

Abstract
Inversions are an important form of structural variation, but are difficult to characterize as their breakpoints often fall within inverted repeats. We have developed a novel method, called ‘Haplotype Fusion’, in which an inversion breakpoint is genotyped by performing Fusion-PCR on single molecules of DNA. Fusing single copy sequences bracketing an inversion breakpoint generates orientation-specific PCR products, as exemplified by a genotyping assay for the int22 hemophilia A inversion on Xq28. This method is suitable for surveying inversion polymorphism at most inverted repeats in the human genome. Furthermore, we demonstrate that inversion events with breakpoints embedded within long (>100kb) inverted repeats can be genotyped by Haplotype Fusion PCR followed by bead-based single molecule haplotyping on repeat-specific markers bracketing the inversion breakpoint. We illustrate this method by genotyping a Yp paracentric inversion sponsored by >300kb long inverted repeats. The generality of our methods for genotyping chromosomal inversions should catalyse our understanding of the contribution of inversions to genomic variation, inherited diseases and cancer.
Introduction
Structural variation within the human genome (deletions, duplications, inversions and translocations) accounts for an appreciable proportion of both causative alleles in genetic disorders with simple inheritance patterns, and susceptibility alleles in infectious diseases 1,2. Therefore, it is to be expected that an as yet unknown proportion of the genetic risk in complex diseases will also be determined by structural variants. Current methods for detecting variants conferring disease susceptibility are biased against identifying structural variants 1. In order to address this deficit, comprehensive surveys of structural variation are required, allied to assays that allow population-scale genotyping in association studies of the variants discovered. Population-scale genotyping of structural variation is also necessary to investigate the role that it has played in recent human evolution 3.
Information on the locations and frequency of copy number polymorphism in the human genome is accruing rapidly, largely from microarray-based comparative genome hybridization studies of increasing resolution 4-6. In addition, a diverse range of quantitative methods allow population-scale multiplexed genotyping of copy number variants 7.
By contrast, in the absence of a similarly high-throughput discovery method for inversions, fewer inversion polymorphisms have been identified. Although recent analysis of dense fosmid end sequencing data from a single individual has been used to identify novel inversion variants on a genome-wide basis 9, this method does not easily scale to the analysis of the tens of samples needed to identify all common (frequency greater than 5%) inversions. Similarly, there are no genotyping methods that allow population-scale genotyping of known inversion variants: the usefulness of SNPs in LD with an inversion is possibly limited by multiple independent origins for the same inversion 10,11. Known inversions are typically assayed singly by laborious interphase FISH or Pulsed Field Gel Electrophoresis 12, neither of which are appropriate for population-scale genotyping. A handful of well-characterised inversions can be genotyped singly by long PCR or RFLP analysis using standard agarose gel electrophoresis 13,14, but this is only practical when inversion breakpoints are not embedded within long (>10kb) inverted repeats. Unfortunately, among the different classes of structural variants, inversions are the most likely to feature rearrangement breakpoints within long, highly-similar duplicated sequences 9.
To address these deficits, we have developed PCR-based single molecule haplotyping methods that enable both surveys for novel inversion variants, and population-scale genotyping of known inversions. We demonstrate our ability to haplotype single DNA molecules which span known or likely inversion breakpoints by developing a novel PCR-based genotyping assay for the haemophilia-causing inversion that disrupts the Factor VIII gene on Xq28 15. Moreover, we demonstrate that these methods are capable of genotyping known inversions with breakpoints embedded in inverted repeats hundreds of kilobases long by developing an assay for the Yp inversion that strongly influences the germline rate of infertility-causing XY translocations 16.
Results
We reasoned that the inversion genotype of an individual can be deduced from their genomic DNA if single molecule haplotyping is performed on the DNA fragments that span the inversion breakpoints (Figure 1).
Figure 1. Strategies for genotyping inversions.

Inverted and wildtype orientations are represented for two inversions. In both cases the inversion breakpoints lie within inverted repeats, shown as red and blue arrows. In the first case, the inverted repeats are shorter than the size of the DNA fragments in the genomic DNA preparation, whereas in the second case the inverted repeats are longer than the DNA fragments. Beneath each inversion are shown the DNA fragments that cover the inverted region. DNA fragments that are informative for genotyping the inversion are shown in green, whereas those that are uninformative are shown in grey. The filled circles above the inverted repeats in the second inversion represent paralogous sequence variants that distinguish between the two repeats; the haplotype of which is diagnostic for the state of the inversion.
