

Abstract
Over the last 20 years, the number of publications outlining the advances in design strategies, growing techniques, and characterization of cocrystals has continued to increase significantly within the crystal engineering field. However, only within the last decade have cocrystals found their place in pharmaceuticals, primarily due to their ability to alter physicochemical properties without compromising the structural integrity of the active pharmaceutical ingredient (API) and thus, possibly, the bioactivity. This review article will highlight and discuss the advances made over the last 10 years pertaining to physical and chemical property improvements through pharmaceutical cocrystalline materials and, hopefully, draw closer the fields of crystal engineering and pharmaceutical sciences.
Short abstract
This review article will highlight and discuss the advances made over the last 10 years pertaining to physical and chemical property improvements through pharmaceutical cocrystals and, hopefully, draw closer the fields of crystal engineering and pharmaceutical sciences.
I. Introduction
A. Common Pharmaceutical Strategies for Altering API Properties
It is well-known that crystalline materials obtain their fundamental physical properties from the molecular arrangement within the solid, and altering the placement and/or interactions between these molecules can, and usually does, have a direct impact on the properties of the particular solid.(1) Currently, solid-state chemists call upon a variety of different strategies when attempting to alter the chemical and physical solid-state properties of APIs, namely, the formation of salts, polymorphs, hydrates, solvates, and cocrystals, as illustrated in Figure 1.
Figure 1.

Pictures displaying the more common solid-state strategies and their respected components.
Currently, salt formation is one of the primary solid-state approaches used to modify the physical properties of APIs, and it is estimated that over half of the medicines on the market are administered as salts.(2) However, a major limitation within this approach is that the API must possess a suitable (basic or acidic) ionizable site. In comparison, cocrystals (multicomponent assemblies held together by freely reversible, noncovalent interactions) offer a different pathway, where any API regardless of acidic, basic, or ionizable groups, could potentially be cocrystallized. This aspect helps complement existing methods by reintroducing molecules that had limited pharmaceutical profiles based on their nonionizable functional groups. In addition, the number of potential nontoxic cocrystal formers (or coformers) that can be incorporated into a cocrystalline reaction is numerous.(3)
It should be made clear that no one particular strategy offers a solution for property enhancement of all APIs. Each API must be examined and evaluated on a case-by-case basis in terms of molecular structure and desired final properties. For this purpose, general guidelines for rational cocrystal design through supramolecular synthesis will be highlighted first.
B. Cocrystals and Salts
To date, a universal and agreeable definition of what constitutes a cocrystal is still unavailable. Within the academic literature, various parameters have been applied to the definition of what is and is not considered a cocrystal (Table 1); however, one broad commonality that is agreed upon is that all cocrystals are crystalline materials comprised of at least two different components (or commonly called multicomponent crystals). Now, one’s opinion as to what constitutes a “component” can be dramatically different, for example, solid, liquid, or gas and/or neutral or ionic species, etc., and this is usually where the differences in definitions arise. Furthermore, the use of “pharmaceutical cocrystal” is commonplace and usually applied when an API is one of the molecules in the multicomponent crystal.
Table 1. Definitions of a Cocrystal.
| author | definition of a cocrystal | ref |
|---|---|---|
| Stahly, G. P. | “a molecular complex that contains two or more different molecules in the same crystal lattice” | (4) |
| Nangia, A. | “multi-component solid-state assemblies of two or more compounds held together by any type or combination of intermolecular interactions” | (5) |
| Childs, S. L. | “crystalline material made up of two or more components, usually in a stoichiometric ratio, each component being an atom, ionic compound, or molecule” | (6) |
| Aakeröy, C. B. | “compounds constructed from discrete neutral molecular species...all solids containing ions, including complex transition-metal ions, are excluded” “made from reactants that are solids at ambient conditions” “structurally homogeneous crystalline material that contains two or more neutral building blocks that are present in definite stoichiometric amounts” | (7) |
| Bond, A. | “synonym for multi-component molecular crystal” | (8) |
| Jones, W. | “a crystalline complex of two or more neutral molecular constituents bound together in the crystal lattice through noncovalent interactions, often including hydrogen bonding” | (9) |
| Zaworotko, M. J. | “are formed between a molecular or ionic API and a co-crystal former that is a solid under ambient conditions” | (10) |
Even though there are limitations with the cocrystal definitions currently found in the literature, we do not see it necessary to complicate the existing debate by generating yet another definition for what constitutes a cocrystal. In this review, the cocrystalline examples presented herein will possess the following criteria:
(1) An API, neutral (example 1, Figure 2), or ionic form (example 2, Figure 2, or a zwitterion), along with a neutral coformer, held together through noncovalent, freely reversible interactions,
Figure 2.

Possible multicomponent systems: cocrystals, salt cocrystals, and salts along with their respective solvate/hydrate forms.
(2) a coformer, which may or may not be pharmaceutically acceptable,
(3) and at least one measured physicochemical property.
A pictorial description of possible multicomponent systems, including cocrystals,(11) salt cocrystals,12,13 and salts along with their respective hydrates and solvates are displayed in Figure 2. When necessary for property or structural comparison, examples of pharmaceutical salts (example 3, Figure 2) will be introduced and discussed. The examples used throughout the paper are representative of the literature reported for the physicochemical properties measured for pharmaceutical cocrystals, but it is not an exhaustive list of all the cocrystal studies available.
C. Crystal Engineering of Pharmaceutical Cocrystals
The number of publications and reviews detailing the topics of crystal engineering and supramolecular synthesis of API-based cocrystals is extensive and continuing to grow.(14) More specifically, researchers usually highlight themes pertaining to functional group compatibility (synthons), for example, acid/N-heterocycle, acid/amide, phenol/N-heterocycle, and/or cocrystal growth strategies such as evaporation,(15) solid-state grinding,(16) sonication,(17) and melting.(18) To complement these topics, we would like to briefly mention the initial steps that should be considered (before a reaction is ever conducted!) when attempting to maximize the experimental effectiveness of generating cocrystalline materials.
In the beginning, an evaluation of the API should be carried out, including but not limited to, number and arrangement of hydrogen bond donors and acceptors,7,19 salt forming ability (pKa’s),(20) conformational flexibility, and solubility requirements.(21) Usually APIs that are rigid, highly symmetrical, possess strong nonbonded interactions, and low molecular weight are more apt to cocrystallize with additional components (coformers or counter-molecules).(4) Frequently, suitable coformers are selected based on hydrogen bonding rules,(22) probable molecular recognition events,(23) and toxicological profiles;2a,24 however, one should not be limited to just these criteria because cocrystals could easily be missed. Finally, as mentioned above, a variety of methods exist for preparing cocrystals. To date, predicting whether or not a cocrystallization reaction will be successful is not yet possible,(25) and thus reactions must be carried out experimentally under varied conditions with different techniques to find available cocrystals.
D. Cocrystal Characterization
Single crystal X-ray diffraction is the preferred characterization technique in determining whether a cocrystalline material has been generated; however, suitable X-ray quality crystals cannot always be produced. Additionally, even if single crystals can be grown of sufficient size and quality, the exact location of the hydrogen atom (determination if proton-transfer has occurred from the acid to the base or not) may be ambiguous.20c,26 Thus, it is advantageous to utilize a variety of solid-state, spectroscopic techniques (Raman, infrared, and solid-state NMR) when attempting to characterize potentially new cocrystalline materials.(27)
Such an exercise was carried out when single-crystal X-ray crystallography and 15N solid-state cross-polarization magic angle spinning (CPMAS) NMR spectroscopy were used to complement one another in the determination of hydrogen bonding interactions and the extent of proton-transfer between a heterocyclic-containing API and a variety of dicarboxylic acids.(28) Three acid−base complexes were obtained and characterized: a sesquisuccinate, a dimalonate, and a dimaleate. Through single-crystal X-ray analysis, measured hydrogen bond distances were used to characterize the materials as one cocrystal (sesquisuccinate), one mixed ionic and zwitterionic complex (dimalonate), and one disalt (dimaleate). These results were confirmed by comparing the 15N chemical shifts of each species to those of the free base. Small shifts, in comparison to the free base, were observed for the sesquisuccinate cocrystal, while the largest shifts, due to complete protonation, were seen from the dimaleate salt. In addition, short contact time CPMAS NMR experiments were used to further characterize the dimalonate and dimaleate complexes as a mixed ionic species and a disalt. For example, if a nitrogen atom had a proton attached to it, then a signal would appear; thus the disalt (dimaleate) displayed two additional peaks in comparison to the free base, while the mixed ionic species (dimalonate) only showed one new peak. The results from the 15N solid-state CPMAS NMR spectroscopy along with single-crystal X-ray crystallography proved sufficient to successfully identify each new form as either a cocrystal, salt, or mixed ionic complex.
Infrared spectroscopy can be a very powerful tool in detecting cocrystal formation, especially when a carboxylic acid is used as a coformer and/or when a neutral O−H···N hydrogen bond is formed between an acid and a base. Distinct differences, within the IR spectra, can be observed between a neutral carboxylic acid moiety and a carboxylate anion. A neutral carboxylate (-COOH) displays a strong C=O stretching band around 1700 cm−1 and a weaker C−O stretch around 1200 cm−1, while a carboxylate anion (-COO−), due to resonance, displays a single C−O stretch in the fingerprint region of 1000−1400 cm−1. Additionally, if a neutral intermolecular O−H···N hydrogen bond has formed between the components, then two broad stretches around 2450 and 1950 cm−1 will be observed.(29) Observations about the state of the carboxylic moiety (neutral or ionic) can also be verified through measuring the C−O and C=O bond distances from the single-crystal X-ray data. A typical C=O bond distance is around 1.2 Å, while the C−O bond distance is around 1.3 Å; however, if deprotonation has occurred then the resonance stabilized C−O bond distances will be very similar.20c,30
Outlined above are a number of different characterization techniques used to help distinguish between cocrystals and salts, and it should be noted that in some cases differentiation between the two may be difficult.(20c) It should also be pointed out that although salts and cocrystals often possess different properties (see sections on stability () and solubility ()), these issues from a development standpoint may not be influential as long as the process can be monitored and closely controlled. However, for intellectual property (IP) rights and regulatory issues, differentiating between a cocrystal and a salt can be important, as discussed in .
II. Physicochemical Property Review
The physical and chemical properties of a cocrystal need to be investigated in the same manner as any other solid form in order to determine developability into a marketed dosage form.(31) Physicochemical properties, such as crystallinity, melting point, solubility, dissolution, and stability, are important when moving a new compound, such as a cocrystal, through early development. The information from these studies can be used to prioritize the available forms, and a flowchart can help organize the process for solid form selection. An example of a flowchart that could be used to choose the best cocrystal candidate is given in Figure 3. It combines a number of properties inherent to drug development; however, it should be used as a guide only since every compound will have its own challenges, and the properties in the flowchart will be unique to the situation at hand.
Figure 3.

Example flowchart for choosing a cocrystal candidate.
The primary focus of this article is centered on highlighting cases where the physicochemical properties of an API have been adjusted through the formation of API-based cocrystals. The properties and questions are
| Melting point: | Does the thermal behavior (melting point) of a cocrystal change with respect to the individual components and can the melting points be estimated within a series of cocrystals? |
| Stability: | Can physical and chemical stability be enhanced upon cocrystallization of an API? |
| Solubility: | Can the solubility of an API be altered by modifying it into a cocrystal? |
| Dissolution: | Are dissolution rates improved by cocrystalline compounds in comparison to the individual APIs? |
| Bioavailability: | Can the bioavailability of an API be improved using cocrystals? |
Related topics, such as scale-up, polymorphism, intellectual property, and lifecycle management are also discussed for the development of pharmaceutical cocrystals.
