

In the last two decades, biogerontologists have found that the pace of aging can be decelerated in mammals by dietary or genetic means. Though still largely unappreciated by the lay and scientific public alike, these discoveries belie the common assumption that human aging probably cannot be slowed and prompt the question of whether we can use our growing knowledge about aging to produce 90-year-old adults who are as healthy and active as today' 50-year-olds. This article explores three related issues: first, why discussions of antiaging medicines are no longer silly; next, why the development of antiaging strategies is making so little headway; and last, whether further work in this area would be worthwhile.
By “aging” I mean a process that converts healthy young adults into less healthy older ones with an increasing risk of illness and death. This definition, which seems fairly innocuous, is careful enough to be controversial, because there are many interesting areas of investigation—studies of childhood development; specific late-life diseases; death or proliferative ennui of cells in culture; leaf abscission; and time-dependent changes in the chemical composition of wines, cheeses, and bones—that are sometimes proposed in various combinations as worthy of gerontological attention and governmental funding. Each of these areas may deserve study, and some of them may eventually provide insights into real aging, but to avoid confusion, I shall discuss only the kind of aging that turns whole young bodies into old ones.
The idea that antiaging interventions might someday be developed makes the assumption, remarkably controversial when carefully scrutinized, that there is an aging process, as contrasted to a set of aging processes that just happen to occur in rough synchrony within any one species and at different rates among different species. In the gerontological community, the majority of deep thinkers (Holliday 1999; Masoro 1995; Partridge and Harvey 1993) are now convinced that there is no aging process, no central clock timing the race through life. The conviction of these “multiplicitists” rests on the profound awe at the complexity of aging, with its effects on proliferating cells, nonproliferating cells, extracellular matrices, cell-to-cell communication, gene expression patterns, cell structures, and the like. It is obvious that each of these age-related forms of disarray can be influenced by many genes, many more or less predictable progressive adaptations to environmental insults, and many forms of stochastic misadventure. Thus the sum of these processes—which we call aging—must be more complicated still. From this perspective, a quest for antiaging medicines resembles the alchemists’ search for the sorcerer' stone; that is, it is clear evidence that the alchemists’ worldview is inadequate.
The Case for an Aging Process
From many fronts, there now is incontrovertible evidence that aging in mammals can be decelerated and that it is not too difficult to do this. In ways that I shall describe, caloric restriction (CR) surely slows aging (Weindruch and Sohal 1997), and methionine restriction may also do so (Orentreich, Matias, DeFelice, et al. 1993). At least three spontaneous mouse mutants (dw, df, and lit) and two induced ones (GHR/BP and p66shc knock-outs) reproducibly increase life span by as much or more than caloric restriction does and do so through metabolic alterations that overlap with CR only partially (Brown-Borg, Borg, Meliska, et al. 1996; Coschigano, Clemmons, Bellush, et al. 2000; Migliaccio, Giorgio, Mele, et al. 1999). Artificial selection for altered body size creates long-lived dogs (Li, Deeb, Pendergrass, et al. 1996; Miller 1999) and mice (Miller, Chrisp, and Atchley 2000), and possibly horses, does so over and over again, and does so in a few dozen to a few hundred generations. Natural selection for adaptation to environmental niches with low intrinsic hazards routinely produces species (and, in some cases, subspecies or races) with longer lives (Austad 1993; Austad and Fischer 1991), including, for example, at least three species of rodents (porcupines, naked mole rats, and capybaras) with lifetimes three-to tenfold longer than those of common household and laboratory rodents. Indeed, nature can churn out long-lived variants from short-lived predecessors quickly and reliably, making bats, tortoises, flying squirrels, elephants, parrots and many other birds, tuna and rockfish, longevitous opossums, and people. This trick seems to be easy to do and, at least in the opossum, takes only an evolutionary eye blink (Austad 1993).
Aging in mice, dogs, horses, and people is easily recognized: a creature with cataracts, weak muscles, poor immunity, incipient deafness, drastically reduced cardiopulmonary reserves, and several early malignancies is clearly old, but we cannot know whether it is 2, 12, 25, or 75 years old until we know to what species it belongs. It seems odd that each of these processes, and many more besides, are timed by independent clocks that just happen to count out 2 years in a mouse and 75 in a human, and it is equally odd to view as pure coincidence the ability of multiple single-gene mutations and at least one dietary modulation to slow all these down in synchrony. My own reading of the evidence is that the aging process is probably timed by a single clock (or perhaps a very small number). We will call this view the “unitarian” thesis.
Aging Is Both Unitarian and Multiplex
Is aging one process or many? When both alternative answers to a question are clearly correct, the question is poorly phrased. An economy, for example, is clearly multiplex, with many interlocking, partially independent, parallel processes and is just as clearly subject to acceleration or deceleration through fairly simple means—a change in interest rates, a war, massive deficit spending—that influence the much larger set of downstream processes. The time spent by gerontologists debating whether aging is a single process or many would be better devoted to trying to figure out the mechanistic links between the master clock whose existence is strongly suggested by the unitarian argument and the many cell-specific, organ-specific, and organism-wide processes that march in crude synchrony at species-specific rates.