We considered that by performing fusion PCR 17,18 on single molecule templates, we should be able to juxtapose sequences that lie on either side of an inversion breakpoint: the haplotypes of these sequences are diagnostic for the inversion. Fusing the sequences allows us to apply a greater range of downstream haplotyping techniques. PCR in oil:water emulsions 19 provides a means to amplify single molecule templates on a far larger scale (103-104 fold greater) than performing single molecule dilutions in a microtitre plate. This is because amplification occurs in millions of aqueous compartments that are stable to thermocycling 20. The number of template molecules is far less than the number of compartments, and so amplification within a compartment derives from a single molecule. Therefore, we developed Haplotype Fusion PCR (HF-PCR) to combine the advantages of fusion PCR and single molecule amplification in emulsions. This method is shown in outline in Figure 2.
Figure 2. Haplotype Fusion PCR.

At the top of the figure is shown a double stranded DNA template containing two sequences on either side of an inversion breakpoint. Three primers shown as black arrows are used in a PCR reaction conducted in an oil:water emulsion. The red tag on the internal primer R1, is an oligonucleotide that is identical to the sequence on the lower strand of the right-hand amplicon. The blue tag on the right-hand outside primer R2, is an oligonucleotide sequence (Zip1) that facilitates subsequent bead-based haplotyping. In early rounds of PCR, a double stranded product encompassing the left-hand amplicon is amplified exponentially, and a single stranded product containing the right-hand amplicon is amplified linearly. After the internal primer is exhausted, the upper strand of the left-hand amplicon acts as a forward primer for amplification of the right-hand amplicon.
In practice, the haplotyping method used to genotype the inversion differs depending on the length of the inverted repeat relative to the size of DNA fragments in the genomic DNA preparation (Figure 1). If the inverted repeat is shorter than the DNA fragment lengths, then the single copy sequences on either side of an inverted repeat can be found on the same single molecule template. Haplotype Fusion PCR can be used to fuse these single copy sequences, and an inversion can be genotyped by determining the identity of the fused PCR product. This is most easily exemplified by generating fused products of different sizes for the two orientations of the inversion. We term this method Unique Sequence Fusion (USF).
If the inverted repeat is longer than the DNA fragments then the inversion can be genotyped by fusing two sequences that contain repeat-specific markers that lie on either side of the inversion breakpoint. Repeat-specific markers that are embedded within a long region of homology are known as paralogous sequence variants (PSVs). We term this method PSV Fusion (PSVF).
Genotyping the int22h Haemophilia inversion
The int22h inversion that is a common cause of Haemophilia A is sponsored by int22h repeats that lie in inverted orientation on Xq28 (see Figure 3). The proximal inverted repeat (int22h1) involved in the pathogenic inversion lies within the Factor VIII gene. There are two distal copies of the int22h repeat, both of which are embedded within a larger segmental duplication. At any one time, only one of these distal int22h repeats (int22h2 and int22h3) lies in inverted orientation to int22h1 21,22. Inversion events between the larger distal segmental duplications are capable of swapping the locations of int22h2 and int22h3 such that either can be found in inverted orientation to int22h1 (both wildtype structures are shown in Figure 3). There are two major classes of haemophilia-causing inversion, Type I and Type II, and these differ in which of the two distal int22h repeats recombines with the int22h1 repeat.
Figure 3. Genotyping the Haemophilia inversion.

Panel A shows the four possible orientations of the genomic region on Xq28 containing the three int22h repeats (purple arrows). Int22h2 and int22h3 are embedded in longer (~50kb) inverted repeats separated by ~70kb of unique sequence (black/white gradient). The portions of this larger segmental duplication that are not found in the int22h repeats are shown as green and red arrows. The four possible orientations refer to the reference ‘wildtype’ sequence, the Int22h2/3 inverted sequence, Type I and Type II inversions. Beneath each orientation are shown the three PCR products (1,2 & 3) amplified using the primers described in the text. The two PCR products fused by Haplotype-Fusion PCR are linked by a dashed line. Panel B shows the position of the PCR primers F1, R1, R2 and R3 on the int22h repeats. The grey arrow labelled F2 shows the binding site complementary to the tail of the R1 primer. Panel 3 shows an agarose gel of the results of Haplotype-Fusion PCR when performed in solution and in emulsions on all possible inversion genotypes. The four inversion genotypes that produce single band patterns in emulsions all produce two band patterns when the fusion PCR is performed in solution, because PCR products can be fused from different DNA templates.