A. Melting Point
The melting point is a fundamental physical property, which is determined by the temperature at which the solid phase is at equilibrium with the liquid phase. Since melting point is a thermodynamic process where the free energy of transition is equal to zero, the value is determined by the ratio of change in the enthalpy of fusion over the change in the entropy of fusion.(32) If available, differential scanning calorimetry (DSC) is the preferred technique for obtaining comprehensive melting point data, over a standard melting point apparatus or Kofler method, because additional thermal data such as the enthalpy of fusion can be determined. For example, the melting point and heat of fusion, both determined from DSC, are necessary when attempting to characterize a polymorphic pair of compounds as monotropic or enantiotropic.(33)
It is standard practice to determine the melting point of a compound as a means of characterization or purity identification; however, within pharmaceutical sciences, the melting point is also very valuable due to its correlations to aqueous solubility and vapor pressure.(34) In fact, the melting point has been directly correlated to the Log of solubility, although assumptions pertaining to the entropy of fusion had to be drawn.(35) Thus, being able to determine the melting point of a particular API before it was synthesized would be very beneficial in order to tailor its aqueous solubility toward a particular function. Unfortunately, correlations relating chemical structure directly to melting point data remain elusive.(36) Given the number of factors contributing to the melting point of a crystalline solid including, but not limited to, the molecular arrangement within the crystal lattice, molecular symmetry, intermolecular interactions, and conformational degrees of freedom for a molecule, one clearly sees the difficulties in attempting to draw strict comparisons from molecular structure to crystalline lattice energy to melting point. The situation only becomes more complex when observing multicomponent systems because each component has its own characteristic properties and those can influence the environment (and intermolecular interactions) around its neighbors. In this section we will examine the thermal behavior of cocrystals in which one component is an API, although findings and trends should be translatable to all cocrystalline materials.
One literature example compares the melting points of 10 cocrystals to the API AMG517 and their respective coformers.(37) Each of the cocrystals displayed a melting point that fell between the melting point of AMG517 and their coformer. A plot was generated using the melting points of the cocrystals and coformers, which displayed a direct proportionality between the two (Figure 4). A correlation coefficient of 0.7849 was determined, meaning that 78% of the variability of melting point of the cocrystal can be attributed to the variability of the coformers melting point. These data show that within the set of AMG517 cocrystals the melting point can typically be tuned according to which coformer is chosen; for example, if a higher melting cocrystal is desired, then a higher melting coformer should be selected and vice versa.
Figure 4.

Melting points of AMG517 cocrystals and their respected coformers (left); melting onset of coformer versus cocrystal (right). Modified table and graphic from ref (37). Copyright 2008 American Chemical Society.
We compiled a larger survey based on reported cocrystal melting points, and these were compared with the melting points of the coformer and API. The purpose was to determine if any correlation can be drawn, with regard to where the melting point of the cocrystal falls: higher, lower, or in between that of the API and coformer. The data are summarized in Table 2. It should be noted that this exercise is not taking into account stoichiometry of components, solvation or hydration, polymorphism, and/or types of intermolecular interactions present within the crystalline lattice.
Table 2. Melting Points of APIs, Coformers, and Their Respective Cocrystals.
| APIa | API MPb (°C) | coformer | coformer MP (°C) | cocrystal MP (°C) | rangec | ref |
|---|---|---|---|---|---|---|
| AMG517 | 230 | (37) | ||||
| AMG517 | trans-cinnamic acid | 133 | 204 | M | ||
| AMG517 | 2,5-dihydroxybenzoic acid | 205 | 229 | M | ||
| AMG517 | 2-hydroxybenzoic acid | 61 | 130 | M | ||
| AMG517 | glutaric acid | 97 | 153 | M | ||
| AMG517 | glycolic acid | 78 | 141 | M | ||
| AMG517 | sorbic acid | 134 | 150 | M | ||
| AMG517 | trans-2-hexanoic acid | 34 | 127 | M | ||
| AMG517 | l-(+)-lactic acid | 46 | 138 | M | ||
| AMG517 | benzoic acid | 122 | 146 | M | ||
| AMG517 | l-(+)-tartaric acid | 171 | 198 | M | ||
| aspirin | 133−135 | (40) | ||||
| aspirin | 4,4′-bipyridine | 111−114 | 91−96 | L | ||
| carbamazepine | 190−192 | (21b) | ||||
| carbamazepine | succinic acid | 187 | 189 | M | ||
| carbamazepine | benzoic acid | 122 | 113 | L | ||
| carbamazepine | ketoglutaric acid, Form A | 115 | 140 | M | ||
| carbamazepine | ketoglutaric acid, Form B | 115 | 134 | M | ||
| carbamazepine | maleic acid | 137 | 158 | M | ||
| carbamazepine | glutaric acid | 98 | 125 | M | ||
| carbamazepine | malonic acid, Form A | 136 | 143 | M | ||
| carbamazepine | oxalic acid | 189 | 158 | L | ||
| carbamazepine | adipic acid | 152 | 137 | L | ||
| carbamazepine | (+)-camphoric acid | 186 | 156 | L | ||
| carbamazepine | 4-hydroxybenzoic acid, Form A | 213 | 172 | L | ||
| carbamazepine | 4-hydroxybenzoic acid, Form C | 213 | 170 | L | ||
| carbamazepine | salicylic acid | 159 | 159 | |||
| carbamazepine | 1-hydroxy-2-naphthoic acid | 192 | 174 | L | ||
| carbamazepine | dl-tartaric acid, Form A | 205 | 170 | L | ||
| carbamazepine | l-tartaric acid | 169 | 160 | L | ||
| carbamazepine | glycolic acid | 149 | 139 | L | ||
| carbamazepine | fumaric acid, Form B | 287 | 189 | L | ||
| carbamazepine | saccharin | 225−227 | 174 | L | ||
| carbamazepine | nicotinamide | 128 | 151−161 | M | (38) | |
| carbamazepine | benzoquinone | 113−115 | 170 | M | ||
| carbamazepine | terephthalaldehyde | 114−116 | 124 | M | ||
| carbamazepine | trimesic acid | >300 | 278 (dec) | M | ||
| carbamazepine | 5-nitroisophthalic acid | 259−261 | 190 (dec) | |||
| chlorzoxazone | 192 | (74) | ||||
| chlorzoxazone | 2,4-dihydroxybenozoic acid, Form 1 | 213 | 179 | L | ||
| chlorzoxazone | 2,4-dihydroxybenozoic acid, Form 1I | 213 | 177 | L | ||
| chlorzoxazone | 4-hydroxybenzoic acid | 213-214 | 182 | L | ||
| ethyl-paraben | 116 | (39) | ||||
| ethyl-paraben | nicotinamide | 128 | 107 | L | ||
| flurbiprofen | 113−114 | (40) | ||||
| flurbiprofen | 4,4′-bipyridine | 111−114 | 155−160 | H | ||
| flurbiprofen | trans-1,2-bis(4-pyridyl)ethylene | 150−153 | 153−158 | H | ||
| fluoxetine HCl | 157 | (12) | ||||
| fluoxetine HCl | benzoic acid | 122 | 132 | M | ||
| fluoxetine HCl | succinic acid | 185−187 | 134 | L | ||
| fluoxetine HCl | fumaric acid | ∼287 | 161 | M | ||
| ibuprofen | 77−78 | (40) | ||||
| ibuprofen | 4,4′-bipyridine | 111−114 | 117−120 | H | ||
| indomethacin | 162 | (46) | ||||
| indomethacin | saccharin | 225−227 | 220 | M | ||
| norfloxacin | 221 | (41) | ||||
| norfloxacin | isonicotinamide | 155−157 | 180−185 | M | ||
| Pfizer 1 | 167 | (28) | ||||
| Pfizer 1 | succinic acid | 185−187 | 143 | L | ||
| piroxicam | 198−200 | (64) | ||||
| piroxicam | saccharin | 225−227 | 220 | M | ||
| Purdue Pharma 1 | 206 | (48) | ||||
| Purdue Pharma 1 | glutaric acid | 98 | 142 | M |
API, active pharmaceutical ingredient.
MP, melting point.
Range: cocrystal has H-higher melting points than API and coformer, M-melting point in between that of the API and coformer, L-lower melting point than API or coformer.
Within the survey 50 cocrystalline samples were analyzed; 26/50 (51%) cocrystals had melting points between those of the API and coformer, while 19/50 (39%) were lower than either the API or coformer, only 3/50 (6%) were higher, and 2/50 (4%) had the same melting point as either the API or coformer. These statistics clearly show that the melting point of an API can be altered through forming cocrystals, and the outcome will usually be a product having a melting point that is in between that of the API and coformer or lower than the API or coformer. Thus, if a lower melting solid is necessary and covalent modifications to the API cannot be achieved, then cocrystallizing the API is a viable pathway.
Within a homologous set of cocrystals (either the API or coformer is kept constant), where single crystal X-ray structures have been determined, comparisons could potentially be drawn between the intermolecular interactions and/or crystal packing existing within the lattice and the thermal behavior of the sample. This knowledge could be beneficial when attempting to determine the behavior of a particular material from its three-dimensional structure. In one example, a set of dipyridyl coformers are cocrystallized with ibuprofen, flurbiprofen, and aspirin producing four cocrystals: (ibuprofen)2(4,4′-bipyridine), (flurbiprofen)2(4,4′-bipyridine), (flurbiprofen)2(trans-1,2-bis(4-pyridyl)ethylene), and (aspirin)2(4,4′-bipyridine).(40) In all cases the compounds crystallize in a 2:1 API/coformer ratio through heteromeric O−H···N hydrogen bonds; however, the overall packing arrangements of the 2:1 supermolecules are different. The first three examples listed all take on a herringbone packing arrangement and have melting points higher than either the API or coformer, while the last example is a channel structure and possess a melting point considerably lower than either of the two reactants. Although this study is on a small number of samples, it shows the impact that crystal packing has on physical properties such as melting point.
As mentioned earlier, correlations have been drawn between a compound’s melting point and its Log of solubility; however, the literature remains sparse when making comparisons to cocrystals. Only one study was found where cocrystals had their melting points and aqueous solubilities determined and compared. In the AMG517 study, the authors note that after a correlation analysis of the solubility parameters, the highest interdependence was the Log of solubility (Log Smax) versus the melting point.(37) A correlation plot of Log Smax versus cocrystal melting point, of the nine AMG517 cocrystals, indicated a 55% correlation of the variability in Log Smax to variability in melting point of the cocrystal (Figure 5). This study, albeit on a homologous set of cocrystals, provides a foundation for attempting to compare the factors influencing a cocrystals melting point and its solubility.
Figure 5.

Solubility and Log Smax values for AMG517 cocrystals (left); Smax as a function of cocrystal melting point (right). Modified table and graphic from ref (37). Copyright 2008 American Chemical Society.
Recently, a second cocrystallization study was conducted on a series of molecules with structures similar to AMG-517 along with a number of structurally diverse coformers.(42) Comparisons were made between the melting points of the cocrystals and coformers as well as between the melting points and the Log solubility values. The conclusions showed low correlations between the melting points of the cocrystals to the coformers and no correlation between the melting points of the cocrystals and the Log solubility. These findings clearly stress the difficulties when attempting to draw lines between series of molecules with different structures, especially as it pertains to solubility and melting point.
Melting point is an important consideration during development. High melting points are usually desirable, but may contribute to poor solubility and are as troublesome as low melting points, which can hinder processing, drying, and stability. Correlation of melting points with other development parameters is an ongoing area of study, and the multicomponent nature of the cocrystals will add another level of complexity to these analyses.
B. Stability
Stability is a heavily studied parameter during the development of a new chemical entity. Different types of stability need to be considered depending on the structure and characteristics of the molecule. Chemical and physical stability data are commonly obtained at accelerated stability conditions to determine developability and shelf life.(43) Water uptake is included from a handling and packaging point of view. The amount of water present can also lead to form changes, degradation, and worse if the effect of the water uptake is not investigated early in the process.(44) Thermal stress studies are also incorporated, and extra work may be warranted for hydrates or thermally labile materials. In the case of cocrystals and salts, solution stability may be a factor due to dissociation of the material resulting in precipitation of the less soluble parent compound or a less soluble form (such as a hydrate in aqueous media). This section will review a variety of stability data reported for cocrystals.
1. Relative Humidity Stress
As with other solid forms, changes over a wide relative humidity (RH) range are a key consideration when developing a cocrystal.44,45 Automated moisture sorption/desorption studies are commonly performed to determine problem areas and to provide direction for more detailed studies when the need arises. X-ray powder diffraction (XRPD) data collected on the solid at the end of the moisture balance experiment provides information on the final form, but not necessarily on any form conversions that may have occurred during the experiment. Significant moisture uptake during the course of the experiment may warrant longer exposure at a specific relative humidity using a relative humidity chamber and subsequent analysis of the sample after equilibration.
Limited water sorption/desorption data were found in the literature for cocrystals. One example for a 1:1 indomethacin/saccharin cocrystal showed minimal uptake (<0.05% water) up to 95% RH.(46) The data suggested no solid-state transformation, although XRPD data of the sample after the moisture balance experiment were not reported. The second example for a 1:1 AMG 517/sorbic acid cocrystal again showed a minimal uptake of 0.7% water at 90% RH.(47) The XRPD data showed only the presence of the AMG 517 sorbic acid cocrystal after the experiment indicating that the initial form was obtained after the sorption/desorption cycle. These studies showed that relative humidity is not a major concern for these cocrystals; however, longer term studies would be advisible to determine the effects of water under more equilibrium conditions. Another system involved a glutaric acid cocrystal of 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide.(48) This material sorbed less than 0.08% up to 95% RH over repeated sorption/desorption cycles. A long-term study at 40 °C/75% RH for 2 months also showed no form change by XRPD and DSC as well as no significant increase in degradation by HPLC. This combined information indicates that the cocrystal will likely be stable under normal processing and storage conditions, which helps reduce risk during development.
