

Abstract
Lipid membranes structurally define the outer surface and internal organelles of cells. The multitude of proteins embedded in lipid bilayers are clearly functionally important, yet they remain poorly defined. Even today, integral membrane proteins represent a special challenge for current large scale shotgun proteomics methods. Here we used endothelial cell plasma membranes isolated directly from lung tissue to test the effectiveness of four different mass spectrometry-based methods, each with multiple replicate measurements, to identify membrane proteins. In doing so, we substantially expanded this membranome to 1,833 proteins, including >500 lipid-embedded proteins. The best method combined SDS-PAGE prefractionation with trypsin digestion of gel slices to generate peptides for seamless and continuous two-dimensional LC/MS/MS analysis. This three-dimensional separation method outperformed current widely used two-dimensional methods by significantly enhancing protein identifications including single and multiple pass transmembrane proteins; >30% are lipid-embedded proteins. It also profoundly improved protein coverage, sensitivity, and dynamic range of detection and substantially reduced the amount of sample and the number of replicate mass spectrometry measurements required to achieve 95% analytical completeness. Such expansion in comprehensiveness requires a trade-off in heavy instrument time but bodes well for future advancements in truly defining the ever important membranome with its potential in network-based systems analysis and the discovery of disease biomarkers and therapeutic targets. This analytical strategy can be applied to other subcellular fractions and should extend the comprehensiveness of many future organellar proteomics pursuits.
The plasma membrane provides a fundamental physical interface between the inside and outside of any cell. Beyond creating a protected compartment with a segregated, distinct, and well controlled internal milieu for the cell, it also mediates a wide variety of basic biological functions including signal transduction, molecular transport, membrane trafficking, cell migration, cell-cell interactions, intercellular communication, and even drug resistance. Plasma membrane-associated proteins, especially integral membrane proteins (IMPs)1 that traverse the lipid bilayer, are key elements mediating these vital biological processes. Consistent with its fundamental importance in both normal cellular functions and pathophysiology, the plasma membrane has also been targeted extensively for biomarker discovery and drug development. In fact, more than two-thirds of known targets for existing drugs are plasma membrane proteins (1).
Despite the potential benefits, profiling the proteome of plasma membranes comprehensively using standard large scale methods including MS-based strategies has been limited and technically quite challenging. Intrinsic hydrophobicity, a wide concentration range of proteins, and other factors have hampered IMP resolution and identification using conventional two-dimensional gel electrophoresis. Gel and gel-free protein separations, including combinations of both, have been reported as an alternative to two-dimensional gel electrophoresis (2–9). Yet most such efforts have focused predominantly on identifying rather soluble proteins from body fluids (i.e. plasma, serum, and cerebrospinal fluid), cell lysates, or cytoplasm. These proteins, unlike IMPs, are relatively abundant and readily susceptible to enzymatic digestion in solution. Various attempts have been made to solubilize and enrich for IMPs, including different detergents, solvents, high pH solutions, and affinity purification (10–22). Even when organellar membranes are enriched through isolation by subcellular fractionation, the yield of proteins identified has been below expectation, especially for multipass transmembrane proteins such as G-protein-coupled receptors.
Here we systematically characterize four analytical approaches to enhance the identification of proteins, specifically those embedded in plasma membranes isolated directly from vascular endothelium in rat lung. Endothelial cells (ECs) constitute the tissue-blood interface that controls many important physiological functions, including tissue homeostasis, nutrition, vasomotion, and even drug delivery. In vivo mapping of the EC plasma membrane proteome provides unique opportunities for extending basic understanding in vascular biology and for directing the delivery of therapeutic and imaging agents in vivo (23–25). But it also presents distinct challenges beyond those generally associated with extraction, solubilization, and identification of IMPs in cells and tissues. ECs form a thin monolayer lining each blood vessel. They constitute a very small fraction of all the cells existing in tissue, thereby making it difficult to isolate sufficiently pure EC plasma membrane fractions for proteomics analysis using conventional subcellular fractionation techniques. Although relatively simple to isolate from tissue and grow in culture, ECs require cues from the tissue microenvironment to maintain their tissue-specific qualities and thus undergo rapid and considerable phenotypic drift after isolation (26).
We have developed a specialized coating procedure using colloidal silica nanoparticles perfused through the blood vessels of the tissue to isolate luminal plasma membranes of the vascular endothelium as they exist natively in tissue (26–28). Our initial survey of these plasma membranes isolated directly from rat lungs used primarily three standard analytical techniques of the time: two-dimensional electrophoresis, Western analysis, and the shotgun method of two-dimensional liquid chromatography-tandem mass spectrometry (24, 26). We identified 450 proteins of which only ∼15% were IMPs. Although at the time this was a notable total number of proteins, more IMPs are expected. In fact, this large scale 2DC study did not identify several well known EC surface marker proteins, including specific enzymes, adhesion molecules, and growth factor receptors.