Why Do We Age at All?
A remarkable proportion of first-year medical students are confident of the same wrong answer to this question: the assertion that evolution promotes genes that cause aging in order to clear away old individuals and make room for their progeny. This belief rests on a misunderstanding of how evolution works: genes that hypothetically accelerate aging would have little chance of taking hold in a population because their owners would, as they became enfeebled and then died, leave fewer offspring than would their colleagues that did not have the fast-aging gene. Evolutionary biologists have indeed developed a clear, consistent, and fairly well-tested understanding of why aging is so common (nearly universal) among plants and animals (Medawar 1952; Williams 1957). The key realization is that the genes that have ill effects only at later ages—ages “late” with respect to the attainment of breeding age—are not exposed very strongly to the pressures of natural selection. Consider, for example, a hypothetical species (the grommit) whose environment is full of hazards. In their environmental niche, even the healthiest young adults have a 50–50 chance each year of starving, being eaten, or succumbing to infection. A genetic mutant that greatly increased the likelihood of dementia, osteoporosis, or cataracts in one-year-old grommits is likely to be weeded out by natural selection because there are many one-year-old grommits, whose chances of reproducing would be diminished by any of these (and many similar) tribulations. But a mutation that caused ill effects in 10-year-old grommits would have very little impact on reproductive success, because with 50 percent annual mortality, very few grommits would live that long. And indeed, a mutation that even slightly increased the reflex speed or muscle strength or immune response in one-year-old grommits would be highly favored, even if as a side effect it caused serious illnesses in 10-year-olds. The consequence is clear: mutations with late-acting deleterious effects tend to accumulate in the genome of every species. If fully evolved grommits are lucky enough to find themselves in a zoo or laboratory in which their food supply is constant and their predators, both infectious and carnivorous, have been banished, they will suddenly be confronted, at age 10 or so, with the fruits of this genetic burden: the losses in skin, bone, muscle, brain, immune, endocrine, and digestive functions that we so readily recognize as aging in our pets and in ourselves.
This argument shows why we age, but why, then, do we not age more slowly? Nature can make 20-year-old turtles and horses and tunas, so why does it shy away from evolving mice that age at the same slow rate? The catch is that genes that could slow aging have reproductive costs. For the same reason, it would not be worthwhile to print the Michigan Daily on stone tablets, because today' issue is not likely to have much value in the next millennium; nor are titanium doors worth the price in an automobile likely to break down in a few dozen years. High-hazard niches, in which a mouse—however resistant to cancers and cataracts and osteopenia it may be—is likely to be eaten within a year or so, do not support the selection of genes that postpone these age effects for decades, because mice that devote their energies to eating and breeding will do better than those that spend valuable capital on eye repair and anticancer surveillance.
Although Nature invests in longevity often, it does so only in safe neighborhoods. Not all rodenty creatures burn their candles at both ends. Rodents that learn to fly or glide become, respectively, bats and flying squirrels, nearly every species of which is remarkably long-lived compared with nonflying animals of the same size (Austad and Fischer 1991). Rodents that evolve protective spines become porcupines, among which most species are quite long-lived (Carey and Judge 2000); the current world record for longevity in a rodent is owned by a Sumatran crested porcupine who made it to 27.25 years. Rodents that learn to live underground in thermocontrolled bunkers also do well—the naked mole rat, a 25-gram denizen of Ethiopia, Kenya, and Somalia, has lived at least 20 years in captivity, with rumors of 30-year-old specimens abounding wherever naked mole rat experts congregate. Nature discovered the backbone once, the flower once, hair once, and the hand once but figures out longevity over and over, always when a species stumbles into a suitably safe niche where flying, body armor, large size, predator-free islands, or (for humans and naked mole rats) environmental control systems mitigate risk and offer a potential payoff for decelerating senescence. This seems to be an easy trick to learn, and it would be of great practical and theoretical interest to find out whether it is performed in the same way—that is, by using genetic alterations of the same set of biochemical pathways—in each phylogenetically distinct instance.
Anti-geric Intervention: Slowing Aging in Laboratory Animals
Mother Nature can slow down aging and so, it turns out, can we. So far, two approaches work for sure: diminished total caloric intake, and changes in genes that regulate the rate of early-life growth.