Longer term cocrystal stability studies with respect to RH have been reported for a small number of cocrystal systems and a range of parameters have been used. As mentioned above for the glutaric acid cocrystal of 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide, 40 °C/75% RH is a common condition to use during development based on ICH guidelines.(43) A pharmaceutical cocrystal of a monophosphate salt with phosphoric acid resulted in no detectable form change or degradation after 8 weeks of storage at 40 °C/75% RH.(13b) A study on nine AMG-517 cocrystals showed no change in the XRPD patterns after one month at 40 °C/75% RH.(37) These types of results certainly suggest adequate physical stability for development; however, each system needs to be evaluated on an individual basis.
Other studies target a range of RH conditions, such as 0, 43, 75, and 98% RH over a period of time (Table 3). In a study of caffeine cocrystals,(49) the focus was to produce a cocrystal that was physically stable at all RH conditions, unlike caffeine, which is known to readily form a hydrate upon exposure to water or water vapor.(50) Six cocrystals were successfully produced: 2:1 caffeine/oxalic acid, 2:1 caffeine/malonic acid, 2:1 caffeine/maleic acid, 1:1 caffeine/maleic acid, and two forms of 1:1 caffeine/glutaric acid. Samples were placed at four RH conditions and analyzed after 1, 3, and 7 days and 7 weeks. The 2:1 caffeine/oxalic acid sample was found to be stable at all RH conditions through 7 weeks. The other cocrystals exhibited behavior similar to caffeine, and one case was found to be worse than caffeine with evidence of dissociation and conversion to caffeine monohydrate. It was noted that the strongest acid used (oxalic acid) resulted in the most stable cocrystal, while the weakest acid (glutaric acid) produced the least stable cocrystal. It is not known at this point if this is a general trend for a homologous series or is isolated to certain compounds. The same set of RH conditions (0, 43, 75, and 98% RH) were used with four cocrystals of theophylline (2:1 theophylline/oxalic acid, 1:1 theophylline/malonic acid, 1:1 theophylline/maleic acid, and 1:1 theophylline/glutaric acid).(51) The 2:1 theophylline/oxalic acid showed better physical stability than theophylline alone, and the remaining cocrystals exhibited the same stability as theophylline after 7 weeks. Dissociation of the materials was again reported, resulting in theophylline monohydrate.
Table 3. Cocrystal Stability With Respect to Relative Humidity (RH)a.
| observed relative humidity stability |
||||||
|---|---|---|---|---|---|---|
| APIs | condition (% RH) | 1 day | 3 days | 1 week | 7 weeks | pKa values of APIs and acids |
| caffeine | 0−75 | √ | √ | √ | √ | 3.6 |
| 98 | X | X | X | X | ||
| theophylline | 0−75 | √ | √ | √ | √ | 8.6 |
| 98 | X | X | X | X | ||
| Cocrystals | ||||||
| C:oxalic acid | 0−98 | √ | √ | √ | √ | 1.27, 4.28 |
| T:oxalic acid | 0−98 | √ | √ | √ | √ | |
| C:malonic acid | 0−75 | √ | √ | √ | √ | 2.83, 5.69 |
| 98 |
√ | √ | √ | X | ||
| T:malonic acid | 0−75 | √ | √ | √ | √ | |
| 98 |
X | X | X | X | ||
| C:maleic acid Form I | 0−75 | √ | √ | √ | √ | 1.83, 6.07 |
| 98 |
X | X | X | X | ||
| C:maleic acid Form II | 0−75 | √ | √ | √ | √ | |
| 98 |
X | X | X | X | ||
| T:maleic acid | 0−75 | √ | √ | √ | √ | |
| 98 |
X | X | X | X | ||
| C:glutaric acid Form I | 0 | √ | √ | √ | √ | 4.31, 5.41 |
| 43 |
√ | X | X | X | ||
| 75−98 |
X | X | X | X | ||
| C:glutaric acid Form II | 0−75 | √ | √ | √ | √ | |
| 98 |
√ | √ | X | X | ||
| T:glutaric acid | 0−75 | √ | √ | √ | √ | |
| 98 | √ | X | X | X | ||
Humidity chambers were set at 0, 43, 75, and 98%, while observations were made at durations of 1 day, 3 days, 1 week, and 7 weeks. C = Caffeine, T = theophylline. The symbol √ represents that the co-crystal was stable at that condition and time. The symbol X represents that the cocrystal exhibited physical instability at that condition and time. Modified table from refs (49) and (51).
While cocrystal hydrate formation was investigated by grinding, a 1:1 theophylline/citric acid cocrystal was compared with a 1:1 caffeine/citric acid cocrystal.(52) The materials were exposed to a variety of RH conditions including 98% RH. It was found that the caffeine/citric acid cocrystal converted to caffeine hydrate upon exposure to 98% RH for 7 days, similar to the other caffeine cocrystals.(49) The theophylline/citric acid cocrystal converted to a cocrystal hydrate upon exposure to 98% RH for 3 days. The theophylline/citric acid cocrystal hydrate was found to be stable and did not change form after 7 days at 0, 43, 75, and 98% RH. This stable hydrate could be an acceptable form for development based on the RH stability results. This example illustrates that cocrystals can exhibit hydrate formation in the solid state, similar to other crystalline forms, and should be investigated during the development process.
The RH stability of chiral and racemic cocrystals has also been reported for caffeine and theophylline using malic and tartaric acids.(53) Samples were exposed to 43, 75, and 98% RH for up to 7 days. The racemic cocrystal was found to be more resistant to hydration than the single enantiomer form in all the systems studied. It is suggested that the stability of the theophylline cocrystals resulted from intermolecular (molecular packing) and intramolecular (conformational strain) factors.
A cocrystal physical stability study can also be compared to the API material to assess any improvement in this property. For a 1:1 carbamazepine/saccharin cocrystal, a physical stability study was performed at 25 °C/60%RH, 40 °C/ambient RH, and 40 °C/75% RH for two weeks.(54) Carbamazepine (Form III) was also included, and the samples were analyzed by XRPD to determine any form change. Both materials appeared to be physically stable under the accelerated conditions used. This study showed comparable physical stability between the two materials, and similar studies could be used to determine any improvement that a cocrystal may impart for a difficult-to-develop compound.
This section illustrates that RH stress of cocrystals is an important step in evaluating these materials for development. RH conditions should be chosen based on the information needed for development of the compound. Hydrate formation as well as dissociation need to be understood early in the process in order to prevent problems during later phases.
2. Thermal Stress
High temperature stress is another common condition used to determine chemical and physical stability based on accelerated stability conditions.(43) Very few reports discuss thermal stress experiments on cocrystals. For the cocrystal of a monophosphate salt with phosphoric acid an 8-week exposure at 60 °C resulted in no detectable degradation or form change.(13b) Two other studies discuss DSC data and the effect of temperature on stability. Paracetamol cocrystals of 4,4′-bipyridine, 1,4-dioxane, N-methylmorpholine, morpholine, N,N-dimethylpiperazine, and piperazine were analyzed by DSC.(55) The paracetamol/4,4′-bipyridine sample was the only cocrystal that did not lose the guest upon heating and exhibit a melting endotherm corresponding to the monoclinic form of paracetamol. Additional data were not included to confirm the conversion, but these types of studies provide valuable information that should be explored in relation to other processes, such as drying and accelerated stability.
Another DSC and high-temperature study was performed on nonstoichiometric cocrystals of l-883555, a phosphodiesterase-IV inhibitor, and l-tartaric acid, with stoichiometries ranging from 0.3:1 to 0.9:1.(56) DSC curves showed a single melting endotherm for stoichiometries close to 0.5:1 tartaric acid/l-883555. Additional thermal events were observed at the other stoichiometries. When the two complexes with the higher stoichiometries (0.7:1 and 0.89:1 tartaric acid/l-883555) were heated above the first observed DSC endotherm, titration and NMR analysis showed values equivalent to the 0.5:1 stoichiometry, indicating a loss of some of the tartaric acid. TG-IR data confirmed CO2 evolution due to tartaric acid. These data indicated that tartaric acid incorporated in excess of 0.5:1 was bound at a different site, and this binding was more labile when compared to the first type of binding occurring within the structure. Variable temperature XRPD also showed a significant contraction of the lattice when heated above the first endotherm, which was attributed to the loss of the tartaric acid. It was hypothesized that the acid content above the 0.5:1 stoichiometry was likely occupying channels within the crystal, which was in agreement with the multiple binding modes established from spectroscopic data. The 0.5:1 stoichiometry was found to be the most thermally stable and was chosen for development based on these data and bioavailability data.
These limited reports show that heating studies can provide valuable information about physical and chemical stability. Information on elevated temperature transitions can give clues about possible problems in long-term stabilities and can provide guidance on drying steps. These results can be translated into a better understanding of the solid-state system and can help develop more robust compounds and processes.
3. Chemical Stability
Chemical stability is commonly investigated early in the development of a new compound and during formulation studies in order to minimize any chemical degradation that may occur. Accelerated stability conditions, such as 40 °C/75% RH and 60 °C/75% RH, are commonly used for early studies on solid materials. Very few reports of chemical stability of cocrystals were found when reviewing the literature. In one example, a pharmaceutical cocrystal of a monophosphate salt with phosphoric acid was reported to have no detectable degradation after 8 weeks of storage at 40 °C/75% RH and 60 °C.(13b) Samples of a glutaric acid cocrystal of 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide were placed at the same conditions for 2 months, and HPLC impurity analysis did not show any significant increases in known degradants.(48)
A direct comparison of a cocrystal to the parent API can provide important information for development. A 1:1 carbamazepine:saccharin cocrystal was compared to carbamazepine (Form III) in a chemical stability study using temperature (5, 40, and 60 °C at ambient humidity) and elevated RH conditions (25 °C/60% RH and 40 °C/75% RH) over 2 months.(54) Degradation was not observed for either material at the elevated temperatures; however, both materials showed similar degradation patterns under the second set of conditions. In this case, a significant improvement in chemical stability was not needed, and the cocrystal proved to be as stable as the marketed carbamazepine form under these conditions.
Although few reports exist, assessing chemical stability of cocrystals is important in understanding the developability of these materials. Assessing chemical stability in the presence of excipients for early simple dosage form development will also provide important information. It is possible that cocrystals may exhibit superior chemical stability when this is an issue with the parent compound, and crystal engineering techniques may provide approaches to prevent known degradation pathways.
4. Solution Stability
Solution stability for this discussion is defined as the ability of the cocrystal components to stay in solution and not readily crystallize. Solution stability can be an important parameter to assess during development, not only for solutions or suspensions, but also for solid dosage forms that will dissolve in the GI tract. A variety of vehicles can be used, including water, simulated gastric fluid (SGF), simulated intestinal fluid (SIF), formulation vehicles, and buffered solutions. In many instances, these experiments can be coupled with solubility or dissolution experiments to get a more complete picture of the behavior and the solid form remaining at the end of the experiment. Because dissociation of a cocrystal can occur, solution stability can be a key consideration for development. However, the results should always be weighed with other properties and needs.
Studies of cocrystals in water can give an indication of possible dissociation and precipitation of another form such as a hydrate. In the study of caffeine cocrystals,(49) the 2:1 caffeine/oxalic acid cocrystal was found to be stable at all relative humidities up to 98% RH for 7 weeks. In order to further test the stability, the material was slurried in water at ambient temperature for 2 days. No change in physical form was observed, demonstrating the stability of the material. In order to determine the form present in aqueous solutions of a 1:1 carbamazepine/saccharin cocrystal, the material was slurried with equal parts carbamazepine dihydrate and saccharin in solution.(54) After 24 h, only the cocrystal was evident based on XRPD data and the carbamazepine dihydrate was not detected. This is likely based on the concentration of the coformer present, which has been investigated for a number of systems.(57) In another example, a carbamazepine cocrystal screen resulting in 24 unique solid phases using 18 coformers studied the formation of carbamazepine dihydrate when the cocrystals were slurried in water for 20−48 h.(21b) Of the 20 cocrystals studied, seven maintained the cocrystal structure and the rest converted to carbamazepine dihydrate. The aqueous solubility of the coformer appeared to be an important parameter for the dihydrate formation. It was noted by the authors that cocrystals containing coformers with relatively high aqueous solubility converted to the dihydrate, while the coformers with relatively low water solubility remained as the cocrystal. Other studies would be needed to determine if this is a general trend or specific to this system. It should be noted that many of the carbamazepine cocrystals were successfully produced directly from water by increasing the concentration of the coformer based on the ternary solubility phase diagrams. The phase diagrams show the conditions where the cocrystal is supersaturated in solution and is the favored solid phase for crystallization; using these conditions will avoid crystallization of the individual components.