Here we comparatively analyze four different MS-based strategies involving two- and three-dimensional separation by combining protein prefractionation via SDS-PAGE with in-gel digestion to produce peptides separated by one- and two-dimensional nano-HPLC before seamless and continuous MS analysis. Each method used multiple replicate measurements to comprehensively identify proteins, especially IMPs, and in doing so achieved a clear statistical definition of completeness that permits meaningful comparisons. Ultimately this analysis greatly expanded the EC plasma membranome to 1,833 proteins of which nearly 30% are membrane-embedded.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Sequencing grade modified trypsin was purchased from Promega (Madison, WI), dithiothreitol and iodoacetamide were obtained from Pierce, ammonium bicarbonate was purchased from Mallinckrodt Baker, Inc. (Phillipsburg, NJ), HPLC grade Burdick & Jackson acetonitrile was purchased from VWR (Westchester, PA), and formic acid was from EMD Chemicals Inc. (Gibbstown, NJ). All other chemical reagents were obtained from Sigma-Aldrich.
Isolation of Luminal Plasma Membranes from Vascular Endothelium in Rat Lung
Sprague-Dawley female rats (150–250 g; Charles River Laboratories) were used for all experiments. Animal procedures were carried out in accordance with the Sidney Kimmel Cancer Institutional Animal Care and Usage Committee standards. Luminal vascular EC plasma membranes were isolated directly from rat lung tissues using a nanoparticle coating procedure as described previously (26–28). Quality controls using Western analysis showed >20-fold enrichment for several known EC and plasma membrane markers as well as >20-fold depletion of markers of other cell types and subcellular organelles (26–29).
Mass Spectrometric Analysis of Membrane Proteins
Sample Preparation
Proteins (40 μg unless otherwise noted) in lung plasma membranes and tissue homogenates were solubilized in cell lysis buffer (CLB; 2 m urea, 0.17 m Tris base, 3 mm EDTA, 1.2% β-mercaptoethanol, and 3% SDS). The solubilized samples were boiled for 5 min and were resolved by SDS-PAGE (PAGEr gel, 8–16% T, 10 × 10 cm; Cambrex Bio Science, Inc., Rockland, ME) and visualized by colloidal Coomassie Blue staining (Invitrogen).
In the early sample preparations, the gel lanes were cut into ∼50 slices for manual digestion performed in a 0.5-ml microcentrifuge tube as described in Wilm et al. (30). After purchasing a robot system (MassPrep Station II from Waters Corp., Milford, MA), ∼70 slices were cut to facilitate the in-gel digestion performed in 96-well plates according to the manufacturer's instructions. All of the gel slices were cut into ∼1-mm2 pieces for the digestions. Digested peptides were extracted from the gel slices three times with 20% acetonitrile and 10% formic acid solution. The extracted peptide fractions were transferred into 0.5-ml microcentrifuge tubes individually or combined into seven fractions before lyophilizing (Heto Vacuum Centrifuge, Appropriate Technical Resources, Inc., Laurel, MD).
Strategies of MS Analysis
GRPC—
For gel plus reversed-phase LC/MS/MS analysis using LCQ (Thermo Fisher Scientific, Inc., Waltham, MA) (see Fig. 1), the lyophilized peptides from each gel slice were resuspended in 10 μl of buffer A (0.1% formic acid and 5% acetonitrile) and loaded onto a self-packed C18 microcapillary column (see supplemental information) manually under a helium pressure cell with approximately 600 p.s.i. The bound peptides were eluted with 5–80% acetonitrile gradients containing 0.1% formic acid over a 60-min period as controlled by Agilent 1100 HPLC quaternary pumps (Agilent, Palo Alto, CA) directly coupled to the LCQ equipped with an ESI nanospray ion source (Micro Sprayer, Mass Evolution, TX). The flow rate was maintained at 200–250 nl/min by a precolumn flow splitter, a 50-μm fused silica capillary tube.
Fig. 1.

Proteomics platforms in this study. Schematic illustration of the methodology used to define the EC plasma membrane proteome.
GRPT—
For gel plus reversed-phase LC/MS/MS analysis using the LTQ (Thermo Fisher Scientific, Inc.) (see Fig. 1), the lyophilized peptides from each gel slice were resuspended in 5 μl of buffer A and injected into a 5-mm trap cartridge (Dionex Corp., Sunnyvale, CA) for desalting using a FAMOS autosampler and a Switchos II system (Dionex Corp.). The desalted peptides were then back-eluted onto the analytical column (PepMap 100, C18) for the separation steps. The bound peptides were separated by a 110-min acetonitrile gradient (5–80% containing 0.1% formic acid) controlled by an UltiMate HPLC system (Dionex Corp.) directly coupled to an LTQ equipped with a Nanospray I ion source (Thermo Fisher Scientific, Inc.). The flow rate was maintained at 200–250 nl/min by an internal precolumn flow splitter.
