

Abstract
The liganded vitamin D receptor is thought to play an important role in controlling cardiac function. Specifically this system has been implicated as playing an anti-hypertrophic role in the heart. Despite this, studies of the VDR in the heart have been limited in number and scope. In the present study we use a combination of real time PCR, Western blot analysis, immunofluorescence and transient transfection analysis to document the presence of functional VDR in both the myocytes and fibroblasts of the heart, as well as in the intact ventricular myocardium. We also demonstrate the presence of 1-α-hydroxylase and 24-hydroxylase in the heart, two enzymes involved in the synthesis and metabolism of 1,25 dihydroxyvitamin D (VD3). VDR is shown to interact directly with the human B-type natriuretic peptide (hBNP) gene promoter, a surrogate marker of the transcriptional response to hypertrophy. Of note, induction of myocyte hypertrophy either in vitro or in vivo leads to an increase in VDR mRNA and protein levels. Collectively, these findings suggest that the key components required for a functional VD3-dependent signaling system are present in the heart and that this putatively anti-hypertrophic system is amplified in the setting of cardiac hypertrophy.
Keywords: vitamin D, cardiac hypertrophy, nuclear receptors, BNP, cardiac myocyte
Introduction
Vitamin D is a secosteroid that functions as a ligand for a nuclear receptor (vitamin D receptor or VDR) to control the transcription of target genes in a positive or negative fashion 1. Vitamin D is either ingested in the diet or generated de novo through scission of cholesterol precursors in the skin by ultraviolet light 2. Vitamin D is activated through two sequential hydroxylation reactions. The first of these, a 25-hydroxylation, takes place largely in the liver to produce 25-hydroxyvitamin D. This molecule circulates bound to a plasma protein and is measured to assess the adequacy of vitamin D stores. The second hydroxylation, a 1-α-hydroxylation, takes place predominantly in the kidney to generate 1,25-dihydroxyvitamin D (VD3). It serves as the principal ligand for the VDR in the nucleus (and extranuclear compartment) of target cells. Another enzyme, the ubiquitously expressed 24-hydroxylase, is responsible for hydroxylating this ligand, leading to its inactivation and subsequent degradation.
Recent studies suggest that VD3, in addition to stimulating absorption of intestinal calcium and promoting mineralization of bone osteoid, may play an important role in controlling cardiac hypertrophy. We have shown that VD3, as well as a number of non-hypercalcemic analogues, act in both atrial and ventricular myocytes 3, 4 to inhibit the activation of phenotypic markers associated with hypertrophy in vitro. Endothelin (ET)-stimulated changes in fetal gene expression and promoter activity, cell size and protein synthesis 4, 5 are partially reversed by VD3 or its non-hypercalcemic analogues. Similar findings have been reported by others using the cultured cardiac HL-1 myocytes 6. In animal studies, vitamin D deficiency in Sprague-Dawley rats leads to both hypertension and cardiac hypertrophy 7 while treatment of Dahl salt sensitive rats with the vitamin D analogue paricalcitol reverses cardiac hypertrophy in that model 8. The VDR knockout mouse displays hypertension, cardiac hypertrophy with enlargement of individual myocytes and elevations in atrial natriuretic peptide (ANP) expression 9. However, the elevated blood pressure precludes assigning the liganded VDR a primary anti-hypertrophic role at the level of the cardiac myocyte. Although VDR has been identified microscopically 10 and functionally 11 in the heart, our understanding of the role of the liganded VDR in the maintenance of cardiac function remains incomplete.
While VDR is clearly present in the heart, we know little of the specific cell types where it is expressed nor of the functional activities associated with the liganding of these receptors. In the present manuscript we demonstrate the presence of VDR and VD3-dependent functional activity in both the myocytes and fibroblasts of the rat heart. We also demonstrate the presence of the 1-α-hydroxylase enzyme in both compartments. Noteworthy, application of a hypertrophic stimulus (ET in vitro and isoproterenol in vivo) leads to an increase in VDR gene expression with little or no effect on expression of the 1-α-hydroxylase. The results indicate that a functional vitamin D-dependent signaling system is present in both cardiac myocytes and fibroblasts and support a growing body of data defining an anti-hypertrophic role for this system in the heart.
Materials and Methods
Materials
ET was purchased from American Peptide (Sunnyvale, CA). Paricalcitol and active hectorol were gifts of Joel Melnick (Abbott Laboratories, Abbott Park, IL). VD3 and 25-hydroxyvitamin D3 were obtained from Calbiochem Inc. (La Jolla, CA). VDR antibody (sc-1008), procollagen I antibody (sc-8787), 24-hydroxylase antibody (a gift from M. Hewison) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (sc-32233) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). 25-hydroxyvitamin D 1-α-hydoxylase antibody (PC290) was from The Binding Site (Birmingham, UK).