Simulated gastric or intestinal fluids (SGF and SIF, respectively) are also common systems for assessing solubility and dissolution rate. For AMG 517 cocrystals, solubility was determined in fasted simulated intestinal fluid (FaSIF) at 25 °C for 24 h.37,47 Four of the cocrystals (trans-cinnamic acid, 2,5-hydroxybenzoic acid, 2-hydroxycaproic acid, benzoic acid) did not dissociate, and XRPD data at the end of the experiment showed no form conversion. It was noted that the maximum solubility of three of these cocrystals was relatively low (<3 μg/mL); therefore, the 24 h experiment may not have allowed enough time for conversion to occur. The benzoic acid cocrystal is the only one that exhibited a higher solubility (21 μg/mL) and did not dissociate. The benzoic acid cocrystal exhibited a bell shaped curve indicative of form conversion, but no conversion was observed by XRPD. DSC data indicated that the benzoic acid was no longer in the crystal, and the concentration of benzoic acid in solution increased over time. One explanation suggested that a free base form, similar in structure to the cocrystal, was formed and was not readily differentiated by XRPD. The remaining five cocrystals (glutaric acid, glycolic acid, sorbic acid, trans-2-hexanoic acid, and l-(+)-lactic acid) dissociated during the experiment and crystalline AMG 517 resulted. The solubility (Smax) of these cocrystals ranged from 9 to 17 μg/mL, which was higher than AMG517 and three other cocrystals (trans-cinnamic acid, 2,5-hydroxybenzoic acid, and 2-hydroxycaproic acid), but less than the solubility of the benzoic acid cocrystal. Dissolution experiments of a 1:1 carbamazepine/saccharin cocrystal in SGF used sieve fractions to investigate the effect of particle size on dissolution.(54) Sieved cocrystal fractions of particles less than 150 μm showed the fastest initial dissolution, and dissolution was essentially complete when the particles were less than 500 μm. Significantly slower dissolution was observed for particles larger than 500 μm. The large particles (500 μm to greater than 1 mm) were found to contain a mixture of the cocrystal and carbamazepine dihydrate by XRPD when exposed to SGF overnight. Conversion to the dihydrate on the surface of the particles was likely responsible for the slower dissolution rate observed for the sample compared to the smaller particle size sample; however, further work on the particle size and conversion to the dihydrate was not conducted.
Buffered solutions are commonly used during development and solubility data at various pH conditions can be valuable. A solubility study at pH 7.4 and two buffer strengths (60 and 200 mM) was conducted on an indomethacin/saccharin cocrystal and compared to the γ-form of indomethacin.(46) At the 60 mM strength, the cocrystal dissolved immediately and was followed by a drop in pH to 6.8 and precipitation of an amorphous material with traces of the α-form of indomethacin. By raising the buffer strength to 200 mM, the cocrystal rapidly dissolved and remained in solution for several hours, giving a 50× increase in solubility over the indomethacin γ-form. XRPD data of the remaining solid did not show peaks indicative of the cocrystal indicating that the phase in equilibrium is not the crystalline indomethacin−saccharin cocrystal. Possible salt formation with the buffer components was not addressed and further characterization of the remaining solid was not reported. Studies are continuing to investigate the ionization of the ligand, complexation phenomenon, and solution chemistry of the system.
Intrinsic dissolution and solubility experiments in water can also yield information on solution stability. Intrinsic dissolution studies of a glutaric acid cocrystal of 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide showed very minor conversion of the cocrystal to the parent compound after 90 min in water at 37 °C.(48) Discs left in the dissolution apparatus for 24 h under the same conditions showed full conversion to the parent compound. In another study, the aqueous solubility of fluoxetine hydrochloride cocrystals made from benzoic acid, fumaric acid, and succinic acid were measured.(12) The benzoic and fumaric acid cocrystals were stable for several hours and XRPD data of the solids at the end of the experiment confirmed that the form had not changed. The succinic acid cocrystal showed a full conversion to fluoxetine hydrochloride after the experiment, in agreement with the powder dissolution data. The powder dissolution profile for this material exhibited high solubility initially with a subsequent decrease in solubility due to the recrystallization of the fluoxetine hydrochloride. Succinic acid exhibited the highest solubility of the three coformers and was the only cocrystal to dissociate. This is similar to what was found in the carbamazepine cocrystal study where cocrystals produced from coformers with the highest solubility tended to dissociate.(21b)
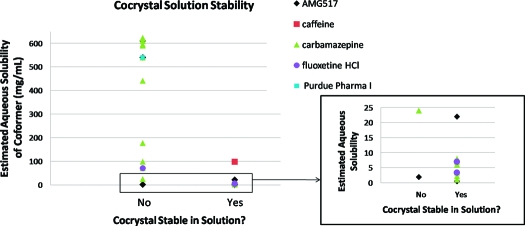
Solution stability results found in the literature were compiled in Table 4 and compared to the aqueous solubility of the coformer used. A total of five APIs were included, but the majority of the data were on carbamazepine. The aqueous solubilities of the coformers were primarily taken from the Handbook of Aqueous Solubility Data(58) when available; solubility data for coformers that were not found were estimated in SSCI’s laboratory. The solution stability studies reported were in water except for one study, which was FaSIF (AMG517). This study was included since it is water based, but it is not a direct comparison of the cocrystal and coformer solubilities. The solution stability and coformer solubility data are compared in Figure 6. This limited data set seems to show that in general, cocrystals comprised of low aqueous solubility coformers do appear to have better solution stability, and especially with coformer solubility values less than 10 mg/mL. One exception is the AMG517/sorbic acid cocrystal which exhibits poor solution stability with a low solubility coformer. This could be due to the use of FaSIF for the studies rather than water where other factors may be contributing to the solubility. It is interesting to note that this cocrystal exhibited an intermediate solubility for this series in FaSIF (12 μg/mL); therefore, cocrystal solubility was not able to explain the deviation in this case. Although this limited data set is interesting, further work would be needed to determine if low solubility coformers result in cocrystals that are more stable in solution. The effect of the cocrystal solubility versus the coformer solubility would also be an interesting factor when investigating solution stability.
Table 4. Cocrystal Solution Stability Compared to Coformer Solubility.
| aqueous solubility of coformer (mg/mL)a | cocrystal stable in solutione | coformer | API | ref |
|---|---|---|---|---|
| 0.5 | Y | trans-cinnamic acid | AMG517 | (37) |
| 1b | Y | 1-hydroxy-2-naphthoic acid | carbamazepine | (21b) |
| 1.9 | N | sorbic acid | AMG517 | (47) |
| 2.2 | Y | salicylic acid | carbamazepine | (21b) |
| 3.4 | Y | benzoic acid | carbamazepine | (21b) |
| 3.4 | Y | benzoic acid | fluoxetine HCl | (12) |
| 3.4 | Y | benzoic acid | AMG517 | (37) |
| 6 | Y | 4-hydroxybenzoic acid | carbamazepine | (21b) |
| 7 | Y | fumaric acid | carbamazepine | (21b) |
| 7 | Y | fumaric acid | fluoxetine HCl | (12) |
| 7.6 | Y | (+)-camphoric acid | carbamazepine | (21b) |
| 22 | Y | 2,5-dihydroxybenzoic acid | AMG517 | (37) |
| 24 | N | adipic acid | carbamazepine | (21b) |
| 71 | N | succinic acid | carbamazepine | (21b) |
| 71 | N | succinic acid | fluoxetine HCl | (12) |
| 89c | Y | 2-hydroxycaproic acid | AMG517 | (37) |
| 98 | Y | oxalic acid | caffeine | (49) |
| 98 | N | oxalic acid | carbamazepine | (21b) |
| 177 | N | dl-tartaric acid | carbamazepine | (21b) |
| 441 | N | maleic acid | carbamazepine | (21b) |
| 540 | N | l-malic acid | carbamazepine | (21b) |
| 540 | N | glutaric acid | AMG517 | (37) |
| 540 | N | glutaric acid | carbamazepine | (21b) |
| 540 | N | glutaric acid | Purdue Pharma 1 | (48) |
| 562c | N | l-(+)-lactic acid | AMG517 | (37) |
| 590 | N | dl-malic acid | carbamazepine | (21b) |
| 595 | N | l-tartaric acid | carbamazepine | (21b) |
| 610 | N | glycolic acid | carbamazepine | (21b) |
| 610 | N | glycolic acid | AMG517 | (37) |
| 620b | N | ketoglutaric acid | carbamazepine | (21b) |
| 623 | N | malonic acid | carbamazepine | (21b) |
| NAd | N | trans-2-hexanoic acid | AMG517 | (37) |
Solubility values reported are at 25 °C when temperature data were available. Data are taken from the Handbook of Aqueous Solubility Data(58) unless otherwise noted.
Solubility values estimated in our laboratory.
Solubility values calculated using ALOGPS.
NA, not available.
Solution stability performed in water for all compounds except AMG517, which was determined in FaSIF.
Figure 6.

Plot of the solution stability of pharmaceutical cocrystals versus the estimated aqueous solubility of the corresponding coformer.
The dissociation of cocrystals and the resulting solid produced presents a possible complication during development of a drug product. A celecoxib/nicotinamide cocrystal was studied in various formulation vehicles (1−10% sodium dodecylsulfate (SDS) and polyvinylpyrrolidone (PVP)) in order to understand the solid form that may result upon contact with simulated GI fluids.(59) Four crystal forms of celecoxib have been reported with Form III being the marketed form.(60) Dissolution and solubility of the celecoxib/nicotinamide cocrystal were dependent on the medium and have been attributed to the dissociation of the cocrystal and recrystallization of celecoxib Form I and III. It was found that the addition of surfactants affected this conversion. When a mixture of SDS with a 1/1 celecoxib:nicotinamide/PVP was exposed to 0.01 N HCl the following was found to occur: (a) a portion of the drug becomes amorphous, presumably stabilized by PVP; (b) the crystalline material is present as a metastable celecoxib Form IV; and (c) the crystalline material present is mostly nonaggregated and has a very small particle size. This resulted in a shelf stable formulation that dissolved rapidly, which could lead to faster absorption, although animal bioavailability studies were not performed. This study highlights the importance of investigating formulation approaches for cocrystals and understanding the possible conversions that could take place upon contact with simulated GI fluids and ultimately in animal or human subjects.
Solution stability is important not only for solution or suspension products, but can also impact other properties such as solubility and dissolution data for other types of dosage forms. The effects of dissociation, recrystallization, ionization, complexation, and solution chemistry are all areas that will need further work to better understand how cocrystals will behave in various media.
C. Solubility
One of the main reasons to investigate cocrystals is to increase the solubility of a poorly soluble compound. For neutral molecules, cocrystals can certainly expand the solid forms possible for development. For a free acid or free base, both salts and cocrystals can be used to improve the solubility; however, it is not always straightforward to determine whether a salt or a cocrystal has been formed(4) and a variety of techniques may be needed to understand the system.
There are a number of considerations when discussing solubility data. The first is equilibrium versus kinetic (or apparent) solubility measurements. Kinetic solubility values are approximate values usually based on one measurement at one time point. Unless preliminary experiments have been performed, it is not known if equilibrium has been reached in the time frame used. For equilibrium solubility, a number of time points and measurements are taken to ensure that the solution has reached equilibrium as evidenced by a plateau in the concentration data. This is sometimes called powder dissolution. The time required to reach the equilibrium solubility can also be a factor for development based on the residence time in the stomach and intestines. It is desirable to have the drug dissolve while it is in the gastrointestinal tract and very long dissolution times may result in less absorption of the drug. Powder dissolution rates can also be dependent on particle size; therefore intrinsic dissolution rate may be a better assessment of this parameter. A second consideration is form changes during the experiment, as discussed previously for solution stability. When form changes occur, the solubility data obtained may not be relevant to the starting compound in the experiment. Form changes can be suggested by solubility data collected at various time points by a precipitous drop in concentration indicating crystallization of a less soluble form (as shown in Figure 7). In these cases, the maximum solubility observed over the time profile (Smax) may be reported along with the time it occurred (it should be noted that the Smax value is likely not an equilibrium solubility value). Any suggested form changes can then be confirmed by analysis of the solid form remaining at the end of the experiment. Another consideration is the media used for the experiments. Acidic or basic media can certainly have an effect on molecules containing acidic or basic groups. However, the native pH produced in an aqueous solution of a molecule with acidic or basic groups may also play a role in solubility.