G2DC—
For gel plus 2D LC/MS/MS using LCQ, the peptides extracted from each gel slice were first pooled into seven groups before lyophilization. The dried digests were resuspended with 30 μl of buffer A and then loaded manually into a two-dimensional (strong cation exchange (SCX) and reversed-phase; see supplemental information) self-packed microcapillary column under a helium pressure cell with approximately 600 p.s.i. The loaded samples were directly introduced into an LCQ equipped with ESI nanospray ion source by eluting the bound peptides with a 2D LC/MS/MS scheme (31) controlled by Agilent 1100 HPLC quaternary pumps. Briefly 17 salt steps (ammonium acetate) were applied stepwise as 0, 10, 17.5, 25, 37.5, 50, 62.5, 75, 100, 125, 150, 175, 200, 225, 250, 375, and 500 mm over 2 min to elute the bound peptides from the SCX column. The initial step was a 100-min run with the gradient of 80 min to 60% buffer B (0.1% formic acid and 80% acetonitrile), 10 min to 100% buffer B, and 10 min to 100% buffer A. The gradients from steps 2 to 16 were 3 min at 100% buffer A, 2 min at 2 to 75% buffer C, 10 min to 15% buffer B, and a 97-min gradient to 45% buffer B. The gradient for step 17 was 2 min at 100% buffer A, 20 min at 100% buffer C, 8 min to 15% buffer B, and 110 min to 45% buffer B. The flow rate was maintained at 200–250 nl/min by a precolumn flow splitter of a 50-μm fused silica capillary.
2DC—
For gel-free 2D LC/MS/MS analysis using an LCQ, plasma membrane and lung tissue homogenate proteins (150 μg) were analyzed as described previously (26). Briefly the SDS-solubilized samples were precipitated by methanol/chloroform to remove the detergent and then resolubilized in 8 m urea buffer. In-solution enzymatic digestions were performed by both Lys-C and trypsin. The resultant peptides were desalted before lyophilization. The dried peptide mixture was resuspended in ∼30 μl of buffer A and then loaded manually into a two-dimensional (SCX and reversed-phase; see supplemental information) self-packed microcapillary column under a helium pressure cell with approximately 600 p.s.i. The loaded samples were directly introduced into an LCQ equipped with an ESI nanospray ion source by eluting the bound peptides for 2D LC/MS/MS analysis (26) similar to G2DC described above.
Data Acquisition
For both the LCQ and the LTQ, MS analysis was carried out in data-dependent mode. A full MS scan of data acquisition was performed at the range of 400–1400 m/z, and one MS scan was followed by three MS/MS scans on the most abundant ions. The temperature of the ion transfer tube of both mass spectrometers was set at 180 °C, and the spray voltage was 2.0 kV. The normalized collision energy was set at 35% for both the LCQ and the LTQ. A dynamic exclusion window was applied for a duration of 3 min for reversed-phase LC/MS/MS and 10 min for 2D LC/MS/MS.
Bioinformatics
Database Search
MS/MS spectra were converted into peak lists using the default settings of Extract_msn in Xcalibur 2.0 (Thermo Fisher Scientific, Inc.), and the protein database search was performed with Sequest algorithms in Bioworks 3.1 (Thermo Fisher Scientific, Inc.) using Linux cluster. The forward and reversed databases used in the searches contained human, rat, and mouse sequences downloaded from UniProt and NCBI RefSeq (non-redundant) protein databases April 2006 (see Table I for entry details). The accession numbers for all proteins identified in the final data set were updated based on the January 2009 databases of UniProt, UniParc, and RefSeq. The false positive rate for protein identifications was determined by the ratio of the number of peptides found only in the reversed database searches to the total number of peptides in both forward and reversed database searches. Searches were performed as tryptic peptides only with two missed cleavages and precursor mass tolerance of 1.5 Da for LTQ data, 2.0 Da for LCQ data, and 0.0 Da for fragment ions in both LCQ and LTQ. No modifications were applied to our database searching. Accepted peptide identifications were based on a minimum ΔCn score of 0.1 and minimum cross-correlation scores of 1.8 (z = 1), 2.5 (z = 2), and 3.5 (z = 3). The peptides identified using these criteria were shown to have much lower mass errors compared with other Sequest scores tested (see supplemental information and supplemental Fig. 3). Protein identification results were extracted from Sequest .out files, filtered, and grouped with DTASelect software (32) using the above criteria and a minimum of two unique peptides from the same measurement.
Table I.