Isoproterenol (ISO)-induced Cardiac Hypertrophy
Wistar rats purchased from Charles River Laboratories (Wilmington, MA) were anesthetized and subcutaneous osmotic minipumps were implanted into the dorsum of the neck. Individual rats received vehicle alone or ISO at the rate of 2.4 mg/kg/day for 7 days (n=8 each group). Animals were weighed and sacrificed by CO2 narcosis followed by bilateral thoracotomy. Left ventricles were isolated and weighed. Left ventricular weight/body weight (LVW/BW) and LVW/tibia length (LVW/TL) were measured as indices of cardiac enlargement. All experiments were approved by the Institutional Animal Care and Use Committee at UCSF.
Cell Culture
Ventricular myocytes and fibroblasts were prepared from 1–2 day-old neonatal Sprague-Dawley rats (Charles River Laboratories) as previously described 4. Both cell types were maintained in Dulbecco’s modified Eagle’s medium (DMEM) H-21 supplemented with 10% enriched calf serum (ECS; Gemini Bioproducts, West Sacramento, CA).
Immunoblotting
Myocytes and fibroblasts were changed from medium containing 10% ECS to medium containing 10% serum substitute (SS)12 for 24 hours. At that point, fresh media containing different concentrations of ET or VD3 was added. Cells were cultured for another 24–48 hours before being lysed in lysis buffer13. Total protein was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to membranes. The membranes were probed with an antibody directed against VDR, 24-hydroxylase, 25-hydroxyvitamin D, 1-α-hydroxylase, procollagen I or GAPDH. Blots were then incubated with horseradish peroxidase-conjugated secondary antibodies and visualized by ECL® reagent (Amersham Life Sciences, Arlington Heights, IL). When VDR competing peptide was included in the experiment, VDR primary antibody and competing peptide were mixed at the weight ratio of 1:5. Before being added to the membrane, the mixture was diluted to a VDR antibody concentration reflecting a 1:200 final dilution. Membranes from parallel experiments were incubated with VDR antibody alone at 1:200 dilution. For tissue analysis, the left ventricle sample was homogenized. Total protein was analyzed by Western blot with anti-VDR, anti-1-α-hydoxylase, anti-24-hydoxylase, or anti-GAPDH antibodies. Signal intensities were quantified using Kodak Scientific Imaging systems.
Immunofluorescence Analysis
Cardiac myocytes or fibroblasts were isolated as described above and plated on 4-well BD Biocoat culture slides coated with fibronectin (BD Bioscience, Franklin Lakes, NJ). Cells were incubated for 48 hours, fixed using Z-Fix (Anatech Ltd, Battle Creek, MI.), followed by permeabilization with 0.1% Triton X-100. The following primary antibodies were used: rabbit anti-VDR, mouse anti-α-actinin (Sigma Aldrich, St. Louis, MO.), and mouse anti-vimentin. Anti-mouse Alexa Fluor 488 (Invitrogen, Carlsbad, CA.) and anti-rabbit Cy3 (Invitrogen) secondary antibodies were used. The slides were then mounted with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Immunofluorescent images were acquired using an Olympus IX-70 inverted fluorescent microscope.
3H-Thymidine Incorporation
After serum starvation in 0.1% FBS for 12 hours, cells were treated with 10−8 mol/L VD3 for 36 hours. During the final 12 hours they were incubated with 3H-thymidine (4 μCi/mL) in thymidine-free Eagle’s minimal essential medium. 3H-thymidine incorporation assay was carried out as previously described 14.
Transfection and Luciferase Assay
Cardiac myocytes were transiently co-transfectedwith a hBNP luciferase reporter 15 and Renilla-Luc, VDR and/or RXR expression vectors 16 using lipofectin reagent (Invitrogen) as previously reported 17. Twenty-four hours following transfection, cells were incubated with VD3 or 25-hydroxyvitamin D for 48 hours with or without ET for the final 24 hours. At that time point, the cells were collected and lysed. Luciferase activity was measured using the Dual-Luciferase® kit (Promega, Madison, WI). BNP luciferase activity was normalized for Renilla luciferase activity.
DNA Immunopreciptation Assay
Cardiac myocytes were transfected with -198 hBNP-luciferase or pcDNA3. The media was then changed to serum free-media, and the cells were treated with 10−8 mol/L VD3 or vehicle and incubated for an additional 24 hours. At that time the cells were treated with 10−7 mol/L ET or vehicle for one hour. The DNA-IP assays were performed using a modification of published methodology 18. Briefly, after treatment, cells were fixed with 1% formaldehyde for 15 min at 37°C, neutralized with 0.125 mol/L glycine for 5 min at room temperature, washed, lysed and sonicated. The supernatant was pre-incubated with protein G sepharose beads, 2 μg salmon sperm DNA, 100 mg/ml bovine serum albumin and shaken at 4°C overnight. At that point, the supernatant was divided, either anti-VDR antibody or normal rabbit IgG was added, and the incubation was continued at 4°C overnight. Immunoprecipitates were collected, then sequentially washed as described. Bound material was then eluted with freshly made elution buffer (1% SDS and 0.1 mol/L NaHCO3). Crosslinking was reversed by heating the elutes at 65°C overnight, DNA was extracted and PCR was performed with the following primer pair: sense 5′-CCGGAATGTGGCTGATAAA-3′ and antisense primer present in the luciferase gene coding sequence 5′-CTTCCAGCGGATAGAATGGC-3′.