Figure 7.

Schematic of a solubility curve due to a form change and precipitation of a less stable form (curve A); at equilibrium, the curve will level out to the solubility of the less soluble form. Curve B represents the less soluble form.
The solubility of cocrystals has been reported in a number of cases and in a variety of media, including water, 0.1 N HCl, phosphate buffer, SGF, and SIF. Most studies report powder dissolution data with multiple time points. In some cases, particle size was controlled by sieving samples, in some there was no reported control, and in others different particle size ranges were used for comparison. This shows the wide range of experimental variables that can be used for solubility testing which can be tailored to obtain the desired information.
Three itraconazole cocrystals (succinic acid, l-malic acid, and l-tartaric acid) were compared with crystalline itraconazole (particles less than 10 μm) and commercial Sporanox beads (amorphous itraconazole).(61) Solutions of 0.1 N HCl were used and sampled over 500 min. The cocrystals all exhibited higher solubility than the crystalline itraconazole. The l-malic and l-tartaric acid cocrystals exhibited solubilities similar to that obtained for the Sporonax beads (approximately 7 × 10−4 M) and the succinic acid was lower (approximately 2 × 10−4 M). The cocrystalline forms achieved and sustained from 4- to 20-fold solubility increases over the crystalline itraconazole.
For cocrystals of fluoxetine HCl, the aqueous solubility (called powder dissolution in the paper) was measured at various time points up to 120 min, and the solutions were analyzed by UV.(12) Samples were sieved to obtain a particle size range of 53−150 μm. The fluoxetine hydrochloride solubility was 11.4 mg/mL. The benzoic acid cocrystal solubility was lower at 5.6 mg/mL and the fumaric cocrystal solubility was higher at 14.8 mg/mL. The succinic acid cocrystal had a peak solubility of 20.2 mg/mL after about 1 min, but then decreased to that of fluoxetine HCl based on the dissociation of the cocrystal to fluoxetine hydrochloride. This system exhibited higher and lower solubilities along with dissociation, making it a very interesting and complex set of cocrystal solubilities.
For nine cocrystals of AMG517,(37) solubility was measured in fasted simulated intestinal fluid (FaSIF), and samples were taken out to 24 h and analyzed by HPLC. Particle size was not controlled or measured for the solids. Smax ranged from 1 μg/mL for trans-cinnamic acid to 21 μg/mL for benzoic acid and log Smax ranged from 0.00 for trans-cinnamic acid to 1.32 for benzoic acid. Six of the nine cocrystals reached maximum solubility within 1−2 h (glutaric acid, glycolic acid, sorbic acid, trans-2-hexanoic acid, lactic acid, benzoic acid) with a noticeable decrease in solubility after that time. The decrease was attributed to formation of the free base hydrate, which was confirmed by analysis of the remaining solids. Four cocrystals with Smax values below 3 μg/mL exhibit no form change after the experiment (trans-cinnamic acid, 2,5-dihydroxybenzoic acid, 2-hydroxycaproic acid, and benzoic acid) possibly due to insufficient time to undergo conversion in the 24 h time frame. As discussed previously, correlation analysis showed the highest interdependence between melting point and log Smax for nine cocrystals with R2 = 0.546. Previous reports of correlations between melting point and log S have been better, but were performed on unicomponent systems with equilibrium solubility values, not multicomponent systems with only maximum solubility values. Association and dissociation of cocrystals in various media are not well understood and may be a contributing factor. It should be noted that the initial solubility advantages of the cocrystals may be sufficient to give an exposure advantage in pharmacokinetic studies even if they dissociate.
The powder dissolution profile of a 1:1 indomethacin/saccharin cocrystal was measured in phosphate buffer (pH 7.4, 60 and 200 mM strengths) and compared to crystalline δ-indomethacin.(46) Samples were sieved to obtain a particle size less than 125 μm. Aliquots were taken over 2−3 days and analyzed by HPLC. The solubility of the δ-indomethacin ranged from 0.72 mg/mL in 60 mM buffer to 1.3 mg/mL in 200 mM buffer with peak dissolution obtained after 250 min; the solids did not change form during the experiment. The cocrystal was found to dissolve instantaneously in the 60 mM buffer followed by precipitation and pH drop to 6.8. Analysis of the solids showed mainly amorphous material with evidence of δ-indomethacin peaks. When the buffer strength was increased to 200 mM, rapid dissolution was again observed and a maximum solubility of 3.7 mg/mL was obtained. Analysis of the remaining solids did not show peaks indicative of the crystalline cocrystal. As noted, it is important to consider ionization of the ligand, complexation, and solution chemistry to understand the solubility behavior of cocrystals.
The dissolution and solubility of a 1:1 celecoxib/nicotinamide cocrystal was found to be medium dependent where transformation of the cocrystal to celecoxib Forms I and III was affected.(59) Cocrystal solids were gently ground with PVP-K-30 alone or with SDS to make a formulation for comparison with the cocrystal solid alone. Small amounts of surfactants (SDS and Triton-X100) were added to 20 mM sodium phosphate buffer or 0.1 N HCl. Samples were equilibrated for up to 60 min in most cases and analyzed by HPLC. The presence of the surfactants in solution resulted in a rapid dissolution of the cocrystal and conversion to large aggregates of celecoxib Form III. These aggregates dissolved more slowly than commercial Form III in a 1% SDS solution. The formulated cocrystal containing SDS and PVP were found to wet rapidly and convert to a mixture of amorphous celecoxib and small particles of celecoxib Form IV. Form IV is up to four times more bioavailable than Form III. This study illustrates the importance of understanding form conversions in solution and their effect on not only solubility and dissolution, but also on bioavailability. It is also a demonstration of how simple formulations can be used to overcome the dissociation of cocrystals and recrystallization of poorly soluble forms.
Two studies investigated the effect of particle size on the powder dissolution of cocrystals. For a 1:1 carbamazepine/saccharin cocrystal, experiments were conducted in SGF using six particle size fractions ranging from <53 μm to >1000 μm.(54) Samples were analyzed out to 120 min. The study focused on the initial rate of dissolution, which has been correlated to increased bioavailability for carbamazepine.(62) As expected, faster initial dissolution was observed with the smaller particles. Dissolution rates for sieve fractions >500 μm slowed to the point where carbamazepine concentrations were significantly lower after 2 h than the smaller particles. Sieved fractions >500 μm were less than 50% dissolved after 60 min; however, fractions <150 μm were greater than 80% dissolved at the same time point. Presence of carbamazepine dihydrate was found in large particle size samples equilibrated overnight in SGF and could be contributing to the low solubility; however, the amount present at 60 min was not reported. Another report on a 1:1 exemestane/maleic acid cocrystal and a 1:1 megestrol acetate/saccharin cocrystal looked at powder dissolution in FaSIF with three particle size fractions (150−300 μm, 106−150 μm, and fines).(63) Agglomeration and wettability were improved by physically mixing the cocrystals with lactose (1:10 drug/lactose). Samples were tested in FaSIF for 30 min, and samples were analyzed by HPLC. The 1:1 exemestane/maleic acid cocrystal was found to be similar to the exemestane fine crystals and did not show improved dissolution, likely due to transformation of the cocrystal to the parent compound. The dissolution profile of the 1:1 megestrol acetate/saccharin cocrystal fines showed a significant improvement, with a six times increase in concentration at 15 min compared to megestrol fines alone; however, a drop in concentration was observed after this time point for the cocrystal fines. The other particle size fractions did not show significant improvement. Transformation studies showed that the exemestane/maleic acid cocrystal transformed much more quickly than the megastrol acetate/saccharin cocrystal and can help explain the observed differences between the two systems. It was also found that the transformation rate for the small crystals in both systems was slower than the large crystals. Understanding the solid transformations occurring in solution was brought up as an important step in developing cocrystals. The significant increase in concentration with the fine 1:1 megestrol acetate/saccharin crystals may have an impact on absorption and ultimately solubility, and particle size reduction of cocrystals should be considered as an additional option during development.
In a number of cases, both salts and cocrystals have been prepared and the solubilities compared, as summarized in Table 5. For norfloxacin, a cocrystal was formed with isonicotinamide and three salts were prepared (succinate, malonate, and maleate).(41) Apparent aqueous solubility was measured after 72 h and the solution was analyzed by UV−vis; the form remaining at the end of the experiment was not reported. Norfloxacin apparent solubility was 0.21 mg/mL, whereas the apparent solubility ranged from 0.59 mg/mL for the isonicotinamide cocrystal to 3.9 mg/mL for the malonate salt and 9.8 mg/mL for the maleate salt. This resulted in a 3× increase in solubility for cocrystal and 20−45× increase for salts. A similar trend was reported for a Pfizer compound (Pfizer 1).(28) It is a weak base with poor aqueous solubility (0.0008 μmol/mL). The goal was to find a form that showed significant increases in solubility and bioavailability compared to the free base. Twenty solid acid−base complexes were found. Three of these were dicarboxylic acid−base complexes: a sesquisuccinate (neutral), a dimalonate (mixed ionic and zwitterionic), and a dimaleate (salt). Aqueous solubility data showed that the sesquisuccinate cocrystal was better than the parent compound (0.79 μmol/mL), the dimalonate mixed ionic state was more of an improvement (3.83 μmol/mL), and the dimaleate salt exhibited the best solubility (10.4 μmol/mL). Details about the solubility measurements were not given.
Table 5. Salt and Cocrystal Solubility Values.
| common compound in series | coformer | salt or cocrystal | solubility | media | ref |
|---|---|---|---|---|---|
| norfloxacin | (mg/mL) | water | (41) | ||
| free base | 0.21 | ||||
| isonicotinamide | cocrystal | 0.58 | |||
| succinic | salt | 6.60 | |||
| malonic | salt | 3.90 | |||
| maleic | salt | 9.80 | |||
| Pfizer 1 | (μmol/mL) | water | (28) | ||
| free base | 0.0008 | ||||
| sesquisuccinate | cocrystal | 0.79 | |||
| dimalonate | mixed ionic and zwitterionic complex | 3.83 | |||
| dimaleate | disalt | 10.4 | |||
| saccharin | (mg/mL) | water | (64, 65) | ||
| haloperidol | free base | <0.10 | |||
| salt | 6.08 | ||||
| mirtazapine | free base | <0.01 | |||
| salt | 2.08 | ||||
| piroxicam | free base | <0.01 | |||
| cocrystal | <0.01 | ||||
| quinine | free base | <0.01 | |||
| salt | 5.40 | ||||
| pseudoephedrine | free base | <0.50 | |||
| salt | >300.00 | ||||
| lamivudine | free base | 70.00 | |||
| salt | 10.56 | ||||
| risperidone | free base | <0.10 | |||
| salt | 2.87 | ||||
| sertraline | free base | <0.10 | |||
| salt | 6.45 | ||||
| venlafaxine | free base | <0.10 | |||
| salt | 30.06 | ||||
| zolpidem | free base | <0.10 | |||
| salt | 11.9 |
Another system took a different approach.64,65 Saccharin was used as the coformer with 11 pharmaceutical compounds. One cocrystal (piroxicam) and 10 salts were formed. In this case the cocrystal did not show improved solubility over piroxicam free base. The reported solubility values are listed in Table 5 displaying a value of <0.01 mg/mL for the cocrystal and piroxicam free base to a range of 2.08 to >300 mg/mL for the various salts. Eight of the 10 salts exhibited a higher solubility than the free base, one was lower than the free base (lamivudine), and one (amlodipine) could not be measured due to hygroscopicity issues. It was interesting to note that the solution pH of the saccharin cocrystal was much lower (3.27) than the salts (5.25−6.25). A follow-up study compared calculated solubility values for salts and cocrystals formed with saccharin.(66) The thermodynamic solubility product (Ksp) was calculated for the saccharin cocrystal (piroxicam) and the nine saccharinate salts based on one solubility value at a known pH and the pK of each component reported in the literature. The calculations showed that in water, salts can form for all 11 compounds in the study including piroxicam which formed a cocrystal. It was suggested that piroxicam was isolated as a cocrystal due to the shift in pK values when chloroform or ethanol solutions were used for crystallization. On the basis of the calculations, the general conclusion that the saccharinate salts were more soluble than the cocrystal was justified despite the pH differences. It was also noted that cocrystal solubility needs to take into account a revised definition of the solubility product as well as any possible complexation in solution and underlying factors related to both solid-state and solution chemistry.
