R, Heyman RA 2003 Orphan nuclear receptor modulators. Curr Top Med Chem 3:1637–1647 [DOI] [PubMed] [Google Scholar]
- Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ 1995 LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9:1033–1045 [DOI] [PubMed] [Google Scholar]
- Teboul M, Enmark E, Li Q, Wikström AC, Pelto-Huikko M, Gustafsson JA 1995 OR-1, a member of the nuclear receptor superfamily that interacts with the 9-cis-retinoic acid receptor. Proc Natl Acad Sci USA 92:2096–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JM, Kliewer SA, Moore LB, Smith-Oliver TA, Oliver BB, Su JL, Sundseth SS, Winegar DA, Blanchard DE, Spencer TA, Willson TM 1997 Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem 272:3137–3140 [DOI] [PubMed] [Google Scholar]
- Mitro N, Mak PA, Vargas L, Godio C, Hampton E, Molteni V, Kreusch A, Saez E 2007 The nuclear receptor LXR is a glucose sensor. Nature 445:219–223 [DOI] [PubMed] [Google Scholar]
- Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ 2000 Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289:1524–1529 [DOI] [PubMed] [Google Scholar]
- Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, Tontonoz P 2001 LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci USA 98:507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ 1998 Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR α. Cell 93:693–704 [DOI] [PubMed] [Google Scholar]
- Repa JJ, Berge KE, Pomajzl C, Richardson JA, Hobbs H, Mangelsdorf DJ 2002 Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors α and β. J Biol Chem 277:18793–18800 [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B 2000 Role of LXRs in control of lipogenesis. Genes Dev 14:2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ 2000 Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 14:2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N 2001 Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21:2991–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulnig TM, Steffensen KR, Gao H, Reimers M, Dahlman-Wright K, Schuster GU, Gustafsson JA 2002 Novel roles of liver X receptors exposed by gene expression profiling in liver and adipose tissue. Mol Pharmacol 62:1299–1305 [DOI] [PubMed] [Google Scholar]
- Shalev A, Pise-Masison CA, Radonovich M, Hoffmann SC, Hirshberg B, Brady JN, Harlan DM 2002 Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFβ signaling pathway. Endocrinology 143:3695–3698 [DOI] [PubMed] [Google Scholar]
- Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang SH, Burant CF, Steffensen KR, Gustafsson JA, MacDougald OA 2005 LXRβ is required for adipocyte growth, glucose homeostasis, and β cell function. J Biol Chem 280:23024–23031 [DOI] [PubMed] [Google Scholar]
- Choe SS, Choi AH, Lee JW, Kim KH, Chung JJ, Park J, Lee KM, Park KG, Lee IK, Kim JB 2007 Chronic activation of liver X receptor induces β-cell apoptosis through hyperactivation of lipogenesis: liver X receptor-mediated lipotoxicity in pancreatic β-cells. Diabetes 56:1534–1543 [DOI] [PubMed] [Google Scholar]
- Wells WW, Xu DP, Washburn MP, Cirrito HK, Olson LK 2001 Polyhydroxybenzoates inhibit ascorbic acid activation of mitochondrial glycerol-3-phosphate dehydrogenase: implications for glucose metabolism and insulin secretion. J Biol Chem 276:2404–2410 [DOI] [PubMed] [Google Scholar]
- Botolin D, Jump DB 2003 Selective proteolytic processing of rat hepatic sterol regulatory element binding protein-1 (SREBP-1) and SREBP-2 during postnatal development. J Biol Chem 278:6959–6962 [DOI] [PubMed] [Google Scholar]
- Linning KD, Tai MH, Madhukar BV, Chang CC, Reed Jr DN, Ferber S, Trosko JE, Olson LK 2004 Redox-mediated enrichment of self-renewing adult human pancreatic cells that possess endocrine differentiation potential. Pancreas 29:e64–e76 [DOI] [PubMed] [Google Scholar]
- Zawalich WS, Matschinsky FM 1977 Sequential analysis of the releasing and fuel function of glucose in isolated perifused pancreatic islets. Endocrinology 100:1–8 [DOI] [PubMed] [Google Scholar]
- Pawar A, Xu J, Jerks E, Mangelsdorf DJ, Jump DB 2002 Fatty acid regulation of liver X receptors (LXR) and peroxisome proliferator-activated receptor α (PPARα) in HEK293 cells. J Biol Chem 277:39243–39250 [DOI] [PubMed] [Google Scholar]
- Wang Y, Botolin D, Christian B, Busik J, Xu J, Jump DB 2005 Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J Lipid Res 46:706–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkera SM, Moore PC, Johnson LM, Poitout V 2003 Palmitate potentiation of glucose-induced insulin release: a study using 2-bromopalmitate. Metabolism 52:1367–1371 [DOI] [PubMed] [Google Scholar]
- Olson LK, Qian J, Poitout V 1998 Glucose rapidly and reversibly decreases INS-1 cell insulin gene transcription via decrements in STF-1 and C1 activator transcription factor activity. Mol Endocrinol 12:207–219 [DOI] [PubMed] [Google Scholar]
- Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW 2004 Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res 64:2070–2075 [DOI] [PubMed] [Google Scholar]
- Brun T, Roche E, Assimacopoulos-Jeannet F, Corkey BE, Kim KH, Prentki M 1996 Evidence for an anaplerotic/malonyl-CoA pathway in pancreatic β-cell nutrient signaling. Diabetes 45:190–198 [DOI] [PubMed] [Google Scholar]
- Roche E, Farfari S, Witters LA, Assimacopoulos-Jeannet F, Thumelin S, Brun T, Corkey BE, Saha AK, Prentki M 1998 Long-term exposure of β-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes 47:1086–1094 [DOI] [PubMed] [Google Scholar]
- Steffensen KR, Gustafsson JA 2004 Putative metabolic effects of the liver X receptor (LXR). Diabetes 53(Suppl 1):S36–S42 [DOI] [PubMed] [Google Scholar]
- Ntambi JM, Miyazaki M, Dobrzyn A 2004 Regulation of stearoyl-CoA desaturase expression. Lipids 39:1061–1065 [DOI] [PubMed] [Google Scholar]
- Joseph JW, Odegaard ML, Ronnebaum SM, Burgess SC, Muehlbauer J, Sherry AD, Newgard CB 2007 Normal flux through ATP-citrate lyase or fatty acid synthase is not required for glucose-stimulated insulin secretion. J Biol Chem 282:31592–31600 [DOI] [PubMed] [Google Scholar]
- Ronnebaum SM, Joseph JW, Ilkayeva O, Burgess SC, Lu D, Becker TC, Sherry AD, Newgard CB 2008 Chronic suppression of acetyl-CoA carboxylase 1 in β-cells impairs insulin secretion via inhibition of glucose rather than lipid metabolism. J Biol Chem 283:14248–14256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YP, Grill VE 1994 Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 93:870–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alstrup KK, Brock B, Hermansen K 2004 Long-term exposure of INS-1 cells to cis and trans fatty acids influences insulin release and fatty acid oxidation differentially. Metabolism 53:1158–1165 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiao M, Niu G, Tan H 2005 Mechanisms of oleic acid deterioration in insulin secretion: role in the pathogenesis of type 2 diabetes. Life Sci 77:2071–2081 [DOI] [PubMed] [Google Scholar]
- Colin S, Bourguignon E, Boullay AB, Tousaint JJ, Huet S, Caira F, Staels B, Lestavel S, Lobaccaro JM, Delerive P 2008 Intestine-specific regulation of PPARα gene transcription by liver X receptors. Endocrinology 149:5128–5135 [DOI] [PubMed] [Google Scholar]
- Assimacopoulos-Jeannet F, Thumelin S, Roche E, Esser V, McGarry JD, Prentki M 1997 Fatty acids rapidly induce the carnitine palmitoyltransferase I gene in the pancreatic β-cell line INS-1. J Biol Chem 272:1659–1664 [DOI] [PubMed] [Google Scholar]
- Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M 2006 Fatty acid signaling in the β-cell and insulin secretion. Diabetes 55(Suppl 2):S16–S23 [DOI] [PubMed] [Google Scholar]
- Boni LT, Rando RR 1985 The nature of protein kinase C activation by physically defined phospholipid vesicles and diacylglycerols. J Biol Chem 260:10819–10825 [PubMed] [Google Scholar]
- Birner-Gruenberger R, Susani-Etzerodt H, Waldhuber M, Riesenhuber G, Schmidinger H, Rechberger G, Kollroser M, Strauss JG, Lass A, Zimmermann R, Haemmerle G, Zechner R, Hermetter A 2005 The lipolytic proteome of mouse adipose tissue. Mol Cell Proteomics 4:1710–1717 [DOI] [PubMed] [Google Scholar]
- Duque M, Graupner M, Stütz H, Wicher I, Zechner R, Paltauf F, Hermetter A 1996 New fluorogenic triacylglycerol analogs as substrates for the determination and chiral discrimination of lipase activities. J Lipid Res 37:868–876 [PubMed] [Google Scholar]
- Prentki M, Madiraju SR 2008 Glycerolipid metabolism and signaling in health and disease. Endocr Rev 29:647–676 [DOI] [PubMed] [Google Scholar]
- Mulder H, Yang S, Winzell MS, Holm C, Ahrén B 2004 Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes 53:122–128 [DOI] [PubMed] [Google Scholar]
- Soupene E, Kuypers FA 2008 Mammalian long-chain acyl-CoA synthetases. Exp Biol Med (Maywood, NJ) 233:507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinozzi PA, Segall L, Prentki M, McGarry JD, Newgard CB 1998 Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long-chain acyl-CoA hypothesis. J Biol Chem 273:16146–16154 [DOI] [PubMed] [Google Scholar]
- Poitout V 2008 Phospholipid hydrolysis and insulin secretion: a step toward solving the Rubik’s cube. Am J Physiol Endocrinol Metab 294:E214–E216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kazanietz MG 2003 Divergence and complexities in DAG signaling: looking beyond PKC. Trends Pharmacol Sci 24:602–608 [DOI] [PubMed] [Google Scholar]
- Straub SG, Shanmugam G, Sharp GW 2004 Stimulation of insulin release by glucose is associated with an increase in the number of docked granules in the β-cells of rat pancreatic islets. Diabetes 53:3179–3183 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.