Since the work of McCay and his colleagues (McCay, Crowell, and Maynard 1935) in 1935, which was replicated and expanded by Masoro and his colleagues (Maeda, Gleiser, Masoro, et al. 1985; Yu, Masoro, and McMahan 1985), we have known that rats and mice that are given about 40 percent less food than they would eat on their own live about 40 percent longer than do fully fed controls. This basic observation has been repeated in about 100 experiments done in dozens of independent laboratories (Weindruch and Walford 1988). The “caloric restriction” (CR) protocol differs from starvation in that CR rodents receive fully adequate amounts of proteins, vitamins and minerals, fatty acids, and other nutrients; that is, their diets are adjusted to provide adequate nutrition but with drastically fewer calories. Their life span is extended because the CR diet postpones whatever diseases are the key causes of death in each species and stock that has been tested. The CR diet does not merely postpone diseases and death but also seems to decelerate aging per se and, in so doing, retards age-related changes in nearly every system and cell type that has been examined. For example, CR slows down the effects of aging on cells that divide frequently (such as bone marrow and gut-lining cells), cells that divide only rarely (bone cells), and cells that do not divide at all (neurons). It retards age changes in extracellular materials (such as collagen and the crystallin proteins of the eye lens), in intracellular processes (age-sensitive patterns of gene expression), and in systems that mediate intercellular communications (endocrine feedback loops). Thus CR seems to slow aging, and indeed, the CR-mediated deceleration of aging is a key element in the argument that aging is sensibly considered a unitary process with varying rates. It will be highly informative to learn more about the ways in which an apparently simple intervention leads to a delay or deceleration of age-related changes in so wide a range of apparently disparate cell types and inter- and intracellular processes.
CR works in rodents and has been shown to extend life in a wide range of other animal types, most of them invertebrates. At least three research groups are now conducting studies designed to determine whether CR will retard aging in a nonhuman primate, the rhesus monkey. The data, though incomplete, so far look fairly promising. The largest CR effects are seen in experimental protocols in which the dietary restriction is begun early in life, for example, in adolescents or young adults, but partial effects have been demonstrated in rodents first exposed to CR about 40 percent through their average life span.
How does CR work? Of the dozen or so plausible ideas, only a few, to date, have been disproved. McCay' original notion was that CR extended the life span by preventing growth to full body size, but this is wrong: CR dramatically extends life span even if begun when the animal is already a full-sized adult. CR rodents are typically very lean, suggesting that the lack of adipose tissue is a key element in their extended youthfulness, but the imposition of CR on a mouse with a genetic mutation that leads to extreme obesity also produces a very long-lived, albeit fat, mouse. The idea that CR slows aging by diminishing the rate at which food is converted to energy (and thus the rate at which metabolism produces toxic, oxidizing by-products) was disproved by studies showing that the small body size of CR rodents balances their lower food intake, so that the amount of fuel used (or oxygen used) per gram of body mass is not lower in CR than in control rodents (McCarter, Masoro, and Yu 1985). Several other theories, though, are still fully plausible, including notions that the long healthy lives of CR rodents are due to their low glucose levels, high insulin sensitivity, mildly elevated glucocorticoid levels, or changes in gene expression that represent adaptive responses to metabolic hardship. Determining the mechanism by which CR slows aging, and discovering a technique for inducing these changes without restricting the number of calories per se, would constitute a major landmark in medical research.
In regard to public health and preventive medicine, we should note that CR does not merely postpone death and does not merely prolong late-life disabilities. Instead, CR rodents remain healthy and active at ages at which all their control littermates have long since died. Studies of exercise capacity, for example, show that when ordinary rats are given access to a running wheel, they typically put in about 1,000 meters/day for their first few months of adult life, but then after the age of eight months, they abruptly slow down and rarely run more than 200 meters/day. In contrast, CR rodents typically run 4,000 to 5,000 meters/day until they are at least two years old (the median life span for controls) and are still running 1,000 meters/day at three years of age. Autopsy studies of CR animals at the end of their life span typically show very low levels of arthritic, neoplastic, and degenerative change, and in functional tests of immunity, memory, muscle strength and the like, they typically resemble much younger animals of the control group.
For about 50 years, the CR protocol was the only established method for producing very old, very healthy rodents. This situation changed in 1996 with the discovery that the Ames dwarf mutant mouse lived about 50 percent (and for females, up to 70 percent) longer than did nonmutant control mice (Brown-Borg et al. 1996). The small stature of these mice results from deficits in their production of growth hormone, thyroid hormones, and prolactin. Since 1996, two laboratories, including my own (Miller 1999), have documented a similar extension of life span in the closely related Snell dwarf mouse, and a third group (Coschigano et al. 2000) has shown exceptional longevity in an engineered mutant that can make growth hormone but cannot respond to it. A fourth mutation with defective activation of the growth hormone pathway, the “little” mutant lit/lit, also shows extended longevity (about 20 percent to 25 percent over controls) when raised on a diet relatively low in fat (Flurkey, Papaconstantinou, Miller, et al. 2001). Snell and Ames dwarf mice, unlike the other two mutants, also are deficient in their production of thyroid hormones and prolactin, one or both of which may contribute to their especially dramatic longevity. The characterization of the pathophysiology of these long-lived mutant mice is much less advanced than studies of the CR effect, but it is already apparent that Snell dwarf mice, at least, are not only long lived but also relatively slow to develop age-related changes in their immune and connective tissues (Flurkey et al. 2001). Thus it seems likely that a thorough characterization will show that these mutants are authentic examples of decelerated or postponed aging. These observations do not contradict the claims that administering growth hormone (GH) to elderly people may help alleviate some of the ill effects of aging, such as loss of muscle mass (Rudman, Feller, Nagraj, et al. 1990), because it is quite plausible that the effects of GH early in life may be different from those it has at advanced ages.