Proteins identified using different databases
The table lists the total number of entries in each database, the total number of proteins identified in each database, the total number of unique proteins identified in each database, and the total number of proteins identified in the final data set in this study.
| Total entries in database used | Total number of proteins identified | Number of groups of sorted proteins | Number of unique proteins in rat database | Number of unique proteins in mouse database | Number of unique proteins in human database | Final identified proteins | |
|---|---|---|---|---|---|---|---|
| Rat | 52,881 | 4,652 | 1,891 | 1,608 | 1,608 | ||
| Mouse | 112,998 | 7,818 | 3,178 | 135 | 135 | ||
| Human | 97,361 | 5,760 | 2,345 | 90 | 90 | ||
| Total | 262,200 | 18,230 | 7,414 | 1,608 | 135 | 90 | 1,833 |
Data Analysis
All MS measurements were imported into Accessible Vascular Targets Database (AVATAR), an in-house, MySQL-based relational protein database that facilitates data mining, including grouping and sorting of proteins by shared peptide sequence. Rat protein identifications were supplemented by searching the mouse and human protein databases. If no rat homologue was present for a given protein identification, then the mouse or human homologue with the highest number of peptides and best sequence coverage was kept. Splice isoforms and all proteins exhibiting identical peptide coverage were manually deleted from the data set to ensure that only one representative protein was kept. We also manually removed the most obvious protein contaminants, including keratins, histones, and most ribosomal proteins, from our final data set.
Calculation of Analytical Completeness
To estimate the number of measurements required to reach 95% analytical completion for a given analytical method, all MS measurements for a given method were analyzed to determine the number of new proteins identified by the addition of each new experiment as described previously (26). The percentage of new proteins identified by each replicate was calculated as the average value (±S.D.) of all possible combinations of MS measurements to eliminate any possible bias introduced by the order of the experiments.
Estimating Relative Protein Quantity
The relative quantity of each protein identified was determined by a normalized label-free quantification method, SIN, recently developed in our laboratory. Briefly we first calculated the spectral index (SI) of each protein. As depicted in Equation 1, three major MS characteristics, spectral count, peptide number, and fragment ion intensity, were all considered.
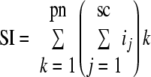 |
(Eq. 1) |
sc is the spectral count for peptide k, i is the fragment ion intensity of each spectrum of peptide k, and pn is the number of peptides identified for that protein.
To rule out the variation introduced by day-to-day sample preparation, instrument performance, and size of the protein, we developed the normalized spectral index (SIN) for each protein identified. The protein SI was normalized by the length of the protein (number of amino acid residues) (L) and the total fragment ion intensity of the n proteins identified in a given MS measurement (Σj= 1n) to yield the normalized spectral index (SIN) for each protein.
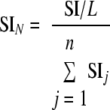 |
(Eq. 2) |
The relative protein quantity (RPQp) of each protein (p) was estimated by converting the SIN values into ng based on Equation 3.
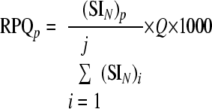 |
(Eq. 3) |
j is the number of all proteins identified with ≥2 unique peptides, and Q is the amount of sample in μg used in a given MS measurement.
Dynamic Range of Detection
The dynamic range for protein detection was measured as the average ratio of the highest RPQ to the lowest RPQ from each measurement of the method.
Enhancement of Protein Coverage
The ratio of the number of peptides detected by each gel-based method, Rpep, relative to 2DC, of common proteins identified by all four methods was calculated as follows.
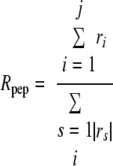 |
(Eq. 4) |
j is the number of proteins identified with more peptides than from 2DC, t is the number of proteins identified with fewer peptides than from 2DC; and r is the log ratio of peptide number relative to that from 2DC of each protein identified.
Western Blot Analysis
Proteins in the samples were first separated by SDS-PAGE and electrotransferred onto nitrocellulose (Bio-Rad). The filter was blocked with 5% milk, immunoblotted with specific primary antibodies, and then incubated with appropriate secondary antibodies according to standard protocols (29). The antibodies were purchased commercially or obtained as gifts from other researchers. They are listed in supplemental Table 1.
RESULTS
In our initial proteomics survey of normal rat lung EC plasma membranes (26), we failed to detect several well known EC markers, many of which are lipid-embedded proteins. IMPs are notorious for their hydrophobicity and resistance to extraction and solubilization, which might contribute to their under-representation in many past proteomics analyses. Here prior to MS analysis, we vigorously solubilized the membrane proteins under denaturing conditions with a strong detergent followed by prefractionation using SDS-PAGE, which is conducive to solubilizing, resolving, and separating IMPs. To further enhance MS-based protein identification by reducing sample complexity, after SDS-PAGE separation each gel lane was cut into at least 50 gel slices before in-gel tryptic digestion and MS analysis by three distinct approaches as illustrated schematically in Fig. 1 and as described under “Experimental Procedures.” Briefly the peptides from each gel slice were subjected to 1D LC/MS/MS analysis, specifically reversed-phase (RP) C18 column separation coupled with either an LCQ or LTQ mass spectrometer (GRPC or GRPT, respectively). These two methods are two-dimensional in terms of protein and peptide separation and thus are categorized collectively as 2D methods. The third approach, G2DC, analyzed the gel-extracted peptides using 2D LC/MS/MS (SCX and RP separation with 17-step elution directly and seamlessly into an LCQ mass spectrometer). This approach utilizes an extra dimension in peptide separation and is categorized as a 3D method. For G2DC, the gel slices after digestion and peptide extraction were pooled into seven fractions prior to MS analysis. Because gel-free 2D LC/MS/MS (2DC), another 2D approach, was used by us previously (26) and has been a popular choice for shotgun proteomics, all results were compared directly with it to gauge relative improvement.