We designed a HF-PCR assay to distinguish between the wildtype orientation of the haemophilia-causing inversion and the pathogenic orientation (either type I or type II). The F1 primer lies in the single copy sequence proximal to the int22h1 repeat. The R1 primer lies just inside the tail of the int22h1 repeat. Thus amplicon 1 amplified by F1 and R1 primers spans the boundary between single-copy and duplicated sequence. The tail of the R1 primer is complementary to a binding site just inside the head of the int22h repeat. Two other reverse primers were designed: R2 lies in single copy sequence distal to the head of the int22h1 repeat and leads to amplification of amplicon 2; R3 lies within the larger distal segmental duplication just adjacent to the heads of the int22h2 and int22h3 repeats and leads to amplification of amplicon 3. The fusion of amplicon 1 and 2 during emulsion PCR is characteristic of the wildtype orientation, whereas the fusion of amplicons 1 and 3 generates a shorter PCR product that is diagnostic for the inverted orientation. When these fusion PCR reactions are performed in solution under standard PCR conditions both fusion products are generated in all individuals (Figure 3). However, when the fusion PCR is performed in emulsions, products from different templates can no longer fuse and only the fusion products expected for that specific inversion genotype are generated. Thus this assay distinguishes between males with and without the inversion, female carriers of the inversion, and females homozygous for the wildtype orientation (Figure 3). To demonstrate the general utility of this assay we genotyped blind a mixed population of ten males with and without the int22h inversion with 100% concordance with the known inversion genotypes (Supplementary Figure 1).
Genotyping the Yp inversion by single molecule haplotyping
The constitutive haploidy of the common 3.6Mb Yp inversion 23 makes it an ideal test locus for our inversion genotyping assays. This inversion has been suggested to result from non-allelic homologous recombination between the ~300kb IR3 inverted repeats 24. It is known from previous studies that the alternative orientations of the inversion (here denoted ‘reference’ – as found in the human reference sequence, and ‘inverted’) are present in different YAC libraries 25,26. From these libraries, we identified four YACs (two from each orientation) that contain the IR3 proximal and distal repeats and thus contain the inversion breakpoints. YAC clones AA4A7/11 and 692B1 contain the proximal IR3 repeat in ‘inverted’ and ‘reference’ orientations respectively. YAC clones AMY8 and 821G7 contain the distal IR3 repeat in ‘inverted’ and ‘reference’ orientations respectively. We localised the breakpoint of the Yp inversion within the ~300kb long IR3 repeats to a ~7kb interval bounded by two repeat-specific base substitutions, known as Paralogous Sequence Variants (PSVs) (manuscript in preparation: DJT, CTS, MEH). Consequently, we can define haplotypes of these PSVs that are diagnostic of Y chromosomes having undergone this Yp inversion.
We used HF-PCR to fuse two short amplicons containing the two PSVs defining the Yp inversion breakpoint interval. The two PSVs are separated by 50bp in the fused ~300bp product. We identified four YACs that contain the four possible PSV haplotypes from different YAC libraries generated from males with and without the Yp inversion. We amplified fused haplotypes from these individual YACs, from pairwise mixtures of these YACs, and from genomic DNA from the same individuals used to generate ‘reference’ and ‘inverted’ YAC libraries.
Having juxtaposed the diagnostic PSVs into a condensed haplotype, we adapted bead-based single molecule genotyping to haplotype the fused PCR product: (see Figure 4). In bead-based single molecule genotyping, paramagnetic beads are coated in an amplified product from a single molecule template by performing PCR within an oil:water emulsion. These amplicon-coated beads can then be genotyped by the addition of a single dye-labelled nucleotide (single base extension) to a primer that is annealed directly flanking the variant site (see Figure 4). In a published protocol (BEAMing) these beads were subsequently analysed by flow cytometry 19. We adapted this protocol to haplotyping by performing two successive rounds of PSV genotyping on beads immobilized within a polyacrylamide gel on a microscope slide and aligning the two images to call haplotypes 27,28. We added a universal primer tag to one of the outside primers of the HF-PCR to allow the amplified fusion product to be subsequently linked to beads coated with a matching double-biotinylated oligonucleotide. This use of a universal bead-bound oligonucleotide represents a substantial cost-saving to the published protocol and would facilitate multiplexing. We demonstrated the successful application of this assay using single YACs and mixtures of YACs as templates for haplotype fusion PCR (Figure 5) before validating it on the two genomic DNAs from individuals with known inversion orientation (Figure 6). Finally, to demonstrate the utility of this assay we genotyped thirteen previously uncharacterised individuals of diverse geographic origin. In twelve cases (92%) it was possible to unambiguously genotype this inversion event as both breakpoint haplotypes were diagnostic of either the inverted or reference orientations (Figure 7). The single genotyping failure contained a mixture of two breakpoint haplotypes, one typically of the reference orientation and the other typical of the inverted orientation. This mixture of the two sets of expected breakpoint haplotypes most likely results from a gene conversion event that homogenises PSV2.
Figure 4. Bead haplotyping.

An emulsion PCR is performed in which the product of a haplotype fusion PCR is amplified onto paramagnetic beads (yellow) loaded with a universal oligonucleotide (blue). For simplicity, only a single double-stranded template labelled with the two bases to be genotyped is illustrated. Emulsion PCR generates beads coated in single-stranded amplified product from single molecule templates. These beads are cast into a thin polyacrylamide gel (grey) on a microscope slide. Sequential rounds of genotyping by single base extension with dye-labelled nucleotides yield two images that can be overlaid and offset to reveal the haplotype on each bead. For each genotyping reaction, the annealing position of the single base extension primer on the bead-immobilised template is shown, together with the dye-labelled nucleotide that is incorporated.