The association between alterations in body size and longevity seen in these mutant mice seems likely to be reproducible in other rodents and other species of mammals. The most dramatic example comes from a comparison of body weight and longevity among breeds of purebred dogs. Using statistics compiled by Norman Wolf from an archive of veterinary records (Li et al. 1996), I have shown by means of linear regression that the relationship between breed weight and mean breed longevity is very strong, with a squared correlation coefficient of 0.56 among the 16 breeds for which records were available (Miller 1999). Thus centuries of breeding to produce dogs whose size suited them for particular tasks—small dogs as pets and rodent hunters, larger dogs for guard and military duty—not only has created the intended wide variations in growth rate and ultimate size but also unintended differences in life span. The high correlation coefficient supports the provocative conclusion that 56 percent of the variation in longevity among extant breeds is due to the effects of genes whose main, selective, purpose is to regulate size and growth rate. The physiological basis for differences in size between related large and small breeds has been investigated in two cases only but in both instances was shown to represent genetic differences in the response to growth hormone—smaller dogs producing less growth hormone and, perhaps as a consequence, living longer. There is also some evidence, so far anecdotal, of exceptional longevity among miniature breeds of horses.
Dogs are dogs and mice are mice, but what about humans? The relationship between size and longevity genes in humans is difficult to tease apart, because socioeconomic advantages can contribute to both large body size and good health outcomes. Nevertheless, there is some evidence that after controlling for the potential confounding effects of nonbiological influences, people of relatively short stature may be relatively long lived, or at least relatively resistant to certain major classes of disease (Davey, Hart, Upton, et al. 2000; Samaras and Storms 1992). For example, on the European island of Krk, some humans have a short stature because of a mutation in the same gene responsible for the Ames dwarf mutation in mice. The limited evidence (Krzisnik, Kolacio, Battelino, et al. 1999) suggests that this particular variety of growth hormone deficiency in humans may, like the dwarf mutations in mice, be associated with exceptional longevity.
Thus three lines of evidence—one genetic, one based on dietary restriction, and one phylogenetic—lead to an important conclusion: although the signs of aging are, to a first approximation, similar in all species of mammals, the pace at which these changes develop can be coordinately regulated and, in some cases, by as simple a change as the modification of a single base of DNA sequence or the restriction of food availability. Fifty years ago an assertion that the rate of aging might be deliberately modified would have been only a hunch or statement of faith, without empirical foundation. Today, however, it is clear that the rate of aging can differ among members of the same species and differ radically between species as closely related as monkeys and people. These new findings lead to an important question: what implications would the development of a simple intervention, such as a pill, that would lead to a dramatic decline in the rate of aging have for public health? What obstacles need to be overcome to achieve this result? And would such a discovery be a good thing or, like television, gunpowder, and the internal combustion engine, a mixed blessing?
What Would an Antiaging Pill Do?
What if we figured out enough about caloric restriction or age-retarding mutations to be able to design strategies that could routinely produce centenarians whose physical and mental health (and life expectancy) resembled that of today' typical retiree? A pill, free of side effects, that could slow down aging to the extent that is now reproducibly attained in calorically restricted laboratory rodents? One can, without undue speculation, produce a credible estimate about what such a pill would do to actuarial tables and life insurance rates. CR typically produces in rodents an increase in mean and maximal longevity of about 30 percent to 40 percent. Similarly, the dwarf mutations of mice lead to an increase in both their mean and maximal life span of about 25 to 70 percent, and the longest-lived small-dog breeds typically outlive average-sized dogs by a similar amount. Restriction of the amino acid methionine, which, like the restriction of calories, also retards growth and extends life span (Orentreich et al. 1993; Richie, Leutzinger, Parthasarathy, et al. 1994), lengthens life by about 30 to 42 percent, and a mutation that alters cellular resistance to irradiation seems to produce mice that live 28 percent longer (Migliaccio et al. 1999). Thus one can, with some confidence, expect that an effective antiaging intervention might increase the mean and maximal human life span by about 40 percent, which is a mean age at death of about 112 years for Caucasian American or Japanese women, with an occasional winner topping out at about 140 years. Claims in the popular press, however, even by accredited gerontologists, suggesting the possibility of 200-to 600-year-old people are not supported by any credible evidence.
Effective antiaging interventions—those that mimic the known results of CR or the genes that produce fox terriers and miniature schnauzers—would be expected to produce 112-year-old people with the same, highly variable set of abilities and disabilities seen in today' 78-year-olds. We can infer that CR, or something like it that worked in people, would not produce 112-year-olds that resemble today' supercentenarians in their precarious states of mental and physical health, because 3.25-year-old CR mice, our current best guides to what we would expect to see in a 112-year-old CR-facsimile person, are still fairly vigorous and admirably free of degenerative changes. Interventions that slow aging—at least the ones we know about so far—do not prolong the period of late-life suffering but instead delay its appearance by increasing the length of healthy adult life.