First we assessed the effect of protein amount on the number of proteins identified for the gel-based methods (Fig. 2a). Although we continued to use 150 μg for the 2DC analysis as described previously (26), only 40 μg of the isolated endothelial plasma membranes was necessary to reach near maximum protein identification for the gel-based techniques (Fig. 2a). Thus, the additional gel prefractionation step appeared to reduce the protein amount required for MS analysis.
Fig. 2.

Analysis of protein and unique peptide identifications by all four methods. a, optimal sample quantity for gel-based method. The number of proteins identified (from supposed non-redundant rat database without further removing redundancy) by GRPC based on one, two, or three unique peptides is shown for 10-, 40-, and 150-μg samples. b, number of replicates required for 95% analytical completeness. The number of newly identified proteins (mean ± S.D.) (y axis) of any given measurement is shown as the percentage of total proteins of the indicated number of measurement(s) (x axis). c, total number of proteins identified by each method individually and by all three gel-based methods. The y axis on the left denotes the number of proteins identified by each method, and the y axis on the right shows the ratio relative to 2DC. d, distribution of proteins identified among the four MS methods including all combinations (Three Gs means GRPC, GRPT, and G2DC). The number of proteins identified is provided inside the pie chart, and the percentage is next to the designation of method(s). e, distribution of the unique peptides detected by each method and all combinations. f, histogram of number of proteins identified by each method according to the number of unique peptides for that protein. The number of proteins (graphed on a log10 scale) is plotted by each binned peptide group. g, peptide space map. After binning each protein according to the number of peptides detected in each method, the percentage of peptides identified in each bin for the four methods was calculated and plotted.
To determine the number of salt elution steps needed for the gel-based 2D LC/MS/MS analysis, we examined the peptide elution profiles of the G2DC. Each of the 17 elution steps separated peptides albeit at different levels (details in supplemental information) (supplemental Fig. 1). Thus, we maintained 17 salt step gradients to ensure maximum peptide separation and resolution necessary to facilitate MS detection.
We previously discovered that each MS measurement of a shotgun proteomics analysis identifies only a subset of proteins. Therefore, multiple MS measurements should be performed to comprehensively define the full proteome (26). We defined analytical comprehensiveness statistically from multiple replicate MS measurements of the same sample. Once a level of 95% completion was reached, further MS measurements produced little new information, and thus each became harder to justify (26). 2DC required a minimum of seven replicate measurements as reported previously (26) to reach 95% completion. The gel-based techniques required only four to five replicate measurements per sample (Fig. 2b). Apparently when the overall sample complexity is reduced by SDS-PAGE prefractionation, less sample and fewer replicate MS measurements are necessary to analyze a sample to a statistically defined end point.
Using four different analytical approaches, multiple replicate measurements within each approach, a protein identification criterion of at least two unique tryptic peptides within a single measurement, and the rat database, we identified 14,526 unique peptides that matched to 4,652 proteins. After further eliminating redundancies by selecting one representative protein among isoforms and homologues to determine the smallest number of proteins that could account for all assigned peptides (see “Experimental Procedures”), we identified 1,608 unique rat proteins. After examining mouse and human databases and removing species and further redundancies as described above, we found another 990 (6.8%) unique peptides assigned to 135 (7.4%) mouse proteins and 383 (2.6%) unique peptides assigned to 90 (4.9%) human proteins (Table I). These proteins did not yet have rat homologues in the database used. Thus, 12% of the total 1,833 proteins identified (supplemental Table 2) required the use of the mouse and human databases. Table II lists the various MS instruments, approaches, sample quantities, and throughput and summarizes the number of proteins identified and peptide assignments with each approach in this study. All positive peptide identifications were determined by our optimized Xcorr scores, 1.8 for singly charged, 2.5 for doubly charged, and 3.5 for triply charged peptides. These scores gave us the maximum number of protein identifications while maintaining a false positive rate ≤1.1% (see “Experimental Procedures,” supplemental information, and supplemental Fig. 3).
Table II.
Summary of methods used in this study
This table lists all methods (column 1), protein prefractionation approaches (column 2), mass spectrometers (column 3), sample quantities (column 4), protein and peptide separation strategies (column 5), number of replicates (column 6), sample throughput (columns 7 and 8), and the number of proteins and peptides identified by each approach (columns 9 and 10).
| Method | Protein separation | MS instrument | Sample quantity | Peptide analysis | No. of repetitive analyses | Throughput/ measurement
|
95% analytical completeness
|
Total unique peptides identified | Protein identification
|
|||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time | Relative to 2DC | Time | No. of proteins/total measurement days | No. of proteins identified | Relative to 2DC | |||||||
| μg/run | days | days | ||||||||||
| 2DC | —a | LCQ | 150 | SCX-RP | 10 | 2 | 1 | 14 | 24 | 2,916 | 332 | 1 |
| GRPC | SDS-PAGE | LCQ | 40 | RP | 5 | 10 | 5 | 50 | 11 | 3,883 | 532 | 1.6 |
| GRPT | SDS-PAGE | LTQ | 40 | RP | 5 | 7 | 3.5 | 35 | 40b | 5,320 | 1,110 | 3.3 |
| G2DC | SDS-PAGE | LCQ | 40 | SCX-RP | 5 | 14 | 7 | 70 | 19 | 10,713 | 1,361 | 4.1 |
No further sample purification for this method.