Figure 5. Single molecule haplotyping on YACs containing single IR3 repeats.

A. The portions of the IR3 repeats (red and blue arrows) encompassing the inversion breakpoint (striped interval) are aligned for both the reference and inverted orientations. Each IR3 repeat is labelled with its respective haplotype of the two PSVs that flank the breakpoint. B. In the first column, the four possible PSV haplotypes apparent within the four IR3-containing YACs are illustrated. Beneath these four haplotypes are shown the pair of PSV haplotypes obtained from mixing IR3-containing YACs from the reference and inverted orientations. Each row shows the results of bead haplotyping conducted using the individual YACs and mixtures of YACs as templates for haplotype fusion PCR and a subsequent haplotyping reaction. The bead haplotyping results are shown as three fluorescent microscope images of the same portion of a much larger image. For each image, the bases corresponding to the different colours are given above the image and the genotype/haplotype observed is given below the image. The first image in each set of three shows the genotype of the first PSV. The second image shows the genotype of the second PSV, and the third image shows the results of overlaying the two images with the second PSV shifted 10 pixels to the right.
Figure 6. Single molecule haplotyping on genomic DNA.

The results of bead haplotyping using genomic DNA as the template for the initial haplotype fusion PCR are shown for individuals known to have reference and inverted orientations of the Yp inversion. See the legend for figure 5 for more details.
Figure 7. Single molecule haplotyping on a panel of genomic DNAs.
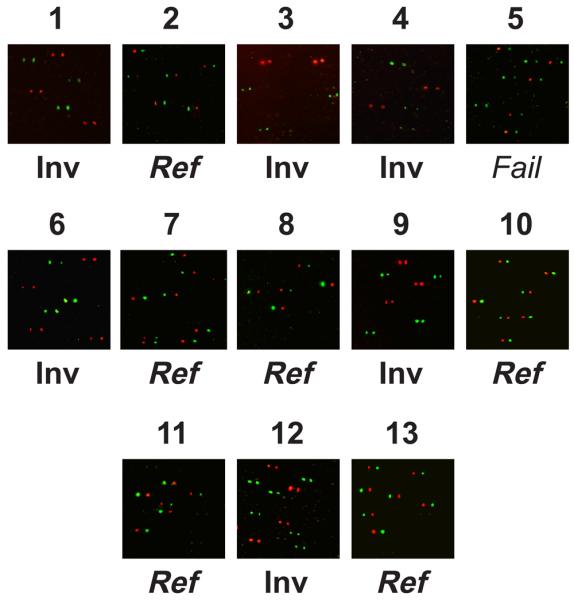
The results of bead haplotyping using genomic DNA as the template for the initial haplotype fusion PCR are shown for thirteen individuals of unknown orientations of the Yp inversion. Each image shows the results of overlaying the two genotyping images with the second PSV shifted 10 pixels to the right. See the legend for figure 5 for more details. Inv = inversion, Ref = reference. Individual 5 failed inversion genotyping due to the unexpected combination of breakpoint haplotypes (see text for more details).
Discussion
We have demonstrated that juxtaposing sequences flanking an inversion breakpoint by Haplotype-Fusion PCR can be used to genotype inversions. We have also successfully shown that bead-based single molecule genotyping 19 can be extended to haplotyping by immobilizing beads coated with amplified product from a single template and performing sequential rounds of genotyping.
In addition to being able to genotype known inversions and validate putative inversions, our USF inversion genotyping method can be used to screen panels of individuals for novel inversion variants by targeting this assay towards highly similar inverted repeats. This is due to the ability of the USF method to scan an entire inverted repeat for an inversion breakpoint. Using the USF method, we have initiated a survey for inversion polymorphisms sponsored by known inverted repeats identified within databases of segmental duplications. In principle, the length of a haplotype that can be condensed by Haplotype-Fusion PCR is limited only by the size of the template DNA. The shearing effect of emulsion preparation is negligible on genomic DNA fragments 100-150kb in length as judged by pulsed field gel electrophoresis (Supplementary Figure 2). Thus it is reasonable to expect that template DNA molecules in emulsions can be at least 100kb in length. The vast majority (97%) of inverted repeats in the segmental duplication databases 29,30 are less than 100kb in length and so are amenable to USF genotyping. By contrast, long PCR methods targeted to unique sequences are typically restricted to inverted repeats 10kb in length. The majority of duplicated sequences known to sponsor recurrent chromosomal rearrangements are greater than 10kb in length 1,2.