Total RNA isolation and Real-time PCR
Total RNA was isolated from cardiac myocytes and fibroblasts, and left ventricles with the RNeasy kit (Qiagen) and reverse transcribed into cDNA. Real-time PCR was carried out with rat ANP (Rn00561661_ml) and GAPDH (Rn99999916_sl) Taqman primers (Applied Biosystems, Foster City, CA) and rat VDR (sense: AGGACAACCGGCGACACT; antisense: CTGTACCTCCTCATCTGTCA), rat 1-α-hydroxylase (sense: CTGCAGAGACTGGGATCAGA; antisense: AAATCCTCCTCAGGCTTTCC) and GAPDH (sense: GACATGCCGCCTGGAGAAAC; antisense: AGCCCAGGATGCCCTTTAGT) SYBR Green primers. ANP, VDR and 1-α-hydroxylase mRNA levels were normalized to GAPDH mRNA expression. Real-time PCR was performed on the ABI Prism 7900HT (Applied Biosystems).
Statistical Analysis
Data was analyzed by one-way ANOVAusing Student-Newman-Keuls post-hoc test.
Results
VDR is expressed in the heart, both in the neonatal rat cardiac myocyte (Fig. 1A) and fibroblasts (Fig. 1B) that populate the myocardium. Immunoreactive protein of the appropriate size (~55 kilodaltons) was identified in extracts of both cell populations as well as inner medullary collecting duct (IMCD) cells, a population which has been shown to respond to vitamin D in previous studies 17. This immunoreactivity was competed effectively by antigenic VDR peptide (Fig. 1C). Interestingly, treatment of either the myocyte (Fig. 1A) or fibroblast (Fig. 1B) cultures with the pro-hypertrophic, vasoactive peptide ET resulted in a significant increase in levels of the VDR protein. A similar stimulation of VDR mRNA transcripts by ET was seen in both myocytes and fibroblasts (Fig. 1D). ET-stimulated VDR mRNA expression was seen as early as 4h, peaked at 14h and gradually declined at 24h in both types of cells (data not shown).
Figure 1.

ET increases VDR protein and mRNA expression in cardiac myocytes and fibroblasts. ET stimulates VDR protein expression in a dose-dependent fashion in cardiac myocytes (n=3–5) (A) and fibroblasts (n=4) (B). (C) VDR immunoreactivity was blocked by VDR competing peptide (CP). The protein from cardiac myocytes (CM), fibroblasts (F), heart tissue (H), and inner medullary collecting duct (IMCD) cells were transferred onto membranes and incubated with VDR antibody alone or with the antibody-competing peptide mixture. (D) ET (10−7 mol/L) increases VDR mRNA levels in cardiac myocytes and fibroblasts. Quiescent cells were treated with ET for 14 hours and total RNA was collected (n=3). *P<0.05, **P<0.001 vs. control.
In an effort to confirm the presence of VDR through independent methodology, we carried out immunocytochemistry of both myocytes and fibroblasts (Fig. 2) in culture. Myocytes were stained with antibody directed against α-actinin to demonstrate sarcomeric structure, as well the anti-VDR antibody, while fibroblasts were stained with anti-vimentin, an antibody which selectively stains these cells 19. In both cases the anti-VDR antibody identified immunoreactivity in the nuclei and to a lesser degree the cytoplasm, of the cells. The pattern of staining in the myocytes was coarser and aggregated in selected subnuclear locations while that in the fibroblasts was intense but homogenous throughout the nucleus.
Figure 2.
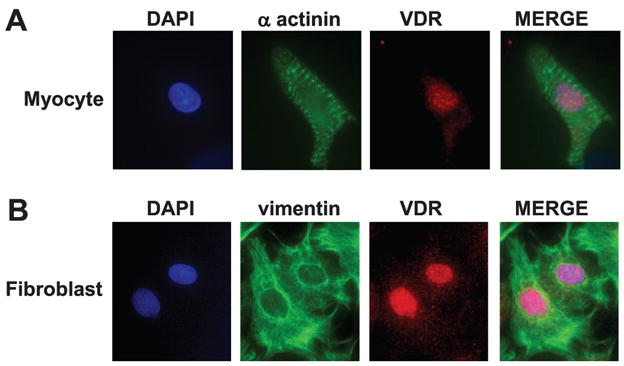
Immunocytochemistry of VDR in cardiac cells. VDR was visualized by immunofluorescence in cardiac myocytes (100×, oil immersion) and in cardiac fibroblasts (60×). (A) Cardiac myocytes were double stained with a polyclonal anti-VDR antibody, visualized with Cy3 (red) conjugated anti-rabbit antibody, and sarcomere-staining, polyclonal anti-α-actinin (green), visualized by AG488 conjugated anti-mouse antibody. VDR co-localized to the nucleus, stained with DAPI (blue). (B) Cardiac fibroblasts were double stained with anti- VDR, visualized with Cy3 conjugated anti-rabbit antibody, and anti-vimentin, visualized with AG488 conjugated anti-mouse antibody. VDR co-localized to the nucleus stained with DAPI (n=3).