Demographic considerations show that aging research has potentially the biggest bang for the buck and may improve public health to a far greater extent than would research that works on only one disease at a time. Calculations based on estimations of life tables based on the hypothetical elimination of specific causes of late-life illness clearly make this point. Figure 1, based on calculations by S. Jay Olshansky (Olshansky, Carnes, and Cassel 1990), illustrates this idea. In 1985, for example, the typical 50-year-old American woman could look forward to another 32 years of life, with a mean age at death of about 82 years. The elimination of all forms of cancer—that is, the hypothetical adjustment of cancer mortality risks to zero at all ages above 50—would increase this woman' life expectancy by only 2.7 years, with death expected, on average, at about age 85. In fact, the complete elimination of all deaths due to cancer, heart diseases, stroke, and diabetes would produce a mean life span of about 96 years, or a change in the mean age of death of only 17 percent (i.e., from 82 to 96 years). The reason that the disease-at-a-time approach is so unproductive is indicated in figure 2: most causes of death show an exponential, rather than a linear, increase in incidence across the last third of the life span. Therefore, eliminating any one of the major lethal illnesses buys only a short respite from the implacable march of the next illness in line. Figure 1 also estimates the longer life span that we would expect if we could decelerate the aging process in humans to the extent that has been routinely feasible in rodents.
FIG. 1.
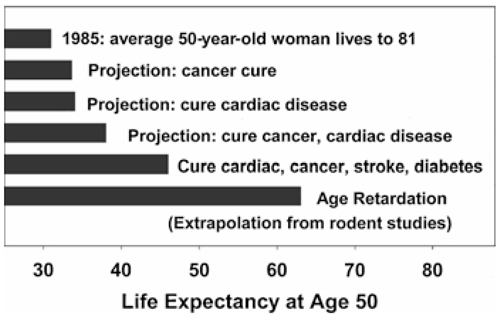
Remaining life expectancy of a 50-year-old Caucasian woman in the United States in 1985, at then-current mortality risk schedule (top bar), or as projected under the assumption that adult mortality risks for specific diseases (cancer, cardiac disease, etc., as indicated) were reduced to zero from 1985 onward. The bottom bar shows projected life expectancy if human adult mortality risks could be reduced to the same extent that caloric restriction reduces them in mice. Data from Olshansky, Carnes, and Cassel 1990
FIG. 2.
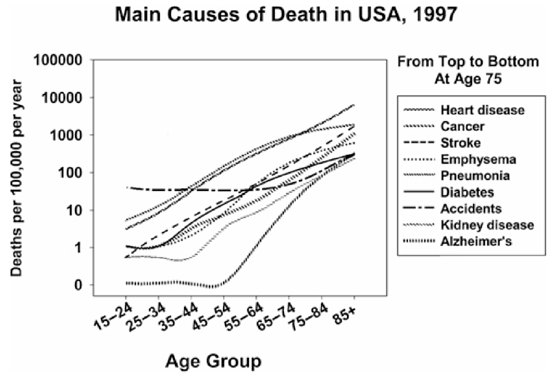
Number of deaths per 100,000 individuals at risk, by age decade, for the nine leading causes of death at age 75. Data from 1997 U.S. Vital Statistics.
It seems reasonable to suppose that the discovery of a technique that can, in a laboratory animal, diminish to near zero the incidence of neoplasia, cardiovascular illnesses, and diabetic changes would prompt a substantial public commitment to working out the mechanism of the intervention and developing analogues that work well in humans. Reasonable but wrong: such an intervention has been clearly established for decades, and yet its investigation receives such a small proportion of governmental research funding that it cannot be seen in a pie chart.
Obstacles to Applied Gerontology
Why is research on aging and longevity—despite its immense potential to prevent late-life illness and prolong active adult life—relegated to an obscure and dusty corner of the research establishment? I can suggest eight reasons:
Most gerontologists who are widely known to the public are unscrupulous purveyors of useless nostrums. Fears of aging, illness, and death make most of us easy prey to confidence hucksters armed with bottles of potions and attractively polished testimonials. Amazon.com lists 57 titles on melatonin and another 20 on DHEA, most at sales rankings well above that for the Journal of Gerontology: Biological Sciences. At present, books like HGH— The Amazing Medically Proven Plan to Reverse Aging are gaining rapidly on last year' champions, The Melatonin Miracle and The Superhormone (aka DHEA). Scientists and their patrons—even those who have legitimate research interests in interventional gerontology—do not wish to be seen hanging out with snake-oil vendors. Perhaps for these reasons, discussions of research on life span extension are carefully skirted in political discourse at the National Institutes of Health and among similar custodians of public funding. One can sometimes get away with cautious circumlocutions (“we do research on the causes of late-life illnesses”), but to be safe, it is clearly better to focus on how to “add life to years” and how “to learn the secrets contributing to a healthy old age.” A president who announces a war on cancer wins political points, but a president who publicly committed the government' resources to research on extending people' life span would be deemed certifiable.