In contrast to the USF method, we have demonstrated that the PSVF inversion genotyping method allows inversions with breakpoints within the longest inverted repeats to be genotyped, albeit with only a proportion of the inverted repeat assayed for inversion breakpoints. The PSVF inversion genotyping method is likely to be most useful for the genotyping of known inversion events with well characterized breakpoints as it requires that an inversion breakpoint is known to fall within an interval defined by two PSVs. To the authors' knowledge, PSVs are not catalogued in the standard genome databases, although candidate PSVs can easily be identified from the human reference sequence by identifying variant sites within alignments of duplicated sequences. PSVs identified in this manner are not necessarily fixed in the population, which means that assays targeted to these variants may not work on all individuals. Gene conversion between duplicated sequences may be one cause of PSVs not being fixed in the population. Therefore it is important that any assay targeted towards PSVs is robust enough to distinguish between gene conversion events and inversion events. Gene conversion is the non-reciprocal transfer of sequence information from one homologous sequence to another. As a result, a single gene conversion event can only generate an apparent recombinant haplotype at one of the two inversion breakpoints. The PSVF inversion genotyping method described here haplotypes both inversion breakpoints, and it is the combination of these two haplotypes that allows the inversion event to be genotyped unambiguously. The PSVF method will identify discrepant pairs of breakpoint haplotypes that result from gene conversion events, and does not confuse them with either state of the inversion. A single gene conversion event was identified in this manner within the cohort that was subjected to blind inversion genotyping by PSVF, thus demonstrating the importance of assaying both inversion breakpoints.
In principle, PSVF inversion genotyping could be used to survey for novel inversions by tiling along a long (>150kb) inverted repeat with overlapping assays, but using the present implementation this is likely to be overly laborious. The existence of hotspots of non-allelic homologous recombination within which most rearrangement breakpoints fall is a common feature of rearrangements sponsored by duplicated sequences 31. These hotspots are typically restricted to a small proportion of a much larger segmental duplication. PSVF inversion genotyping will be particularly useful for long inverted repeats which harbour hotspots for rearrangements. Moreover, the high-throughput identification and sequencing of fosmid clones harbouring inversion breakpoints 9 should greatly increase the rate at which these hotspots are characterized.
How do our methods for inversion genotyping compare to alternative methods? First, in using a standard genomic DNA preparation as input for these assays, our methods are immediately applicable to a far greater range of samples than are either cytogenetic or Pulsed-Field Gel Electrophoresis methods. Second, long-range PCR is limited by the processivity of DNA polymerases and is often not robust over distances of 10kb or more, whereas HF-PCR is only limited by the size of the DNA fragments in the preparation. As a result, our methods are applicable to all sizes of inverted repeat and not just the subset that can be PCR-amplified in their entirety. Finally, our assay methods are quick, robust and scaleable to high-throughput (emulsions can be prepared in 96-well microtitre plates – data not shown). These features are all pre-requisites for the population-scale genotyping required to elucidate the functional impact of any given inversion. In addition, as amplification is presently only occurring in a small proportion (0.1% to 1%) of aqueous compartments within an emulsion, HF-PCR should be readily amenable to multiplexing, using different sets of primers that do not compete for resources due to amplification in separate compartments.
Under some circumstances, it may be possible to genotype inversions by typing nearby markers (e.g. SNPs) in high linkage disequilibrium (LD) with the inversion 3,32. This approach may be confounded if an inversion arises recurrently on different haplotype backgrounds. The probability of any mutation being recurrent is a function of its germline mutation rate. For example, a given microsatellite allele almost always appears on different haplotypes as a result of microsatellites typically having a high mutation rate (10−3-10−4 per generation). By contrast, SNPs are often in very high LD with other SNPs due to their much lower mutation rate (10−7-10−8 per generation). In the absence of population-scale genotyping studies little evidence has accrued for the recurrence or otherwise of neutral inversions 33, however, it is clear that pathogenic inversions can arise recurrently 10,11. Furthermore, the mutation process that gives rise to a majority of inversions (Non-Allelic Homologous Recombination) typically operates on long (>10kb) duplicated sequences at rates of 10−3 to 10−5 per generation 34. Thus many inversions are likely to be more akin (in terms of the likelihood of arising recurrently) to microsatellites, than to SNPs, and as a consequence the association between an inversion and flanking markers may be lower than for other variants. Moreover, it is worth remembering that LD-based ‘indirect’ genotyping always reduces statistical power (as compared to direct genotyping) to detect an association between phenotype and genotype when r2<1.
The methods developed in this study have application beyond genotyping inversions. A logical extension of these methods would be to the detection of recombinant haplotypes that are diagnostic of other types of chromosomal rearrangement (deletions, duplications and translocations) that arise either in the germline or somatic tissues. In addition, these methods are applicable to any genome sequence in which duplicated sequences have been mapped. Assays that facilitate the population-scale genotyping of all types of structural variation can be expected to greatly increase our understanding of the evolutionary history and medical consequences of this important class of genomic variation.