To assess the effects of the liganded VDR on the non-myocyte population in the heart, we treated cells with VD3 for 36–48 hrs prior to assessing proliferative (i.e. 3H-thymidine incorporation) as well as synthetic effects (i.e. procollagen I levels) in these cultures. As shown in Fig. 3, VD3 treatment resulted in a significant reduction in both 3H-thymidine incorporation (panel A) as well as procollagen I levels (panel B), demonstrating that VD3 has important biological effects in these cells and suggesting that it may have anti-fibrotic activity in the interstitial compartment of the myocardium.
Figure 3.
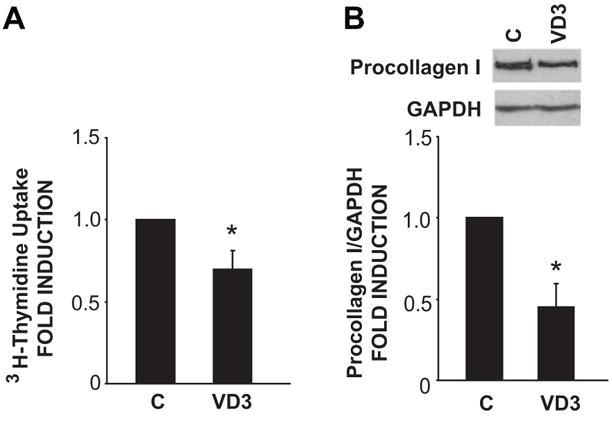
VD3 inhibits fibroblast proliferation and procollagen I synthesis. (A) VD3 reduces [3H]-thymidine incorporation in cultured ventricular fibroblasts. Values are normalized to controls in each experiment (n=4). (B) VD3 inhibits procollagen I synthesis in cultured ventricular fibroblasts. Cells were treated with VD3 for 48 hours in SS media. Procollagen I protein was detected by Western blot (n=4). Values are normalized to control in each experiment. *P<.05 vs. control.
To confirm the biological activity of VD3 in the myocyte cultures, we transfected a hBNP luciferase reporter into these cells. BNP gene expression and BNP promoter activity serve as surrogate markers of hypertrophy 13, 20, 21. We then treated them with VD3 or one of two non-hypercalcemic analogues of VD3 (i.e. paricalcitol or active hectorol), followed by the prohypertrophic peptide ET. As shown in Fig. 4A treatment of cells with 10−7 mol/L ET resulted in a greater than two-fold increase in promoter activity in these experiments. As shown in the same panel, pre-treatment with VD3 or the non-hypercalcemic analogues of VD3 resulted in a reduction in basal hBNP promoter activity and a significant reduction in the ET-dependent stimulation of promoter activity. In each case, the effective dose range was similar among the three VDR ligands. We also tested the ability of the VD3 precursor 25-hydroxyvitamin D to suppress basal hBNP promoter activity. As shown in Fig. 4B, 25-hydroxyvitamin D also led to a reduction in hBNP promoter activity, although, perhaps, with somewhat lower potency (compare effect of 10−10 mol/L VD3 in panel A with effect of 10−10 mol/L 25-hydroxyvitamin D in panel B). The ability of the precursor to mirror activity of VD3 implies that these cultures may have the requisite enzymatic machinery to convert 25-hydroxyvitamin D to VD3 (i.e. 1-α-hydroxylase). As shown in Fig. 4C, this was confirmed by Western blot analysis and real time PCR which demonstrated immunoreactive 1-α-hydroxylase protein and mRNA in the myocyte cultures. In this case, protein and mRNA levels were unaffected by ET treatment. Expression of 1-α-hydroxylase was also seen in cultured fibroblasts and was not changed following ET treatment (VD3 1.14+/− 0.17 fold induction vs. control, mean +/− SD, n=3).
Figure 4.