Calculated based on four replicates.
Having defined the number of replicates needed to reach 95% analytical completeness, we performed a series of comparisons of the overall effectiveness of each method to identify proteins as shown in Fig. 2, c–g. Of the four MS methods, the 3D approach G2DC identified the greatest total number of proteins (1,360; 74.2% of the total) followed by GRPT (1,109; 60.5%), and GRPC (531; 29%) (Fig. 2c). 2DC detected the least (331; 18.1%). Addition of the SDS-PAGE prefractionation step apparently enabled G2DC to detect >4 times the number of proteins identified by 2DC (both techniques used similar 2D LC/MS/MS). Although 2DC has been a popular choice, SDS-PAGE combined with 1D LC/MS/MS recently has gained favor. Interestingly for all the gel-resolved samples, the 2D LC/MS/MS was superior to the 1D LC/MS/MS. G2DC identified 2.5 times more proteins than GRPC.
The three gel-based methods, when combined, more than quintupled the protein identifications relative to 2DC (Fig. 2c). A mere 10% of the total proteins were identified commonly by all four approaches (Fig. 2d), whereas 53.2% were found uniquely by a single method (28.8% by G2DC, 21.1% by GRPT, 1.7% by GRPC, and 1.6% unique to 2DC). Overall the gel-based methods accounted for >98% of the total protein identifications. Likewise 95% of the total unique peptides were detected by the three gel-based methods of which 39% were detected by G2DC alone (Fig. 2e). Although the number of protein identifications was very different among the methods, the profiles based on calculated protein size and isoelectric point were rather similar (supplemental Fig. 2).
To assess the relative quality of protein identifications, the identified proteins were binned by the number of peptides detected for each protein by each method. G2DC identified the greatest number of proteins in nearly all bins (Fig. 2f). Relative to 2DC, G2DC exhibited the highest -fold increases, ranging from 1.7- to 5.7-fold (data not shown). A peptide space map (Fig 2g) also showed that G2DC dominated the total peptide space. Overall G2DC accounted for ∼41% of all unique peptides, whereas 2DC accounted for the least, just 10%. G2DC also clearly dominated GRPC (16%). It appears that the increases in protein identifications by the gel-based methods and the extra dimension gained from G2DC in particular enhanced peptide detection.
To evaluate protein coverage impartially among the four methods, we binned the 187 proteins common to all four methods by number of peptides detected by each method, ranging from two to >100 peptides (Fig. 3a). G2DC identified more common proteins with ≥11 peptides than any other method. More than 91% of the common proteins detected by G2DC contained ≥6 peptides, and about 68% contained ≥11 peptides (Fig. 3b). No common proteins were identified with only two peptides by G2DC (100% with ≥3 peptides) (Fig. 3, a and b). By comparison, 2DC detected only two peptides for 10% of the common proteins, and just 30% were identified with ≥11 peptides. The peptide space map (Fig. 3c) revealed that G2DC accounted for >40% of the total peptide space for these common proteins. A direct plot of the number of peptides identified by each gel-based method relative to 2DC for each common protein (Fig. 3d) showed again nearly complete dominance by G2DC with greatly enhanced protein coverage. Only G2DC showed enhanced peptide detection for almost all common proteins. The overall peptide enhancement ratio relative to 2DC was 160.7 for G2DC compared with <4 for the other gel-based methods (see Equation 4 under “Experimental Procedures”). When we normalized peptide numbers per protein by the median value of peptide number detected by 2DC, we found that GRPC and GRPT showed little profile shift. Only G2DC showed substantial change in its peptide distribution profile with nearly 90% of the common proteins exceeding the 2DC median peptide value (Fig. 3e). Thus, it is quite clear that G2DC provided the best protein coverage.
Fig. 3.

Analysis of protein coverage by all four methods. The 187 proteins common to all four MS methods were grouped according to the number of unique peptides detected for that protein. a, histogram of the number of proteins identified by each method (as indicated). b, number of proteins identified by G2DC versus 2DC according to the number of peptide detected, shown as percentage of total common proteins. c, peptide space map of common proteins. d, ratio of number of unique peptides detected in each protein by each gel-based method relative to the number of unique peptides detected by 2DC (log10 scale). The calculated ratio, Rpep, for each gel-based method is indicated (see Equation 4 under “Experimental Procedures”). e, distribution of peptides detected for each common protein. The number of unique peptides detected by each method for each common protein, normalized by median number for 2DC (log10 scale), is plotted against common proteins. The number of proteins with <7 unique peptides (the median for 2DC) is shown next to the orange arrow, the number of proteins with >7 unique peptides is next to the blue arrow, and the number of proteins with ≥7 unique peptides is shown as a percentage (top panel of each method). f, box plots of the distribution of the number of unique peptides in common proteins detected by each method. All values, including smallest values, first quartiles (25% of the data), medians, third quartiles (75% of the data), greatest values, and outliers are graphed on a log2 scale. Raw values for the medians are indicated.