Materials and Methods
DNA samples
Genomic DNA from males with known Y-chromosomal haplotypes were a kind gift of Mark Jobling. YAC clones 692B1 and 821G7 come from the CEPH YAC library 35 (‘reference’). YAC clones AA4A7/11 and AMY8 come from libraries constructed from CGM1 36 and OXEN DNA 37, respectively (‘inverted’). Genomic DNA of individuals of diverse geographic ancestry were obtained from ECACC. Genomic DNA from haemophilia A patients was obtained during the National Haemophilia A Mutation Database project and the intron 22 inversion previously typed using the long PCR method 13.
Quantifying the template:compartment ratio
Water-in-oil emulsions prepared as described below contain spherical aqueous compartments with an average diameter of 15μm 20. An emulsion made from 200μl oil and 100μl aqueous phases would therefore be expected to contain 5.7 × 107 aqueous compartments. The target region of the fusion PCRs lie on the X or Y chromosomes of male individuals, so emulsions made using 200ng genomic DNA contain approximately 30,000 amplifiable template molecules. Consequently only 1 in around 1900 aqueous compartments contains a suitable template molecule for fusion PCR and so amplification derives from a single molecule in the overwhelming majority of cases.
Pulsed Field Gel Electrophoresis
An emulsion was prepared as described below using 100μl of 0.20μg/μl male genomic DNA (Novagen) as the aqueous phase. Following hexane treatment to disrupt the emulsion, 10μg of the DNA was electrophoresed in a 1% agarose gel in a Bio-Rad CHEF pulsed field electrophoresis cell alongside 10μg genomic DNA, 2μg 5kb DNA size standard (Bio-Rad) and approximately 1/3 block of lambda DNA ladder (Bio-Rad). Electrophoresis was performed in 0.5X TBE, at 6V/cm with switch times ramped from 3 to 7 seconds over the course of 24 hours. The gel was stained with 1μg/ml ethidium bromide and was visualized under UV light.
Haplotype fusion PCR
Yp inversion
PCR reactions were prepared in a total volume of 100μl, 1X Phusion HF buffer (Finnzymes), an additional 1mM MgCl2, 250μM dNTPs, 1μM primers InvF1 (Sigma; 5′ TGCCTGAGGAAGCCCTGTAGTTAG 3′) and InvR2 (Sigma; 5′ GGAGCACGCTATCCCGTTAGACCCCTCCATTTTAGAGACAGGTGC 3′), 10nM primer InvFusionR1 (Sigma; 5′ GATATATAAATGTTGCAATGGGGGAGCATGAGCTGCAGGAGGCCTTTGC 3′), 200ng (6 × 104 copies) genomic DNA, 16 units Phusion DNA polymerase (Finnzymes). This aqueous phase was added dropwise over ~50 seconds to 200μl oil phase (4.5% v/v Span 80, 0.4% v/v Tween 80 and 0.05% Triton X-100 dissolved in light mineral oil (all Sigma)), in a 2ml Corning Cryo-Vial whilst stirring with a magnetic bar (8 × 3mm with a pivot ring; VWR International) at 1000rpm, as described previously 20. Emulsions were stirred for a total of 5 minutes before being overlaid with 30μl mineral oil (Sigma) and thermocycled (98°C for 30 seconds; 33 cycles of 98°C for 10 seconds, 63°C for 30 seconds and 72°C for 15 seconds; 72°C for 5 minutes).
After thermocycling, emulsions were disrupted by the addition of hexane (Aldrich). 2μl of each clean PCR product was subjected to reamplification in a total volume of 50μl using primers InvF1 an InvR2 (1X Phusion HF buffer, 200μM dNTPs, 300nM primers and 1 unit Phusion DNA polymerase) under the same cycling conditions. PCR products were concentrated by ethanol precipitation, and resuspended in 18μl 1x Phusion HF buffer, to which 1.6 units Proteinase K (Sigma) were added. Reactions were incubated at 56°C for 1 hour to digest the polymerase and 95°C for 10 minutes to denature the Proteinase K, after which DNA was quantified by gel electrophoresis.