Solubility is a very important parameter to obtain during development of a new compound. As expected, the limited examples in this section show that solubility may be improved using cocrystals, but not in all cases. For a poorly soluble compound, especially if it does not have ionizable groups, it is worth trying cocrystals to see if an improvement can be obtained. In the case of cocrystals versus salts, there appears to be a trend that salts may offer a greater solubility advantage, as shown with limited data in Table 5, but more work needs to be performed to determine if this is a general trend. Solubility measurements of cocrystals will continue to present interesting challenges for pharmaceutical scientists.
D. Intrinsic Dissolution
Intrinsic dissolution measures the rate of dissolution without the effect of particle size. This is accomplished by pressing a disk or pellet, commonly using a Woods apparatus in a dissolution vessel.(67) Solution concentration is measured over time to determine the dissolution rate (in mg/cm2·min). The disk needs to remain intact during the experiment, so compression pressures may be critical for poorly compressible powders. It is also important that there is no form change upon pressing the pellet or during the dissolution study. XRPD data can be obtained on the initial disk and the remaining disk after completion of the experiment to determine any major form changes that may affect the dissolution data.
There are a limited number of intrinsic dissolution studies on cocrystals. Data for the glutaric acid cocrystal of 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide(48) were collected in water over 90 min and showed that the cocrystal dissolution was approximately 18 times faster than the parent compound. XRPD of the remaining solid showed mainly the glutaric acid cocrystal, with only minor peaks for the parent material, indicating that the results were not skewed by significant form changes over the course of the experiment. The intrinsic dissolution rates for fluoxetine HCl cocrystals were also measured in water, Figure 8.(12) The dissolution of the 2:1 fluoxetine HCl/succinic acid cocrystal was too fast to measure an accurate value for the dissolution rate, but a 3−fold increase over fluoxetine HCl was estimated based on early time points. The 1:1 fluoxetine HCl/benzoic acid cocrystal was roughly half-that of the API and the 2:1 fluoxetine HCl/fumaric acid cocrystal was approximately the same as that of the API. These results show that cocrystals can enhance the dissolution rate, but can also show no improvement or a slower dissolution rate. It was interesting to note that the aqueous solubility of the guest molecule appears to correlate with the aqueous dissolution rates of the corresponding cocrystal, with benzoic acid being the least soluble (0.34 g/100 g), fumaric acid being intermediate in solubility (0.61 g/100 g), and succinic acid being the most soluble (7.5 g/100 g). Other examples will be needed to determine if this is a general trend or specific to this system.
Figure 8.

Intrinsic dissolution profile of fluoxetine HCl and its cocrystals measured in water at 10 °C. (Top ⧫) fluoxetine HCl/succunic acid cocrystal; (middle ◼) fluoxetine HCl; (middle ▲) fluoxetine HCl/fumaric acid cocrystal; (bottom ●) fluoxetine HCl/benzoic acid cocrystal. Figure modified from ref (12).
In a third study, measurements were obtained on 1:1 exemestane/maleic acid and 1:1 megestrol acetate/saccharin cocrystals in FaSIF.(63) The 1:1 exemestane/maleic acid cocrystal showed essentially the same dissolution rate as the exemestane alone; however, analysis of the remaining solid material showed a conversion to exemestane during the experiment. The 1:1 megestrol acetate/saccharin cocrystal was 3−4 times higher than megestrol acetate, but there were significant variations in the data. Analysis of the remaining solid showed only a small amount of conversion to megestrol acetate. These data show that the initial dissolution rate of megestrol acetate was improved by using the cocrystal.
As discussed for solubility and solution stability, intrinsic dissolution is an important parameter to investigate, but it may become more complicated with cocrystals. Various factors need to be considered and extra experiments may be needed to correctly obtain and interpret intrinsic dissolution data on cocrystals.
E. Bioavailability
Bioavailability is a measurement of the rate and extent of the active drug that reaches systemic circulation.(68) Animal bioavailability is an important parameter to consider when preparing new forms of a compound, and studies can be set up in a number of different ways to obtain specific information for development. Species for animal studies can include rodents, rabbits, dogs, pigs, and primates. These studies are usually performed during early development and can be small studies (4−6 animals) to determine pharmacokinetic data quickly on a new form. Usually the same animals are used for all forms/formulations with a washout period, typically, a week in length, between the doses. This gives a direct comparison within the same animals for all the materials in the study.
A study with both the parent material and the cocrystal will give a direct assessment of bioavailabiliy improvements due to the cocrystal. In a relative or comparative bioavailability study, the amount of drug in the blood is measured after oral administration of the original form and then the cocrystal. Blood samples are taken after dosing to investigate blood levels over time. It is important to note that the materials used in the study need to be formulated in the same way if a direct comparison is desired. Most dosing is done orally and formulation possibilities include powder in a capsule, powder and excipient in a capsule, or liquid formulations (solutions or suspensions). It is important to differentiate between solutions and suspensions since dissolution of the solid in a solution could significantly improve bioavailability. For suspensions it is helpful to know how much of the compound may actually be dissolved in order to determine how that may affect the results. Absolute bioavailability includes not only the cocrystal formulation, but an IV formulation as well to determine maximum exposure based on the IV data and a comparison with the oral formulation.
A limited number of animal bioavailability studies have been reported using cocrystals. One study on the pharmaceutical cocrystal of a monophosphate salt with phosphoric acid mentions that excellent in vivo performance was observed, but no details about the study are given.(13b) A hemitartaric acid cocrystal of l-883555 was given orally to rhesus monkeys.(56) Four animals were used and a dose of 3 mg/kg (based on the parent compound) was administered. A methocel formulation was used, but it was not clear if a solution or suspension was produced. The cocrystal was found to be 15 times more bioavailable than the parent compound based on the maximum blood concentration (Cmax) values.
A dog study compared a 1:1 carbamazepine/saccharin cocrystal with the marketed immediate release tablets of carbamazepine(54) (Tegretol containing carbamazepine form III). Four dogs were used and samples were taken up to 12 h post dose. The ground cocrystal (particle size <53 nm) was mixed with lactose in a dry blending step and placed into capsules containing a dose of 200 mg of carbamazepine. The pharmacokinetic parameters (AUC, Cmax, Tmax) were all found to be similar to the marketed product. This study noted that cocrystals can serve as a model for addressing physicochemical problems of a pharmaceutical compound (i.e., stability, solubility, dissolution), but will not overcome issues of metabolism or pharmacology.
For dog studies of a 1:1 2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide/glutaric acid cocrystal, a simple powder in capsule formulation was used at two dose levels (5 and 50 mg/kg).(48) The cocrystal was compared directly to the crystalline API (2-[4-(4-chloro-2-fluorphenoxy)phenyl]pyrimidine-4-carboxamide). Six dogs were used and samples were collected for 36 h post dose. The mean Cmax values for the 5 mg dose were 89 and 25 ng/mL for the cocrystal and parent compound, respectively. For the 50 mg dose, the Cmax values were 278 and 89 ng/mL for the cocrystal and parent compound, respectively. The cocrystal was found to be over three times more bioavailable than the parent material in these studies using only powder in a capsule as a formulation. It should be noted that the increase in dose was not proportional to the increase in exposure. Intrinsic dissolution experiments showed the cocrystal to be 18 times more soluble than the parent material in pure water and after 90 min the pellet showed very minor XRPD peaks due to the parent material. Exposure of the pellets to water for 24 h showed complete conversion to the parent material. Even though it is possible that the cocrystal may have dissociated in the animal studies, the increase in bioavailability and exposure was still significant.
The 1:1 AMG 517/sorbic acid cocrystal(47) was compared to the parent free base in a rat bioavailability study using 10% (w/v) Pluronic F108 in OraPlus suspensions of free base (500 mg/kg) and cocrystal (10, 30, 100, 500 mg/kg). A significant increase in bioavailability was observed for the cocrystal. It was found that a 30 mg/kg dose of the cocrystal resulted in comparable exposure to a 500 mg/kg dose of the free base. This is another example where the cocrystal was found to dissociate in FaSIF, but displayed significantly higher solubility than the parent compound before it dissociated. This higher solubility may increase exposure if it stays in solution while it passes through the small intestine.
Although the number of examples is limited, cocrystals can significantly increase the bioavailability of poorly soluble compounds. There is not always a direct in vitro/in vivo correlation and even examples of dissociated cocrystals in in vitro studies can provide improved performance in animal studies.
III. Additional Development Factors
A. Scale-up
In order for pharmaceutical cocrystalline materials to move from small scale screening exercises to a viable option for development and ultimately drug products, scalable (kilo to multikilo) processes offering high cocrystal purity and yields must be devised and established. Cocrystal quantities of milligrams to a few grams are typically produced depending on the types of characterization and/or initial physicochemical studies (rate dissolution, solubility, bioavailability, etc.) the materials are subjected to. A recent survey of the open literature showed that slow evaporation and grinding are the two most common techniques for cocrystal growth; however, these approaches possess obvious limitations upon scale-up, and thus additional routes must be formulated.(69)
In one account, the preparation of a carbamazepine/saccharin cocrystal was produced on a 30 g scale through solution crystallization.(54) Within this procedure, the components were dissolved in a mixture of ethanol and methanol (∼3:1) and refluxed at 70 °C for 1 h. The temperature was then lowered, and the precipitate was filtered, dried, and characterized as the 1:1 cocrystal of carbamazepine and saccharin. Interestingly, under the reaction conditions selected, seeding was not necessary to produce the desired cocrystalline form in high purity.
Recently a detailed report on the scalable solution-crystallization process of a carbamazepine/nicotinamide cocrystal was described.(69) Outlined within the crystallization methodology are three criteria to be examined: (a) determine an appropriate solvent system; (b) identify the pathway, through multicomponent solid−liquid phase equilibrium diagrams, to produce the desired form; (c) determine a mechanism to induce nucleation and control the desaturation kinetics of the process. Through a seeding strategy in ethanol, the cocrystal was successfully produced on a 1 L scale with yields in excess of 90%.
The construction and use of ternary phase diagrams should be considered when scaling up cocrystals from solution because information about the relationship between equilibria of the solid phases and solvent choices is obtainable. It is critical to determine and map the solubilities of the individual components since the phase region where the most thermodynamically stable cocrystal is located will be altered based on whether or not they possess similar solubilities in a given solvent.14,70,71
As pharmaceutically based cocrystalline materials move further into development and become viable options as marketed products, scaleable cocrystal processes must be evaluated and optimized.
B. Cocrystal Polymorphism
Searching for polymorphs of a particular compound is commonplace in solid-state pharmaceutics. Since polymorphic compounds can potentially possess drastically different physical and chemical properties, considerable efforts are taken to identify and characterize all forms during development. It is not surprising that polymorphic cocrystals can also exist(72) and possess varying physicochemical properties between forms. Detailed below are three examples of API-based polymorphic cocrystals and one example of an extensive polymorph screen on a cocrystalline material.
A conformational polymorphic set of 1:1 cocrystals was observed when a chloroform solution of caffeine and glutaric acid were allowed to slowly evaporate.(73) The two different morphologies, rods (Form I) and blocks (Form II), crystallize concomitantly with identical molecular connectivities; however, differences arise in the torsional angles of the methylene carbons on the acid. Interestingly, each of the forms could be produced individually through mechanical grinding. In the absence of solvent or when a fairly nonpolar solvent was used, Form I was formed; however, when a more polar solvent was utilized, Form II resulted. Form II was found to be more stable with respect to humidity, as Form I displayed partial conversion to Form II after 1 day at 43% RH, with full conversion to Form II after 1 day at 75% RH.(49) This example clearly shows the differences in stability that polymorphic cocrystalline forms can possess.
In another example, when an equimolar mixture of chlorzoxazone and 2,4-dihydroxybenzoic acid are evaporated from tetrahydrofuran (Form I) or ethyl acetate (Form II), 1:1 polymorphic cocrystals are generated.(74) Upon mechanical grinding of the individual components Form II was only produced, and solvent-assisted grinding or heating a sample of Form I results in full conversion to Form II.