Senators’ and voters’ parents died of specific diseases. Cancer, kidney diseases, AIDS, lung diseases, and Alzheimer' all have lobbies raising significant amounts of private funds for research and, more important, convincing legislators to allocate public funds to disease-specific research programs. Lobbyists for basic aging research, though doing the Lord' work, are thinner on the ground. To a great extent, the problem is one of public education: cancer and Alzheimer' are officially viewed as diseases and are thus in the realm of the theoretically curable, like polio, but aging is proverbially inevitable, its opponents as deluded as Canute. Those of us who have become convinced that aging, whether or not it is considered a disease, is the root of nearly all late-life illnesses, a fulcrum from which to shift the timing of all the real diseases on the list, have not yet convinced those who have the power to do much about aging.
Aging experiments in mammals take more than four years to finish. Young scientists need to write a lot of papers to get a postdoctoral fellowship, to get a job, and to get tenure. No responsible mentor will advise a smart and ambitious protégé to go into biogerontology research (except possibly in malleable, but questionably relevant, model organisms that have the grace to die in a few weeks).
Worse yet, aging experiments in mammals usually do not require fancy equipment or “cutting edge” methodology. Smart beginners can sometimes be seduced by the opportunity to learn the hottest new methods and use expensive new equipment, but unfortunately, many gerontological questions of interest require only the ability to distinguish live from dead mice. Learning to make this distinction is not adequate preparation for getting an academic or industrial position, and students know this.
Pharmaceutical firms can and do make excellent profits selling antiaging medicines that do not work. U.S. law permits over-the-counter sales of medicines without proven efficacy as long as the sales pitch does not explicitly state that the substances are to be used to treat disease. Pharmaceutical firms are, to some extent, in business to make money, and if it is legal and more or less harmless to pour tap water into bottles labeled “Authentic Ann Arbor Springs Health Water,” they see little reason to spend valuable marketing money on proving that the stuff retards aging.
A pharmaceutical firm cannot test, let alone sell, actual antiaging medicines in the expected lifetime of the firm' CEO. The good-guy CEO of PharmaInc, who wants to develop an agent that slows aging, has a problem: it will be extremely difficult, and very time-consuming, to develop convincing evidence that a candidate drug delays aging in people. Although some interventions slow aging in rodents, there is no evidence that these would work if applied beyond approximately the middle of the life span. Caloric restriction has its largest effect if begun at juvenile or adolescent stages; it has a reproducible but smaller effect when applied at about midlife, but no detectable effect if applied at later ages. But even if a potential anti-geric compound appears in animal models to postpone or decelerate aging when administered to middle-aged volunteers, documentation that it has an effect on longevity or late-life illnesses in humans is, at minimum, a 20-year exercise, and an expensive one with no certainty of success.
We do not know how to measure aging. Research on medicines to help hypertensives depended heavily on the invention of a device to measure blood pressure. Research on antipyretics requires a thermometer, and research on antiobesity agents requires a scale. Aging is harder to measure than blood pressure, temperature, and weight, indeed so hard to measure that many authorities (such as the multiplicitists) are convinced that aging does not exist and thus cannot be measured. Informally, many researchers, like most members of the lay public, can use either visual clues or functional testing (such as uphill races to avoid the maws of charging beasts) to sort acquaintances into age classes with a good deal of accuracy, but rigorous attempts to develop test batteries that do better than chronological age at identifying those middle-aged persons who resemble younger ones in many respects have had little success so far. Traits that (a) can be measured innocuously in middle age, (b) predict remaining life span and/or time before the occurrence of some adverse late-life event of interest, and (c) predict the outcome of other age-sensitive tests in widely diverse experimental arenas (e.g., an immune test that predicts cataracts and muscle weakness and pulmonary compliance as well as life span) will deserve to be classified as biomarkers of aging and would be highly valuable as surrogate variables in tests for anti-geric interventions. No such biomarkers exist yet. Obtaining public or private money for such testing is very very difficult, and there is no organized program to develop such a biomarker battery. The absence of a well-validated method to measure biological age is a major impediment to the testing of agents thought likely to slow aging.
To be honest, we do not know what biochemical pathways control the rate of aging. Some scientific obstacles must still be overcome, an area beyond the scope of this article (see Miller 1997).
A Ninth Obstacle to Manipulating Longevity
A ninth obstacle is preventing the discovery of effective anti-geric interventions, which deserves a separate heading and special consideration: gerontologiphobia. There is an irrational public predisposition to regard research on specific late-life diseases as marvelous but to regard research on aging, and thus on all late-life diseases together, as a public menace bound to produce a world filled with nonproductive, chronically disabled, unhappy senior citizens consuming more resources than they produce. No one who speaks in public about longevity research goes very far before encountering the widespread belief that research on extending the life span is unethical, because it will create a world with too many old people and not enough room for young folks. Pointing out that such an argument could also be used, with equally fallacious force, against research on heart attacks, diabetes, and cancer (whose goals, like those of gerontology, are to allow people to live longer and healthier lives) does little to dispel this belief. It also does little good to point out that a similar argument could have been made 200 years ago against penicillin, plumbing systems, and surgical anesthesia, each of which helps produce people who remain healthy and productive to the age of 40 and sometimes even beyond.