Haemophilia Inversion
A similar protocol to that used for the Y inversion was employed, but with 1μM primers F1 (Sigma; 5′ ATTTACCTCCCCTGCCATGGAAATC 3′), R2 (Sigma; 5′ ACAATCTGGAGGTGAATGCTTAGTTC 3′) and R3 (Sigma; 5′ CATTTAACCAAAAAGCACTTGTAAGCCA), and 10nM primer R1 (Sigma; 5′ CCAGAATTTGGCCCATAGCCTGCTGTGTTAAGCATTGAGACAACACCAAC 3′). Thermocycling conditions were as with the Y Inversion but using 40 cycles and a 66°C annealing step. Following disruption of the emulsion by hexane, fused PCR products were reamplified in 50μl of aqueous solution containing 1X JumpStart Polymerase PCR buffer (Sigma; 10mM Tris-HCl pH8.3, 50mM KCl, 1.5mM MgCl2, 1 × 10−3% (w/v) gelatine), 200μM dNTPs, 600nM primer F1, 300nM primers R2 and R3, 1μl 100x diluted PCR product and 1.25 units JumpStart polymerase (Sigma). Cycling conditions were: 94°C for 2 minutes, 35 cycles of 94°C for 30 seconds, 62°C for 30 seconds and 72°C for 15 seconds; 72°C for 10 minutes). 15μl of each product of this second amplification was electrophoresed in a 2.5% agarose gel alongside 5μl HyperLadder IV Quantitative DNA Marker (Bioline). Gels were visualized by ethidium bromide staining under UV light.
Bead-based PCR in emulsion
For each reaction, approximately 6 × 107 MyOne streptavidin-coated paramagnetic beads (Dynal Biotech) were prepared by rinsing three times in TTL buffer (1M LiCl, 100mM Tris-HCl pH8.0, 0.1% Tween 20) followed by incubation in TTL buffer containing 40μM double biotinylated Zip1 primer (Integrated DNA Technologies; 5′ Bio-Bio-GGAGCACGCTATCCCGTTAGAC 3′) with stirring for 20 minutes at 37°C. To saturate any streptavidin to which no oligo had become attached, beads were incubated subsequently in TTL buffer containing 400μM biocytin, again with stirring for 20 minutes at 37°C. Beads were rinsed twice in TT buffer (250mM Tris-HCl pH8.0, 0.1% Tween 20), twice in 1X JumpStart Polymerase PCR buffer (Sigma) and resuspended in 100μl PCR aqueous phase, consisting of 1X JumpStart DNA Polymerase buffer (Sigma), an additional 1mM MgCl2, 200μM dNTPs, 500nM primer InvF1, 50nM primer InvR2, 5.7 × 106 copies of haplotype fusion PCR product and 25 units of JumpStart polymerase (Sigma). Emulsions were prepared as described above, and thermocycling was performed (94°C for 2 minutes; 100 cycles of 94°C for 30 seconds, 60°C for 1 minute, 72°C for 45 seconds; 72°C for 5 minutes). Following thermocycling, emulsions were disrupted with hexane and beads were captured magnetically.
40μM thick 6% polyacrylamide gels containing the magnetic beads were prepared as described 27,38, except that polymerization was initiated by addition of 1.25μl 5% TEMED (Sigma) and 1.875μl 0.5% ammonium persulphate solution (Sigma). Slides were inverted during polymerization to allow beads to settle onto the surface of the gel. Bead-bound double stranded DNA was denatured and primer annealing and single base extension reactions were performed as described 27 using Perkin Elmer Cy3 and Cy5-labelled dNTPs, except that 25 units Klenow fragment DNA polymerase (NEB) were used per slide for the single base extension reaction, in 1X NEB buffer 2 (50mM NaCl, 10mM Tris-HCl pH7.9, 10mM MgCl2, 1mM DTT). Coverslips were attached to the slides, and these were visualized by fluorescence microscopy on an Ariol SL-50 system (Applied Imaging).
Supplementary Material
Acknowledgements
This work was funded by the Wellcome Trust. The authors would like to thank Mark Jobling for the kind gift of DNA, Mark Ross for insights into Xq28, John McCafferty and Will Howat for help with imaging, John Collins and Alison Coffey for PFGE guidance, and Philipp Holliger and Zoryana Oliynyk for advice on emulsion preparation.