Piroxicam is known to exist in two tautomeric forms in the solid state, a nonionized tautomer and a zwitterionic tautomer. From a cocrystallization screen, two 1:1 polymorphic cocrystals were formed between piroxicam and 4-hydroxybenzoic acid.(6) Interestingly, the hydrogen bonding patterns are different between forms (this is arguably the first report of cocrystal synthon polymorphism),(75) as well as, in one form the piroxicam nonionized tautomer exists, while in the other the zwitterionic tautomer is observed.
Unlike the previous two examples, in the case of the 1:1 cocrystal of carbamazepine and saccharin (Form I), an extensive polymorphic screen was conducted on one form.(54) By means of high-throughput crystallization, 480 experiments including saccharin and carbamazepine in various solvents and solvent mixtures were performed, yielding 156 solid materials which were characterized by XRPD or Raman spectroscopy. No polymorphic forms of the cocrystal were observed. Additionally, solvent-assisted mechanical grinding experiments were tried, using 24 different solvents. The remaining solids were characterized by XRPD, resulting, once again, in only the original cocrystal form. Finally, slurry conversion experiments utilizing seven different solvents at ambient temperatures for four days were attempted. The remaining solids were characterized by XRPD, displaying only the initial cocrystal form. Furthermore, dissociation of the cocrystal was not observed from the seven solvents, giving additional insight into the solution stability of the cocrystalline material. It should be noted, however, that a second polymorph of a 1:1 cocrystal of carbamazepine/saccharin (Form II) was found using a polymer heteronuclei crystallization media, and the hydrogen bonding patterns for the two forms are shown in Figure 9.(76)
Figure 9.

Polymorphic cocrystals of 1:1 carbamazepine/saccharin.(77)
Polymorphism of cocrystals will attract more attention as cocrystals continue to gain momentum during the development of new pharmaceuticals. As with salts, cocrystal polymorphs offer additional options to alter properties, increase patent protection, and improve marketed formulations.
C. Intellectual Property (IP) and Lifecycle Management
Patents have become a critical element of drug development, especially when covering solid forms. There have been limited reports on cocrystals and IP,78,79 but more information on this area is expected as the field grows. Pharmaceutical patents can cover a number of different areas, including composition of matter (molecular structure, solid form, or formulation), method of use (medical indication), and manufacturing processes. In order for an invention to qualify for patent coverage, it must satisfy three criteria: novelty, utility, and nonobviousness. For solid form patent applications directed to pharmaceuticals in general, utility is not usually problematic, and the examples in this paper readily show examples of utility of new cocrystals above and beyond the therapeutic effect of the API. For a solid form to be novel, it cannot appear in the prior art either expressly or inherently. For most pharmaceutical cocrystals, prior art is limited since the coformer would likely not be published in connection with the crystallization of the API. This lack of prior art is an advantage from the IP perspective. The third area is being nonobvious, which may be viewed, in at least some circumstances, as correlating with predictability. Crystal engineering is certainly an advantage when trying to decide on possible coformers, but there is no guarantee that a cocrystal will form. Computational analyses are also not able to reliably predict the structure or properties of cocrystals at this point in time, which adds to the nonobvious nature of solid forms in general and especially cocrystals.
Cocrystals represent a broad patent space since there is a large number of coformers available based on the possible compounds in the EAFUS (Everything Added to Food in the US) and GRAS (Generally Regarded as Safe) lists.(24) However, because of the lack of predictability in the field, it is expected that in many circumstances patent coverage will be narrow.
Cocrystals can also play a role in lifecycle management.(78) Lifecycle management can involve drug product improvements along with new solid forms. Early in the development process, chemical structures are patented and additional IP protection can be obtained by patenting different solid forms throughout development. If an approved drug product contains a new patented solid form, especially where the solid form offers a commercial advantage over the original form, the solid form patent might provide meaningful IP protection after the expiration of the original patent. However, a solid form that was not found by the innovator, but was found and patented by a competitor, could significantly alter this strategy. Cocrystal screens for potential blockbuster drugs could end up being very large in order to protect, not only the cocrystals found, but also any polymorphs, hydrates, solvates, or other solid forms of the individual cocrystals.
From a regulatory point of view for generic products, cocrystals may present an interesting option. Currently, when generic pharmaceutical companies use polymorphs and hydrates as alternatives to ethical drugs, they file Abbreviated New Drug Applications (ANDAs), which requires the submission of minimal bioavailability and clinical data and does not require proving safety or efficacy. New salts of an API, however, use a slightly different regulatory pathway, a so-called 505(b)(2) application, and require more testing and clinical data than an ANDA submission. The classification of cocrystals as a generic has not yet been addressed.(78) Cocrystals contain nonionic interactions like hydrates, but they also contain substances with possible toxicity issues, similar to salts. The decision on how to regulate cocrystals for generic products may affect their use in the generic industry.
Cocrystals will raise IP and regulatory questions as more of these compounds are developed, moved later into development, and eventually marketed. As with any solid form, they will offer their own challenges and many issues will need to be dealt with on a case by case basis.
IV. Conclusions
One can clearly see a place for cocrystals within the pharmaceutical market. On the basis of the limited examples available, some general comments can be made on the physicochemical properties of cocrystals. Melting points are altered for most cocrystals, with approximately 51% resulting in melting points between those of the API and coformer and 39% resulting in lower melting points. Correlations with other parameters, such as solubility, are limited and will be complex due to the multicomponent nature of the cocrystals. In many cases, improved stability, such as resistance to hydrate formation, has been shown for cocrystals. However, general trends are not evident and each system needs to be evaluated to determine if improvements are obtained. Improved solubility for poorly soluble compounds has been achieved using cocrystals. Limited studies suggest that salts will provide a larger increase in solubility if they are available. For poorly soluble neutral compounds, cocrystals are a very feasible approach to improving solubility. Cocrystals can provide higher and lower dissolution rates compared to the API. Dissociation is an important consideration in analyzing data from these experiments. It was shown that significant increases in bioavailability are possible with cocrystals, even when dissociation of the cocrystal is suspected based on in vitro studies.
Other aspects of development, such as polymorphism and scale-up, will need to be examined for cocrystals. Processes to produce cocrystals on a large scale will likely require different approaches, such as those based on ternary solubility phase diagrams. Cocrystals will provide additional options for IP, regulatory, and lifecycle management for new and old drugs and will provide additional challenges as they continue through the development process.
To date, no cocrystalline drug products appear to be on the market, although there is no doubt that cocrystals are present in pharmaceutical drug pipelines and it is only a matter of time before this imagination becomes a reality.
Acknowledgments
The authors would like to thank Brett Cowans, Sarah Bethune, Eyal Barash, and the anonymous scientific reviewers for their helpful comments and suggestions for the manuscript.
References
- a Seddon K. R.; Zaworotko M. J.. Crystal Engineering: The Design and Application of Functional Solids; NATO-ASI Series; Kluwer: Dordrecht, The Netherlands, 1999; Vol. 539. [Google Scholar]; b Datta S.; Grant D. J. W. Nat. Rev. Drug Discovery 2004, 3, 42–57. [DOI] [PubMed] [Google Scholar]; c Making Crystals by Design-from Molecules to Molecular Materials, Methods, Techniques, Applications; Grepioni F.; Braga D. Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- a Handbook of Pharmaceutical Salts: Properties, Selection and Use; Stahl P. H.; Wermuth C. G. Eds.; Verlag Helvetica Chimica Acta: Zürich, 2002. [Google Scholar]; b Bighley L. D.; Berge S. M.; Monkhouse D. C.. Salt forms of drugs and absorption. In Encyclopedia of Pharmaceutical Technology; Swarbick J.; Boylan J. C. Eds; Marcel Dekker, Inc.: New York, 1996; Vol. 13, pp 453−499. [Google Scholar]; c Gu C. H.; Grant D. J. W.. Handbook of Experimental Pharmacology: Stereochemical Aspects of Drug Action and Disposition; Eichelbaum M.; Testa B.; Somogyi A. Eds.; Springer: Berlin, 2003. [Google Scholar]
- Molecules from the “generally regarded as safe” GRAS list, pharmaceutical excipients, amino acids, food additives
- Stahly G. P. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar]
- Bhogala B. R.; Nangia A. New J. Chem. 2008, 32, 800–807. [Google Scholar]
- Childs S. L.; Hardcastle K. I. Cryst. Growth Des. 2007, 7, 1291–1304. [Google Scholar]
- Aakeroy C. B.; Salmon D. J. CrystEngComm 2005, 7, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A. B. CrystEngComm 2007, 9, 833–834. [Google Scholar]
- Jones W.; Motherwell W. D.; Trask A. V. MRS Bull. 2006, 341, 875–879. [Google Scholar]
- Vishweshwar P.; McMahon J. A.; Bis J. A.; Zaworotko M. J. J. Pharm. Sci. 2006, 95, 499–516. [DOI] [PubMed] [Google Scholar]
- Cocrystals are also generated using non-traditional low-temperature crystallization techniques; a Kirchner M. T.; Boese R.; Gehrke A.; Bläser D. CrystEngComm 2004, 63, 360–366. [Google Scholar]; b Kirchner M. T.; Das D.; Boese R. Cryst. Growth Des. 2008, 8, 763–765. [Google Scholar]; c Kirchner M. T.; Boese R.; Billups W. E.; Norman L. R. J. Am. Chem. Soc. 2004, 126, 9407–9412. [DOI] [PubMed] [Google Scholar]
- Example: API·HCl/coformer.Childs S. L.; Chyall L. J.; Dunlap J. T.; Smolenskaya V. N.; Stahly B. C.; Stahly G. P. J. Am. Chem. Soc. 2004, 126, 13335–13342. [DOI] [PubMed] [Google Scholar]
- Example: API/fumerate/fumaric acid.; a Pop M.; Sieger P.; Cains P. W. J. Pharm. Sci. 2008, 95, 1820–1834Example: API/phosphate/phosphoric acid.. [DOI] [PubMed] [Google Scholar]; b Chen A. M.; Ellison M. E.; Peresypkin A.; Wenslow R. M.; Variankaval N.; Savarin C. G.; Natishan T. K.; Mathre D. J.; Dormer P. G.; Euler D. H.; Ball R. G.; Ye A.; Wang Y.; Santos I. Chem. Commun. 2007, 419–421. Example: API/saccharinate/saccharin. [DOI] [PubMed] [Google Scholar]; c Velaga S. P.; Basavoju S.; Boström D. J. Mol. Struct. 2008, 889, 150–153. [Google Scholar]
- A few examples, but definitely not a complete list; a Oswald I. D. H.; Allan D. R.; McGregor P. A.; Motherwell W. D. S.; Parsons S.; Pulham C. R. Acta Crystallogr., Sect. B 2002, 58, 1057–1066. [DOI] [PubMed] [Google Scholar]; b Almarsson Ö.; Zaworotko M. J. Chem. Commun. 2004, 188, 9–1896. [DOI] [PubMed] [Google Scholar]; c Rodriguez-Spong B.; Price C. P.; Jayasankar A.; Matzger A. J.; Rodríguez-Hornedo N. Adv. Drug Delivery Rev. 2004, 56, 241–274. [DOI] [PubMed] [Google Scholar]; d Blagden N.; de Matas M.; Gavan P. T.; York P. Adv. Drug. Delivery Rev. 2007, 59, 617–630. [DOI] [PubMed] [Google Scholar]
- One general example: Weyna D. R.; Shattock T.; Vishweshwar P.; Zaworotko M. J.. Cryst. Growth Des 2009, 9, 1106−1123.