VD3, 25-hydroxyvitamin D, paracalcitol and active hectorol inhibit -1595 BNP promoter activity in cardiomyocytes. (A) -1595 BNP-Luc and Renilla-Luc were cotransfected with VDR/RXR expression vectors into the myocytes. Twenty-four hours following transfection, cells were treated with different doses of VD3, paracalcitol, active hectorol or vehicle for 48 hours and ET for 24 hours prior to collecting cells (n=3). BNP luciferase activity was normalized for Renilla luciferase activity. (B) Ventricular myocytes were transfected with -1595 BNP-Luc, Renilla-Luc and VDR/RXR expression vectors for 24 hours and then treated with different doses of 25-hydroxyvitamin D for 48 hours (n = 5). (C) 1-α-hydroxylase expressed in cardiomyocytes as assessed by Western blot and real time PCR (n=3). *P<0.05, **P<0.01 vs. Control; +P<0.01 vs. ET alone.
To determine whether the VDR was directly associated with the hBNP promoter, we carried out DNA immunoprecipitation analysis of myocyte cultures, previously transfected with the -198 hBNP-luciferase reporter, and then treated them with VD3 in the presence or absence of ET. As shown in Fig. 5A, this construct responded to VD3 treatment in a qualitatively similar fashion to the longer promoter construct used above. Promoter activity was reduced by ~30% by VD3 treatment alone and by >80% when the cells were co-transfected with VDR and retinoid X receptor (VDR’s heterodimeric partner) prior to VD3 treatment. Fig. 5B exhibits a direct association of VDR with the hBNP promoter. This association was undetectable at baseline but increased following treatment with the VDR ligand. Treatment with ET resulted in an increase in VDR association with the promoter and this was further amplified by addition of the VDR ligand.
Figure 5.

Liganded VDR inhibits hBNP promoter activity through direct interaction. (A) -198 hBNP-Luc and Renilla-Luc were co-transfected with or without VDR/RXR expression vectors into cardiac myocytes. Twenty four hours post-transfection, cells were treated with vehicle or VD3 for 48 hours. BNP/Renilla activities were measured and pooled data (n=3) are shown. *P<0.05, **P<0.01 vs. Control. (B) Myocytes were transfected with -198 hBNP luciferase or pcDNA3 plasmid. Cells were treated with VD3 followed by ET. DNA immunoprecipitation assay was carried out as described in Methods (n=3).
The ET-dependent induction of VDR was an unexpected finding and raised the possibility that the activation of hypertrophy by ET might be the driving force behind the increased expression of the putatively anti-hypertrophic VDR (i.e. a closed loop feedback system). To test this further, we investigated cardiac VDR expression in a rat model of myocardial hypertrophy following 7 days of continuous isoproterenol infusion. This infusion reliably generates significant increases in cardiac mass and hypertrophy-dependent gene expression (e.g. ANP) over this time interval 22. As shown in Fig. 6A, isoproterenol-treated rats experienced a significant increase in left ventricular size when normalized either to body weight or tibial length. This was accompanied by a significant increase in ANP gene expression, demonstrated in Fig. 6B, reflecting activation of the fetal gene expression in the hypertrophied myocardium 23. We also observed a significant increment in VDR mRNA and protein levels in the isoproterenol-treated animals (Fig. 6C) but no increase in the expression of 1-α-hydroxylase, 24-hydroxylase or GAPDH protein (Fig. 6D) or the 1-α-hydroxylase mRNA (1.04 +/− 0.25 fold induction relative to control; mean +/− SD, n=4).
Figure 6.

Isoproterenol (ISO) induces cardiac hypertrophy and stimulates VDR expression in the Wistar rat. (A) Rats were infused with ISO or vehicle for 7 days. Animals were euthanized and body weight (BW), left ventricular weight (LVW), and tibial length (LT) were measured (n=7). (B) Left ventricular samples from control and ISO-infused rats were homogenized in extraction buffer and total RNA was isolated. ANP mRNA/GAPDH mRNA levels were measured by real-time PCR (n=5). (C) VDR expression assessed by Western blot and real time PCR. Representative immunoblot and pooled data (n=7) for VDR protein and mRNA measurements are presented. (D) 24-hydroxylase and 1-α-hydroxylase expression assessed by Western blot (n=7). **P<0.01 vs. control.
Discussion
The key findings of the present report are 1) the demonstration of VDR in both cardiac myocytes and fibroblasts, as well as ventricular myocardium, using a combination of biochemical, immunofluorescence and functional assays, 2) the demonstration of 1-α-hydroxylase in the same sources and 24-hydroxylase protein in the intact heart, 3) the demonstration that VDR has the capacity to bind directly to the BNP gene promoter and 4) the finding that activation of myocyte hypertrophy, either in vitro or in vivo, is associated with a significant increase in VDR expression.