References
- 1.Hogervorst FB, et al. Large genomic deletions and duplications in the BRCA1 gene identified by a novel quantitative method. Cancer Res. 2003;63:1449–53. [PubMed] [Google Scholar]
- 2.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 3.Stefansson H, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 4.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 5.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 6.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armour JA, Barton DE, Cockburn DJ, Taylor GR. The detection of large deletions or duplications in genomic DNA. Hum Mutat. 2002;20:325–37. doi: 10.1002/humu.10133. [DOI] [PubMed] [Google Scholar]
- 8.Schouten JP, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 10.Bunge S, et al. Homologous nonallelic recombinations between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur J Hum Genet. 1998;6:492–500. doi: 10.1038/sj.ejhg.5200213. [DOI] [PubMed] [Google Scholar]
- 11.Rossiter JP, et al. Factor VIII gene inversions causing severe hemophilia A originate almost exclusively in male germ cells. Hum Mol Genet. 1994;3:1035–9. doi: 10.1093/hmg/3.7.1035. [DOI] [PubMed] [Google Scholar]
- 12.Osborne LR, et al. A 1.5 million-base pair inversion polymorphism in families with Williams-Beuren syndrome. Nat Genet. 2001;29:321–5. doi: 10.1038/ng753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Q, Nozari G, Sommer SS. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood. 1998;92:1458–9. [PubMed] [Google Scholar]
- 14.Small K, Iber J, Warren ST. Emerin deletion reveals a common X-chromosome inversion mediated by inverted repeats. Nature Genetics. 1997;16:96–99. doi: 10.1038/ng0597-96. [DOI] [PubMed] [Google Scholar]
- 15.Antonarakis SE, et al. Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood. 1995;86:2206–12. [PubMed] [Google Scholar]
- 16.Jobling MA, et al. A selective difference between human Y-chromosomal DNA haplotypes. Curr. Biol. 1998;8:1391–1394. doi: 10.1016/s0960-9822(98)00020-7. [DOI] [PubMed] [Google Scholar]
- 17.Wetmur JG, et al. Molecular haplotyping by linking emulsion PCR: analysis of paraoxonase 1 haplotypes and phenotypes. Nucleic Acids Res. 2005;33:2615–9. doi: 10.1093/nar/gki556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yon J, Fried M. Precise gene fusion by PCR. Nucleic Acids Res. 17:4895–(1989. doi: 10.1093/nar/17.12.4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dressman D, et al. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–22. doi: 10.1073/pnas.1133470100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghadessy FJ, Ong JL, Holliger P. Directed evolution of polymerase function by compartmentalized self-replication. Proc Natl Acad Sci U S A. 2001;98:4552–7. doi: 10.1073/pnas.071052198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagnall RD, Giannelli F, Green PM. Polymorphism and hemophilia A causing inversions in distal Xq28: a complex picture. J Thromb Haemost. 2005;3:2598–9. doi: 10.1111/j.1538-7836.2005.01566.x. [DOI] [PubMed] [Google Scholar]
- 22.Ross MT, et al. The DNA sequence of the human X chromosome. Nature. 2005;434:325–37. doi: 10.1038/nature03440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogt PH, et al. Report of the third international workshop on Y chromosome mapping 1997. Cytogenet. Cell Genet. 1997;79:2–16. doi: 10.1159/000134680. [DOI] [PubMed] [Google Scholar]
- 24.Skaletsky H, et al. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–37. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- 25.Williams G. Mapping studies of the centromeric region of the human Y chromosome. University of Oxford; UK: 1998. D. Phil. thesis. [Google Scholar]
- 26.Cooper KF, Fisher RB, Tyler-Smith C. Structure of the sequences adjacent to the centromeric alphoid satellite DNA array on the human Y-chromosome. J. Mol. Biol. 1993;230:787–799. doi: 10.1006/jmbi.1993.1201. [DOI] [PubMed] [Google Scholar]
- 27.Mitra RD, et al. Digital genotyping and haplotyping with polymerase colonies. Proc Natl Acad Sci U S A. 2003;100:5926–31. doi: 10.1073/pnas.0936399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shendure J, et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science. 2005;309:1728–32. doi: 10.1126/science.1117389. [DOI] [PubMed] [Google Scholar]
- 29.Bailey JA, et al. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–17. doi: 10.1101/gr.187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.She X, et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature. 2004;431:927–30. doi: 10.1038/nature03062. [DOI] [PubMed] [Google Scholar]
- 31.Lupski JR. Hotspots of homologous recombination in the human genome: not all homologous sequences are equal. Genome Biology. 2004;5:242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feuk L, et al. Discovery of human inversion polymorphisms by comparative analysis of human and chimpanzee DNA sequence assemblies. PLoS Genet. 2005;1:e56. doi: 10.1371/journal.pgen.0010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broman KW, et al. In: Science and Statistics: A Festschrift for Terry Speed. Goldstein DR, editor. 2003. pp. 237–245. (IMS Lecture Notes - Monograph Series). [Google Scholar]
- 34.Shaffer LG, Lupski JR. Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet. 2000;34:297–329. doi: 10.1146/annurev.genet.34.1.297. [DOI] [PubMed] [Google Scholar]
- 35.Cohen D, Chumakov I, Weissenbach J. A first-generation physical map of the human genome. Nature. 1993;366:698–701. doi: 10.1038/366698a0. [DOI] [PubMed] [Google Scholar]
- 36.Brownstein BH, et al. Isolation of single-copy human genes from a library of yeast artificial chromosome clones. Science. 1989;244:1348–51. doi: 10.1126/science.2544027. [DOI] [PubMed] [Google Scholar]
- 37.Monaco AP, et al. Isolation of the human sex determining region from a Y-enriched yeast artificial chromosome library. Genomics. 1991;11:1049–53. doi: 10.1016/0888-7543(91)90031-9. [DOI] [PubMed] [Google Scholar]
- 38.Mitra RD, Church GM. In situ localized amplification and contact replication of many individual DNA molecules. Nucleic Acids Res. 1999;27:e34. doi: 10.1093/nar/27.24.e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.