A recent set of news articles about progress in extending the life span of worms elicited on the New York Times’ op-ed page a letter from Thomas Lynch, a professional undertaker-poet, calling for a halt to aging research as a danger to the public. His opinion is, in part:
The news, lately reported, that the life span of humans might be doubled in the next century is cause for sober and deliberate contemplation…. People are living longer but suffering diminishing returns on their death-care portfolios. Given adequate financing, given lucky breaks, how far could we go? … Let me hazard that “too far” is one possible answer. We are a species, and especially a country, for whom, like drunks with drink or politicians with other people' money, enough is never going to be enough. It was true of the arms race, deficit spending and TV violence, the impeachment hearings and high-tech stocks—our appetites are insatiable, our habits tend toward gluttony …. To the medicos, maybe “thanks but no thanks” is the thing we should say—as if we had learned to spread the wealth or leave it to our children, or failing that, to leave well enough alone.
(Lynch 1999)
While in a certain respect it is cheering to note that biogerontology has progressed to the point that the leading figures in the mortuary business have begun to feel alarmed, this cheer is muted by the realization that so many members of the educated public, despite frequently being interested in promoting their own longevity, view improving the longevity of their fellows as vaguely distasteful and somehow unwise.
In this context, therefore, we should point out that the current, alarming population crisis and depletion of nonrenewable resources has come about without the slightest aid from biogerontologists, who have not yet discovered anything that actually improves public health or prevents disease. It follows inexorably that placing obstacles in the path of aging research will not help resolve the population crunch. Proposals that do address the root causes of the problem—solutions based on access to birth control information and hardware, strong incentives for resource conservation, and changes in social attitudes toward optimal family size, the scheduling of reproductive effort, and the proper role of the human female—typically elicit strong opposition from powerful religious, economic, and political factions. The currently fashionable concern about the hypothetical ill effects of hypothetical future advances in biogerontology seems, from this perspective, a diversionary tactic to draw attention away from the authentic malefactors whose political connections are better than those of us lowly biogerontologists. Perhaps there are some who, after sober and deliberate contemplation, feel that our Malthusian ills are best addressed by strategies that constrain the productive life span of healthy adults rather than by controlling the supply of new people. Were I a member of such a group, I would suggest that it devote its energies to removing seat belts from automobiles, insulin and antibiotics from pharmacies, and antismoking campaigns from schools, because compared with these interventions, picking on biogerontologists has a pretty low yield. The gerontologiphobic position (“Lynchism”) seems to me indefensible, but it is common enough to present a formidable obstacle to progress in aging research.
Conclusion
If we—the good guys who favor preventive medicine and medical research—accept the idea that it would be worthwhile to prevent late-life illness and thus prolong the period of healthy active life, then we ought to note that researchers are very far from developing any method that can, even in laboratory rodents, prevent cancer, osteoporosis, immunosenescence, heart or kidney disease, cataracts, or indeed any other of the troublesome concomitants of old age. We can, however, routinely retard all these tribulations at the same time, at least in laboratory mammals, by well-validated methods. We do not know how these interventions work and are not likely to figure this out without a substantial investment of money and brainpower. Those of us who are still enjoying life in our forties and fifties and even beyond and still making contributions to the public weal should be grateful to our predecessors who invented plumbing, X-rays, disinfectants, antibiotics, and insulin. We biogerontologists wish to do our share to earn the gratitude of the coming generations, and we now have some good reason for optimism. But the obstacles blocking the development of the hypothetical discipline of applied gerontology are at this point about 85 percent political and 15 percent scientific, and they will not be overcome by biologists alone.