Previous studies have suggested a role of the VD3/VDR system in the control of cardiac function. Our group has shown previously that VD3 as well as less hypercalcemic analogues of VD3 demonstrate anti-hypertrophic activity in cultured neonatal rat cardiac myocytes 5, 14, 16. The VDR gene knockout mouse displays significant cardiac hypertrophy and activation of hypertrophy-dependent gene expression, which can be linked directly to increased myocyte size 9. Moreover, Bodyak et al. recently demonstrated that the VD3 analogue paricalcitol reduces cardiac hypertrophy, without affecting blood pressure, in the Dahl Salt Sensitive rat 8. A variety of clinical studies lend additional support. Vitamin D deficiency in early childhood is associated with significant cardiomyopathy and congestive heart failure 24. Several independent studies in adult patients have linked congestive heart failure to reduced levels of circulating 25-hydroxyvitamin D levels 25. Finally, Park and coworkers reported that low circulating levels of VD3 in patients with chronic renal failure on dialysis are linked to the presence of ventricular hypertrophy 26. Remarkably, treatment of these patients with exogenous VD3 resulted in amelioration of the hypertrophy.
Immunoreactive VDR has been described in human heart and cultured cardiac HL-1 cells 6. To our knowledge, ours is the first report which demonstrates VDR expression in both the myocyte and cardiac fibroblast and the first to document hypertrophy-dependent stimulation of VDR expression. The presence of VDR in both myocytes and fibroblasts implies that the potential exists for widespread effects of the liganded receptor in the heart (e.g. suppression of myocyte hypertrophy and interstitial fibrosis). It also engenders a need to be cautious in interpreting effects of vitamin D on the heart in the whole animal. Such effects could result from direct interactions with the cardiac myocyte, indirect hemodynamic effects (e.g. reductions in blood pressure) or indirect paracrine effects resulting from ligand-dependent interactions with neighboring fibroblasts.
The presence of 1-α-hydroxylase in myocytes and fibroblasts implies that the heart has the capacity to synthesize the bioactive VD3 metabolite independent of its production in the kidney, using circulating plasma 25-hydroxyvitamin D as substrate 27. Similar 1-α-hydroxylase immunoreactivity has been demonstrated in inflammatory cells 28, breast 29, colon 30 and prostate cancer cells 31. This is an important consideration since it places cardiac production of VD3 outside those regulatory controls that govern renal production of the ligand. In the intact heart, local tissue VD3 levels would then be a function of endogenous synthesis (i.e. cardiac 1-α-hydroxylase activity and circulating plasma 25-hydroxyvitamin D levels), delivery of circulating plasma VD3 to the heart and degradation of VD3 (e.g. through 24-hydroxylase activity).
The fact that VDR binds directly to the hBNP gene promoter indicates that at least some of the effects of VD3 operate directly at the level of target gene expression in suppressing the hypertrophic phenotype, rather than indirectly (e.g. through alterations in hemodynamics or inhibition of inflammatory markers associated with hypertrophy). Following treatment with ET, VDR binds to the hBNP promoter in the absence of exogenous ligand, a finding compatible with either ligand-independent activity of this receptor 32 or alternatively, low level production of VD3 in these cultures. In any case, addition of exogenous ligand clearly amplified association of VDR with the promoter. Interestingly, treatment with ET resulted in increased VDR binding, likely reflecting the hypertrophy-dependent increment in VDR available for binding in these cultures.
Finally, the induction of VDR expression with hypertrophic stimuli in vitro or in vivo might be predicted to have important implications in terms of the regulation of the hypertrophic process. In both the neonatal rat myocyte, a traditional model for studying myocyte hypertrophy in vitro 23, and the in vivo model of isoproterenol-induced hypertrophy 22, we observed an induction of VDR gene expression. The induction appears to be somewhat selective for the VDR in the nuclear receptor gene family. Other family members, most notably the peroxisome proliferators activator receptor α and thyroid hormone receptor are down regulated in the cardiac myocyte with the development of hypertrophy 33, 34. We and others have suggested that VD3 or its less hypercalcemic analogues suppress myocyte hypertrophy in vivo 8 and in vitro 4. Amplification of VDR expression in hypertrophy suggests that the myocyte is attempting to close a negative feedback loop that would serve to dampen the magnitude of hypertrophic growth. Interference with the activation of this feedback mechanism would be predicted to amplify the hypertrophic response and conceivably, favor progression to cardiac decompensation and heart failure.
Perspective
Recent studies suggest that vitamin D may play an important physiological role in controlling cardiac hypertrophy. The present study adds support for this hypothesis in demonstrating that the key components of the vitamin D-dependent signaling system (i.e. VDR, 1-α-hydroxylase and 24-hydroxylase) are present in cardiac myocytes and fibroblasts. It also demonstrates for the first time that levels of the VDR are upregulated during hypertrophy, implying activation of a counter-regulatory mechanism to control growth in the hypertrophied heart. Collectively, these data suggest that the use of vitamin D, vitamin D analogues or drugs that modulate vitamin D metabolism could be beneficial in the management of disorders associated with cardiac hypertrophy.
Acknowledgments
The authors are grateful to Martin Hewison for providing the anti-24 hydroxylase antibody used in these studies.