To investigate random effects as a source of the differences observed between the gel-based methods and 2DC, we analyzed the number of peptides assigned to the 187 common proteins by each technique. After logarithmic transformation of the data, analysis of variance demonstrated highly significant differences among the four methods in the number of unique peptides detected for each common protein (F, 36.525; p = 2.2 × 10−16). Tukey's post hoc test (R statistics language, version 2.5.0) showed significant differences between comparisons of G2DC and any other method after multiple comparison adjustment (p < 0.00001 for all G2DC paired tests; p > 0.4 for all other paired tests). These results showed little evidence that random sampling error could account for these differences. Box plots with quartiles further illustrated the significant differences in distributions of unique peptides among the four methods (Fig. 3f). The median value of peptides detected for each protein by G2DC was 15 peptides, more than twice that of the other methods. Clearly the 3D strategy (G2DC) significantly enhanced peptide detection for each protein. Ultimately the G2DC analysis greatly improved overall protein coverage well beyond that detected from GRPT or GRPC analysis.
IMPs are key components of the surface of any cell. They are also abundant proteins, constituting at least 20% of all open reading frames in the genome (33). They are recognized as a special challenge for current standard shotgun proteomics methods (see “Discussion”). In this study, 476 proteins were identified as IMPs with one or more predicted transmembrane helices: 235 with a single transmembrane helix (STM) and 241 with multiple transmembrane helices (MTM) (Fig. 4a). G2DC detected the greatest number of IMPs (377). It also identified more proteins than any other method in both the STM (180) and MTM (197) categories (Fig. 4a). 2DC detected the least number of proteins in all three categories (73, 41, and 32, respectively). Each gel-based method detected 2–6-fold more IMPs than 2DC (Fig. 4b). The gel-based approaches also identified 3- to almost 5-fold more putative lipid-anchored proteins than 2DC (7, 24, 29, and 33 for 2DC, GRPC, GRPT and G2DC, respectively) (Fig. 4b). Even when normalized to all proteins identified as soluble, these gains in detection still remained quite substantial. Only six IMPs were detected by 2DC alone, whereas 471 (99%) were identified by the three gel-based methods (Fig. 4c). In all cases, G2DC provided the greatest improvements. These differences may be predictable because soluble proteins are inherently amenable to in-solution enzymatic digestions (thereby ideal for the 2DC analysis). Hence protein prefractionation by SDS-PAGE appeared to enhance IMP detection by MS/MS analysis. Perhaps more importantly, the 2D LC/MS/MS analysis in G2DC again showed considerable benefits over just 1D LC/MS/MS analysis of the gel; G2DC showed up to 3 times more IMPs than GRPC and 2 times more than GRPT (Fig. 4, a and b).
Fig. 4.

Analysis of integral membrane protein identifications. The total number of soluble proteins, lipid-anchored proteins, all IMPs, and IMPs with STM or MTM identified by each approach (a) and their ratio relative to 2DC (b) are shown. c, heat map representing all proteins identified by all four methods. Proteins are clustered by type of membrane association and color-coded by number of unique peptides detected for each method. Each row represents a specific protein. d, peptide space map of IMPs. e, distribution of unique peptides of IMPs among the four MS methods individually and by all combinations. f, the bar chart represents the number of unique peptides from soluble proteins and IMPs detected by each method individually and by all combinations. g, histogram of the number of IMPs identified by each method in each binned group of number of predicted transmembrane domains. h, box plots depict the distributions, by method, of unique peptides per IMP identified in common to all four method shown in a log2 scale. Raw values for medians are shown. AP, anchored protein; TMH, transmembrane helices; PMP, peripheral membrane protein; TM, transmembrane domains.
To gauge which approach optimally detects IMPs, we compared peptides detected per IMP by each method. A peptide heat map (Fig. 4c) showed the dominance of G2DC not only in unique protein identifications for all protein types including IMPs but also in enhanced protein coverage (redness) for those proteins also detected by other methods. Furthermore the peptide space plots for the identified IMPs showed that the gel-based methods together accounted for 92.4% of all the detected unique peptides (Fig. 4d) of which G2DC alone accounted for 43% (Fig. 4e). Notably >27% of the unique peptides detected by G2DC alone were assigned to IMPs (Fig. 4f). As expected, G2DC detected the greatest number of IMPs in all groups based on the predicted number of transmembrane domains per protein, especially the proteins containing >9 transmembrane domains (zero proteins detected by 2DC, three by GRPC, 11 by GRPT, and 19 by G2DC) (Fig. 4g). Lastly both the non-parametric Kruskal-Wallis rank sum test and the Wilcoxon signed rank test confirmed significant differences among all four methods (Kruskal-Wallis χ2, 430.74; p = 4.86 × 10−93; and p < 10−6 for all paired tests).