- One general example:Frišc̆ić T.; Jones W. Cryst. Growth Des 2009, 9, 1621–1637. [Google Scholar]
- Frišc̆ić T.; Childs S. L.; Rizvi S. A. A.; Jones W. CrystEngComm 2009, 11, 418–426. [Google Scholar]
- Blagden N.; Berry D. J.; Parkin A.; Javed H.; Ibrahim A.; Gavan P. T.; De Matos L. L.; Seaton C. C. New J. Chem. 2008, 32, 1659–1672. [Google Scholar]
- a Desiraju G. R. Angew. Chem., Int. Ed. Engl. 1995, 34, 2311. [Google Scholar]; b Steiner T. Angew. Chem., Int. Ed. 2002, 41, 48–76. [DOI] [PubMed] [Google Scholar]; c Aakeröy C. B.; Schultheiss N.. Assembly of Molecular Solids via Non-covalent Interactions, In Making Crystals by Design-from Molecules to Molecular Materials, Methods, Techniques, Applications; Grepioni F.; Braga D. Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]; d Shattock T. R.; Arora K. K.; Vishweshwar P.; Zaworotko M. J. Cryst. Growth Des. 2009, 8, 4533–4545. [Google Scholar]
- a Kartum G.; Vogel W.; Andrussov K.. Dissociation Constants of Organic Acids in Aqueous Solution; Butterworth & Co. Publishers: London, 1961. [Google Scholar]; b Aakeröy C. B.; Desper J.; Urbina J. F. Chem. Commun. 2005, 2820–2822. [DOI] [PubMed] [Google Scholar]; c Childs S. L.; Stahly G. P.; Park A. Mol. Pharmaceutics 2007, 4, 323–338. [DOI] [PubMed] [Google Scholar]
- a Chiarella R. A.; Davey R. J.; Petterson M. L. Cryst. Growth Des. 2007, 7, 1223–1226. [Google Scholar]; b Childs S. L.; Rodríguez-Hornedo N.; Reddy L. S.; Jayasankar A.; Maheshwari C.; McCausland L.; Shipplett R.; Stahly B. C. CrystEngComm 2008, 10, 856–864. [Google Scholar]; c Chadwick K.; Davey R.; Sadiq G.; Cross W.; Pritchard R. CrystEngComm 2009, 11, 412–414. [Google Scholar]; d ter Horst J. H.; Deij M. A.; Cains P. W. Cryst. Growth Des. 2009, 9, 1531–1537. [Google Scholar]
- a Etter M. C. J. Chem. Phys. 1991, 95, 4601–4610. [Google Scholar]; b Etter M. C. Chem. Res. 1990, 23, 120–126. [Google Scholar]
- Cambridge Structural Database (CSD); ; a Allen F. H. Acta Crystallogr., Sect. B 2002, 58, 380–388. [DOI] [PubMed] [Google Scholar]; b Bruno I. J.; Cole J. C.; Edgington P. R.; Kessler M. K.; Macrae C. F.; McCabe P.; Pearson J.; Taylor R. Acta Crystallogr., Sect. B 2002, 58, 389–397. [DOI] [PubMed] [Google Scholar]; c Fábián L. Cryst. Growth Des. 2009, 9, 1436–1443. [Google Scholar]
- a GRAS Notices: http://www.cfsan.fda.gov/∼rdb/opa-gras.html; b Food additive status list: http://www.cfsan.fda.gov/∼dms/opa-appa.html
- Computational attempts are ongoing to predict cocrystal formation.Issa N.; Karamertzanis P. G.; Welch G. W. A.; Price S. L. Cryst. Growth Des 2009, 9, 442–453. [Google Scholar]
- a Taylor R.; Kennard O. Acta Crystallogr., Sect. B 1983, B39, 133–138. [Google Scholar]
- Newman A. W.; Childs S. L.; Cowans B. A.. Salt Cocrystal Form Selection, In Preclinical Development Handbook; John Wiley and Sons: Hoboken, 2008; pp 455−481. [Google Scholar]
- Li Z. J.; Abramov Y.; Bordner J.; Leonard J.; Medek A.; Trask A. V. J. Am. Chem. Soc. 2006, 128, 8199–8210. [DOI] [PubMed] [Google Scholar]
- Aakeröy C. B.; Salmon D. J.; Smith M. M.; Desper J. Cryst. Growth Des. 2006, 6, 1033–1042. [Google Scholar]
- a Aakeröy C. B.; Hussain I.; Desper J. Cryst. Growth Des. 2006, 6, 474–480. [Google Scholar]
- Solid-State Chemistry of Drugs, 2nd ed.; Bryn S. R.; Pfeiffer R. R.; Stowell J. G. Eds.; SSCI, Inc.: West Lafayette, IN, 1999. [Google Scholar]
- a Jain A.; Yang G.; Yalkowsky S. H. Ind. Eng. Chem. Res. 2004, 43, 7618–7621. [Google Scholar]; b Jain A.; Yalkowsky S. H. J. Pharm. Sci. 2006, 95, 2562–2618. [DOI] [PubMed] [Google Scholar]
- Burger A.; Ramberger R. Mikrochim. Acta 1979, 259–271. [Google Scholar]
- Abramowitz R.; Yalkowsky S. H. Pharm. Res. 1990, 7, 942–947. [DOI] [PubMed] [Google Scholar]
- Jain N.; Yalkowsky S. H. J. Pharm. Res. 2001, 90, 234–252. [DOI] [PubMed] [Google Scholar]
- a Katritzky A. R.; Jain R.; Lomaka A.; Petrukhin R.; Maran U.; Karelson M. Cryst. Growth Des. 2001, 1, 261–265. [Google Scholar]; b Dunitz J. D.; Gavezzotti A. Angew. Chem., Int. Ed. 2005, 44, 1766–1787. [DOI] [PubMed] [Google Scholar]
- Stanton M. K.; Bak A. Cryst. Growth Des. 2008, 8, 3856–3862. [Google Scholar]
- Fleischman S. G.; Kuduva S. S.; McMahon J. A.; Moulton B.; Bailey Walsh R. D.; Rodríguez-Hornedo N.; Zaworotko M. J. Cryst. Growth Des. 2003, 3, 909–919. [Google Scholar]
- Nicoli S.; Bilzi S.; Santi P.; Caira M. R.; Li J.; Bettini R. J. Pharm. Res. 2008, 97, 4830–4839. [DOI] [PubMed] [Google Scholar]
- Bailey Walsh R. D.; Bradner M. W.; Fleischman S.; Morales L. A.; Moulton B.; Rodríguez-Hornedo N.; Zaworotko M. J. Chem. Commun. 2003, 186–187. [DOI] [PubMed] [Google Scholar]
- Bassavoju S.; Boström D.; Velaga S. P. Cryst. Growth Des. 2006, 6, 2699–2708. [Google Scholar]
- Stanton M. K.; Tufekcic S.; Morgan C.; Bak A. Cryst. Growth Des. 2009, 9, 1344–1352. [Google Scholar]
- International Conference on Harmonization (ICH) Q1A, Stability testing of new drug substances and products. http://www.ich.org/LOB/media/MEDIA419.pdf
- Reutzel-Edens S. M. and Newman A. W.. The Physical Characterization of Hygroscopicity in Pharmaceutical Solids, In Polymorphism; Hilfiker R. Ed.; Wiley-VCH:Weinheim, 2006; pp 235−258. [Google Scholar]
- Newman A. W.; Reutzel-Edens S. M.; Zografi G. J. Pharm. Sci. 2008, 97, 1047–1059. [DOI] [PubMed] [Google Scholar]
- Basavoju S.; Bostrom D.; Velaga S. P. Pharm. Res. 2008, 25, 530–541. [DOI] [PubMed] [Google Scholar]
- Bak A.; Gore A.; Yanez E.; Stanton M.; Tufekcic S.; Sysed R.; Akrami A.; Rose M.; Surapaneni S.; Bostick T.; King A.; Neervannan S.; Ostovic D.; KoparkaR A. J. Pharm. Sci. 2008, 97, 3942–3956. [DOI] [PubMed] [Google Scholar]
- McNamara D.; Childs S. L.; Giordano J.; Iarriccio A.; Cassidy J.; Shet M. S.; Mannion R.; O’Donnell E.; Park A. Pharm. Res. 2006, 23, 1888–1897. [DOI] [PubMed] [Google Scholar]
- Trask A. V.; Motherwell W. D. S.; Jones W. Cryst. Growth Des. 2005, 5, 1013–1021. [Google Scholar]
- a Bothe H.; Cammenga H. K. Thermochim. Acta 1980, 40, 29–39. [Google Scholar]; b Griesser U. J.; Burger A. Int. J. Pharm. 1995, 120, 83–93. [Google Scholar]
- Trask A. V.; Motherwell W. D. S.; Jones W. Int. J. Pharm. 2006, 320, 114–123. [DOI] [PubMed] [Google Scholar]
- Karki S.; Frišěiæ T.; Jones W.; Motherwell W. D. S. Mol. Pharm. 2007, 4, 347–354. [DOI] [PubMed] [Google Scholar]
- Frišěiæ T.; Fabian L.; Burley J. C.; Reid D. G. Chem. Commun. 2008, 1644–1646. [DOI] [PubMed] [Google Scholar]
- Hickey M. B.; Peterson M. L.; Scoppettuolo L. A.; Morisette S. L.; Vetter A.; Guzman H.; Remenar J. F.; Zhang Z.; Tawa M. D.; Haley S.; Zaworotko M. J.; Almarsson O. Eur. J. Pharm. Biopharm. 2007, 67, 112–119. [DOI] [PubMed] [Google Scholar]
- Oswald I. D. H.; Allan D. R.; McGregor P. A.; Motherwell W. D. S.; Parsons S.; Pulham C. R. Acta Crystallogr., Sect. B 2002, B58, 1057–1066. [DOI] [PubMed] [Google Scholar]
- Variankaval N.; Wenslow R.; Murry J.; Hartman R.;; Helmy R.; Kwong E.; Clas S.; Dalton C.; Santos I. Crystal Growth Des. 2006, 6, 690–700. [Google Scholar]
- Zhang G. G. Z.; Henry R. F.; Borchardt T. B.; Lou X. J. Pharm. Sci. 2007, 96, 990–995. [DOI] [PubMed] [Google Scholar]
- Yalkowsky S.; He Y.. Handbook of Aqueous Solubility Data; CRC Press: Boca Raton, 2003. [Google Scholar]
- Remenar J. F.; Peterson M. L.; Stephens P. W.;, Z.; Zimenkov Y.; Hickey M. B. Mol. Pharm 2007, 4, 386–400. [DOI] [PubMed] [Google Scholar]
- Lu G. W.; Hawley M.; Smith M.; Geiger B. M.; Pfund W. J. Pharm. Sci. 2005, 95, 305–317. [DOI] [PubMed] [Google Scholar]
- Remenar J. F.; Morissette S. L.; Peterson M. L.; Moulton B.; MacPhee J. M.; Guzman H. R.; Almarsson O. J. Am. Chem. Soc. 2003, 125, 8456–8457. [DOI] [PubMed] [Google Scholar]
- Myer M. C.; Straughn A. B.; Mhatre R. M.; Shah V. P.; Williams R. L.; Lesko L. J. Pharm. Res. 1998, 15, 1787–1791. [DOI] [PubMed] [Google Scholar]
- Shiraki K.; Takata N.; Takano R.; Hayashi Y.; Terada K. Pharm. Res. 2008, 25, 2581–2592. [DOI] [PubMed] [Google Scholar]
- Bhatt P. M.; Ravindra N. V.; Banerjee R.; Desiraju G. R. Chem. Commun. 2005, 1073–1075. [DOI] [PubMed] [Google Scholar]
- Banerjee R.; Bhatt P. M.; Ravindra N. V.; Desiraju G. R. Cryst. Growth Des. 2005, 5, 2299–2309. [Google Scholar]
- Cooke C. L.; Davey R. J. Cryst. Growth Des. 2008, 8, 3483–3485. [Google Scholar]
- Intrinsic dissolution and Woods apparatus, U.S. Pharmacopeia, 2008; Vol. 1, p 526
- Bermajo M.; Gonzalez-Alvarez I.. How and where are drugs absorbed? in Preclinical Development Handbook; John Wiley and Sons: Hoboken, 2008; pp 249−280. [Google Scholar]
- Sheikh A. Y.; Abd Rahim S.; Hammond R. B.; Roberts K. J. CrystEngComm 2009, 11, 501–509. [Google Scholar]
- Nehm S. J.; Rodriguez-Spong B.; Rodriguez-Hornedo N. Cryst. Growth Des. 2006, 6, 592–600. [Google Scholar]
- Chiarella R. A.; Davey R. J.; Petterson M. L. Cryst. Growth Des. 2007, 7, 1223–1226. [Google Scholar]
- It is suggested that approximately 30 polymorphic sets of cocrystals have been deposited into the CSD.Badu N. J.; Reddy S. L.; Aitipamula S.; Nangia A. Chem. Asian J. 2008, 3, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Jones W.; Samual Motherwell W. D.; Trask A. V. Chem. Commun. 2004, 890–891. [DOI] [PubMed] [Google Scholar]
- Childs S. L.; Hardcastle K. I. CrystEngComm 2007, 9, 364–367. [Google Scholar]
- Sreekanth B. R.; Vishweshwar P.; Vyas K. Chem. Commun. 2007, 2375–2377. [DOI] [PubMed] [Google Scholar]
- Porter III W. W.; Elie S. C.; Matzger A. J. Cryst. Growth Des. 2008, 8, 14–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Images created using Mercury 2.1
- Trask A. V. Mol. Pharm. 2007, 4, 301–309. [DOI] [PubMed] [Google Scholar]
- Thayer A. M. Chem. Eng. News 2007, 85, 17–30. [Google Scholar]