References
- Austad SN. Retarded Senescence in an Insular Population of Virginia Opossums Didelphis virginiana. Journal of Zoology. 1993;229:695–708. [Google Scholar]
- Austad SN, Fischer KE. Mammalian Aging, Metabolism, and Ecology: Evidence from the Bats and Marsupials. Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 1991;46:B47–53. doi: 10.1093/geronj/46.2.b47. [DOI] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf Mice and the Ageing Process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Carey JR, Judge DS. Longevity Records: Life Spans of Mammals, Birds, Amphibians, Reptiles, and Fish. Odense, Denmark: Odense University Press; 2000. [Google Scholar]
- Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of Growth Parameters and Life Span of GHR/BP Gene-Disrupted Mice. Endocrinology. 2000;141:2608–13. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- Davey SG, Hart C, Upton M, et al. Height and Risk of Death among Men and Women: Aetiological Implications of Associations with Cardiorespiratory Disease and Cancer Mortality. Journal of Epidemiology & Community Health. 2000;54:97–103. doi: 10.1136/jech.54.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Life Span Extension and Delayed Immune and Collagen Aging in Mutant Mice with Defects in Growth Hormone Production. Proceedings of the National Academy of Science. 2001;98:6736–41. doi: 10.1073/pnas.111158898. U.S.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. Understanding Ageing. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- Krzisnik C, Kolacio Z, Battelino T, Brown M, Parks JS, Laron Z. The “Little People” of the Island of Krk—Revisited: Etiology of Hypopituitarism Revealed. Journal of Endocrine Genetics. 1999;1:9–19. [Google Scholar]
- Li Y, Deeb B, Pendergrass W, Wolf N. Cellular Proliferative Capacity and Life Span in Small and Large Dogs. Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 1996;51:B403–8. doi: 10.1093/gerona/51a.6.b403. [DOI] [PubMed] [Google Scholar]
- Lynch T. Why Buy More Time? New York Times. 1999. (March 14)
- Maeda H, Gleiser CA, Masoro EJ, Murata I, McMahan CA, Yu BP. Nutritional Influences on Aging of Fischer 344 Rats: II. Pathology. Journal of Gerontology. 1985;40:671–88. doi: 10.1093/geronj/40.6.671. [DOI] [PubMed] [Google Scholar]
- Masoro EJ. Aging: Current Concepts. In: Masoro EJ, editor. Handbook of Physiology. Vol. 11. New York: Oxford University Press; 1995. pp. 3–21. Aging. [Google Scholar]
- McCarter R, Masoro EJ, Yu BP. Does Food Restriction Retard Aging by Reducing the Metabolic Rate? American Journal of Physiology. 1985;248:E488–90. doi: 10.1152/ajpendo.1985.248.4.E488. [DOI] [PubMed] [Google Scholar]
- McCay CM, Crowell MF, Maynard LA. The Effect of Retarded Growth upon the Life Span and upon Ultimate Body Size. Journal of Nutrition. 1935;10:63–79. [Google Scholar]
- Medawar PT. An Unsolved Problem of Biology. London: H.K. Lewis; 1952. [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, et al. The p66shc Adaptor Protein Controls Oxidative Stress Response and Life Span in Mammals. Nature. 1999;402:309–13. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Miller RA. When Will the Biology of Aging Become Useful? Future Landmarks in Biomedical Gerontology. Journal of the American Geriatrics Society. 1997;45:1258–67. doi: 10.1111/j.1532-5415.1997.tb03781.x. [DOI] [PubMed] [Google Scholar]
- Miller RA. Kleemeier Award Lecture: Are There Genes for Aging? Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 1999;54:B297–307. doi: 10.1093/gerona/54.7.b297. [DOI] [PubMed] [Google Scholar]
- Miller RA, Chrisp C, Atchley WR. Differential Longevity in Mouse Stocks Selected for Early Life Growth Trajectory. Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2000;55:B455–61. doi: 10.1093/gerona/55.9.b455. [DOI] [PubMed] [Google Scholar]
- Olshansky SJ, Carnes BA, Cassel C. In Search of Methuselah: Estimating the Upper Limits to Human Longevity. Science. 1990;250:634–40. doi: 10.1126/science.2237414. [DOI] [PubMed] [Google Scholar]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low Methionine Ingestion by Rats Extends Life Span. Journal of Nutrition. 1993;123:269–74. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- Partridge L, Harvey PH. Methuselah among Nematodes. Nature. 1993;366:404–5. doi: 10.1038/366404a0. [DOI] [PubMed] [Google Scholar]
- Richie JP, Jr, Leutzinger Y, Parthasarathy S, Malloy V, Orentreich N, Zimmerman JA. Methionine Restriction Increases Blood Glutathione and Longevity in F344 Rats. Federation of American Societies for Experimental Biology Journal. 1994;8:1302–7. doi: 10.1096/fasebj.8.15.8001743. [DOI] [PubMed] [Google Scholar]
- Rudman D, Feller AG, Nagraj HS, et al. Effects of Human Growth Hormone in Men over 60 Years Old. New England Journal of Medicine. 1990;323:1–6. doi: 10.1056/NEJM199007053230101. [DOI] [PubMed] [Google Scholar]
- Samaras TT, Storms LH. Impact of Height and Weight on Life Span. Bulletin of the World Health Organization. 1992;70:259–67. [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Sohal RS. Seminars in Medicine of the Beth Israel Deaconess Medical Center: Caloric Intake and Aging. New England Journal of Medicine. 1997;337:986–94. doi: 10.1056/NEJM199710023371407. 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindruch R, Walford RL. The Retardation of Aging and Disease by Dietary Restriction. Ill.: Thomas: Springfield; 1988. [Google Scholar]
- Williams GC. Pleiotropy, Natural Selection, and the Evolution of Senescence. Evolution. 1957;11:398–411. [Google Scholar]
- Yu BP, Masoro EJ, McMahan CA. Nutritional Influences on Aging of Fischer 344 Rats: I. Physical, Metabolic, and Longevity Characteristics. Journal of Gerontology. 1985;40:657–70. doi: 10.1093/geronj/40.6.657. [DOI] [PubMed] [Google Scholar]