Sources of Funding. Supported by HL45637 to D.G.G., F32 HL086158 to D.J.G. from NIH, AHA 0825140F to W.N. from the American Heart Association and a grant from Abbott Laboratories.
Footnotes
Disclosures: None.
References
- 1.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 2.Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. doi: 10.1056/NEJMra070553. [DOI] [PubMed] [Google Scholar]
- 3.Li Q, Gardner DG. Negative regulation of the human atrial natriuretic peptide gene by 1,25-dihydroxyvitamin D3. J Biol Chem. 1994;269:4934–4939. [PubMed] [Google Scholar]
- 4.Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest. 1996;97:1577–1588. doi: 10.1172/JCI118582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J, Garami M, Cao L, Li Q, Gardner DG. 1,25(OH)2D3 suppresses expression and secretion of atrial natriuretic peptide from cardiac myocytes. Am J Physiol. 1995;268:E1108–1113. doi: 10.1152/ajpendo.1995.268.6.E1108. [DOI] [PubMed] [Google Scholar]
- 6.Nibbelink KA, Tishkoff DX, Hershey SD, Rahman A, Simpson RU. 1,25(OH)2-vitamin D3 actions on cell proliferation, size, gene expression, and receptor localization, in the HL-1 cardiac myocyte. J Steroid Biochem Mol Biol. 2007;103:533–537. doi: 10.1016/j.jsbmb.2006.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weishaar RE, Simpson RU. Vitamin D3 and cardiovascular function in rats. J Clin Invest. 1987;79:1706–1712. doi: 10.1172/JCI113010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bodyak N, Ayus JC, Achinger S, Shivalingappa V, Ke Q, Chen YS, Rigor DL, Stillman I, Tamez H, Kroeger PE, Wu-Wong RR, Karumanchi SA, Thadhani R, Kang PM. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci U S A. 2007;104:16810–16815. doi: 10.1073/pnas.0611202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang W, Kong J, Chen S, Cao LP, Qiao G, Zheng W, Liu W, Li X, Gardner DG, Li YC. Cardiac hypertrophy in vitamin D receptor knockout mice: role of the systemic and cardiac renin-angiotensin systems. Am J Physiol Endocrinol Metab. 2005;288:E125–132. doi: 10.1152/ajpendo.00224.2004. [DOI] [PubMed] [Google Scholar]
- 10.Tishkoff DX, Nibbelink KA, Holmberg KH, Dandu L, Simpson RU. Functional vitamin D receptor (VDR) in the t-tubules of cardiac myocytes: VDR knockout cardiomyocyte contractility. Endocrinology. 2008;149:558–564. doi: 10.1210/en.2007-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walters MR, Wicker DC, Riggle PC. 1,25-Dihydroxyvitamin D3 receptors identified in the rat heart. J Mol Cell Cardiol. 1986;18:67–72. doi: 10.1016/s0022-2828(86)80983-x. [DOI] [PubMed] [Google Scholar]
- 12.Bauer RF, Arthur LO, Fine DL. Propagation of mouse mammary tumor cell lines and production of mouse mammary tumor virus in a serum-free medium. In Vitro. 1976;12:558–563. doi: 10.1007/BF02797439. [DOI] [PubMed] [Google Scholar]
- 13.Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339:321–328. doi: 10.1056/NEJM199807303390507. [DOI] [PubMed] [Google Scholar]
- 14.Chen S, Gardner DG. Retinoic acid uses divergent mechanisms to activate or suppress mitogenesis in rat aortic smooth muscle cells. J Clin Invest. 1998;102:653–662. doi: 10.1172/JCI3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LaPointe MC, Wu G, Garami M, Yang XP, Gardner DG. Tissue-specific expression of the human brain natriuretic peptide gene in cardiac myocytes. Hypertension. 1996;27:715–722. doi: 10.1161/01.hyp.27.3.715. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Costa CH, Nakamura K, Ribeiro RC, Gardner DG. Vitamin D-dependent suppression of human atrial natriuretic peptide gene promoter activity requires heterodimer assembly. J Biol Chem. 1999;274:11260–11266. doi: 10.1074/jbc.274.16.11260. [DOI] [PubMed] [Google Scholar]
- 17.Chen S, Olsen K, Grigsby C, Gardner DG. Vitamin D activates type A natriuretic peptide receptor gene transcription in inner medullary collecting duct cells. Kidney Int. 2007;72:300–306. doi: 10.1038/sj.ki.5002274. [DOI] [PubMed] [Google Scholar]
- 18.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000;14:2314–2329. doi: 10.1101/gad.827900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grimm D, Huber M, Jabusch HC, Shakibaei M, Fredersdorf S, Paul M, Riegger GA, Kromer EP. Extracellular matrix proteins in cardiac fibroblasts derived from rat hearts with chronic pressure overload: effects of beta-receptor blockade. J Mol Cell Cardiol. 2001;33:487–501. doi: 10.1006/jmcc.2000.1321. [DOI] [PubMed] [Google Scholar]
- 20.Gardner RS, Chong KS, Morton JJ, McDonagh TA. A change in N-terminal pro-brain natriuretic peptide is predictive of outcome in patients with advanced heart failure. Eur J Heart Fail. 2007;9:266–271. doi: 10.1016/j.ejheart.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Gardner DG. Natriuretic peptides: markers or modulators of cardiac hypertrophy? Trends Endocrinol Metab. 2003;14:411–416. doi: 10.1016/s1043-2760(03)00113-9. [DOI] [PubMed] [Google Scholar]
- 22.Boluyt MO, Bing OH, Lakatta EG. The ageing spontaneously hypertensive rat as a model of the transition from stable compensated hypertrophy to heart failure. Eur Heart J. 1995;16(Suppl N):19–30. doi: 10.1093/eurheartj/16.suppl_n.19. [DOI] [PubMed] [Google Scholar]
- 23.Chien KR, Knowlton KU, Zhu H, Chien S. Regulation of cardiac gene expression during myocardial growth and hypertrophy: molecular studies of an adaptive physiologic response. Faseb J. 1991;5:3037–3046. doi: 10.1096/fasebj.5.15.1835945. [DOI] [PubMed] [Google Scholar]
- 24.Uysal S, Kalayci AG, Baysal K. Cardiac functions in children with vitamin D deficiency rickets. Pediatr Cardiol. 1999;20:283–286. doi: 10.1007/s002469900464. [DOI] [PubMed] [Google Scholar]
- 25.Zittermann A, Schleithoff SS, Koerfer R. Vitamin D insufficiency in congestive heart failure: why and what to do about it? Heart Fail Rev. 2006;11:25–33. doi: 10.1007/s10741-006-9190-8. [DOI] [PubMed] [Google Scholar]
- 26.Park CW, Oh YS, Shin YS, Kim CM, Kim YS, Kim SY, Choi EJ, Chang YS, Bang BK. Intravenous calcitriol regresses myocardial hypertrophy in hemodialysis patients with secondary hyperparathyroidism. Am J Kidney Dis. 1999;33:73–81. doi: 10.1016/s0272-6386(99)70260-x. [DOI] [PubMed] [Google Scholar]
- 27.Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, Hewison M. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86:888–894. doi: 10.1210/jcem.86.2.7220. [DOI] [PubMed] [Google Scholar]
- 28.Fritsche J, Mondal K, Ehrnsperger A, Andreesen R, Kreutz M. Regulation of 25-hydroxyvitamin D3-1 alpha-hydroxylase and production of 1 alpha, 25-dihydroxyvitamin D3 by human dendritic cells. Blood. 2003;102:3314–3316. doi: 10.1182/blood-2002-11-3521. [DOI] [PubMed] [Google Scholar]
- 29.Friedrich M, Rafi L, Mitschele T, Tilgen W, Schmidt W, Reichrath J. Analysis of the vitamin D system in cervical carcinomas, breast cancer and ovarian cancer. Recent Results Cancer Res. 2003;164:239–246. doi: 10.1007/978-3-642-55580-0_17. [DOI] [PubMed] [Google Scholar]
- 30.Cross HS, Bareis P, Hofer H, Bischof MG, Bajna E, Kriwanek S, Bonner E, Peterlik M. 25-Hydroxyvitamin D(3)-1alpha-hydroxylase and vitamin D receptor gene expression in human colonic mucosa is elevated during early cancerogenesis. Steroids. 2001;66:287–292. doi: 10.1016/s0039-128x(00)00153-7. [DOI] [PubMed] [Google Scholar]
- 31.Chen TC, Wang L, Whitlatch LW, Flanagan JN, Holick MF. Prostatic 25-hydroxyvitamin D-1alpha-hydroxylase and its implication in prostate cancer. J Cell Biochem. 2003;88:315–322. doi: 10.1002/jcb.10342. [DOI] [PubMed] [Google Scholar]
- 32.Ross TK, Darwish HM, Moss VE, DeLuca HF. Vitamin D-influenced gene expression via a ligand-independent, receptor-DNA complex intermediate. Proc Natl Acad Sci U S A. 1993;90:9257–9260. doi: 10.1073/pnas.90.20.9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinugawa K, Yonekura K, Ribeiro RC, Eto Y, Aoyagi T, Baxter JD, Camacho SA, Bristow MR, Long CS, Simpson PC. Regulation of thyroid hormone receptor isoforms in physiological and pathological cardiac hypertrophy. Circ Res. 2001;89:591–598. doi: 10.1161/hh1901.096706. [DOI] [PubMed] [Google Scholar]
- 34.Karbowska J, Kochan Z, Smolenski RT. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell Mol Biol Lett. 2003;8:49–53. [PubMed] [Google Scholar]