To further rule out random effects causing these differences among the methods, we performed an analysis of variance on the 49 IMPs common to all four methods. We found highly significant differences among the four methods in the number of unique peptides (F, 23.245; p = 6.5 × 10−13). Tukey's post hoc test showed significant differences between G2DC and any other method after multiple comparison adjustment (p < 0.00001 for all G2DC paired tests; p > 0.05 for all other paired tests). Box plots demonstrated considerable discrepancy in distributions of unique peptides among the four methods (Fig. 4h). Each gel-based method exhibited some improvement in IMP coverage over 2DC. G2DC was by far the best in peptide detection with a median of 14 peptides per common IMP, nearly triple that of 2DC and double that of the other gel-based methods.
To appraise the detection sensitivity limit and dynamic range for each technique, we used the normalized spectral index (SIN) to estimate the quantity (in ng) of each protein (see Equations 1–3 under “Experimental Procedures”) and plotted the frequency of proteins detected below 100 ng. As shown in Fig. 5a, the lower limit of meaningful protein identification was estimated at ≥2 ng for 2DC, whereas each gel-based method exhibited much better sensitivity. Again G2DC appeared to be the best with the most sensitive detection at <0.1 ng for both soluble proteins (blue bars) and IMPs (red bars). Of the proteins detected at <0.1 ng by G2DC, >93% were not detected by 2DC even though 2DC used more sample (150 versus 40 μg). Hence G2DC revealed improved sensitivity by at least 1 order of magnitude over 2DC. Again the 3D method provided considerable benefit over the three 2D analysis methods with obvious enhancement in detection sensitivity. Thus, both the SDS-PAGE prefractionation and the 2D LC/MS/MS analysis contributed to the expanded dynamic range of the shotgun proteomics analysis.
Fig. 5.

Relative sensitivity of each technique. a, soluble proteins and IMPs from the original data were binned based on their estimated relative quantity (see “Experimental Procedures,” Equations 1–3). The number of proteins identified within each group is shown as the percentage of total proteins for each method. b, all proteins identified by the three gel-based methods were graphed in the order of their averaged estimated quantity (in ng) (in red following the x axis on the bottom). The distribution of their CVs is shown as a trend line of a period of 100 data points (in black following the x axis on the top).
Next we averaged the estimated quantity of each protein identified by the three gel-based methods (Fig. 5b). The coefficient of variation (CV) of each protein was also calculated to assess the deviation introduced by both intra- and intermethodological comparisons. The results indicated that, by combining the three gel-based methods, we could detect proteins over at least 5 orders of magnitude (0.007–155.4 ng). Although each data point was averaged from three different methods, the data generated showed low variance (CV < 1; mean CV = 0.72) consistent with good rigor in MS analysis as well as proper normalization. We detected at least 30% lower variance if we calculated the CV separately for each method (mean CV = 0.43 for GRPC, mean CV = 0.51 for GRPT, and mean CV = 0.50 for G2DC).
It was clear that G2DC outperformed the other methods in protein identifications (especially IMPs), protein coverage, sensitivity, and dynamic range. Nevertheless a considerable number of the proteins identified were unique to the other methods (Fig. 2d). To determine the extent of enhanced comprehensiveness with two or more methods, we compared the numbers of total proteins and IMPs detected in all combinations of approaches (Fig. 6). G2DC alone (Fig. 6, orange arrow) or combined with any of the other three methods produced consistently more proteins than any single or dual combination of 2DC, GRPC, and GRPT. In particular, GRPT plus G2DC identified >95% of total proteins in all categories (96% for total, 96% for IMPs, 95% for STM proteins, and 97% for MTM proteins). Combining three or even all four methods produced little improvement beyond GRPT plus G2DC (as illustrated by the plateau in Fig. 6, blue arrow). Combining G2DC with GRPT exceeded 95% analytical comprehensiveness.
Fig. 6.

Proteins identified by combined MS approaches. The percentage of the total number of proteins identified is plotted against the individual method and all combinations of methods. Upper left panel, total proteins; upper right panel, all IMPs; lower left panel, STM proteins; and lower right panel, MTM proteins. Orange arrows, G2DC alone; blue arrows, G2DC joined with GRPT. Three Gs means GRPC, GRPT, and G2DC.
As mentioned previously, our initial survey of the EC plasma membrane proteome failed to detect several well known EC markers, many of which are IMPs. To begin to understand why so many IMPs escaped detection by 2DC, we investigated the sample preparation procedures for 2DC. Most membrane proteins are best solubilized by ionic detergents such as SDS. However, SDS is not compatible with trypsin digestion so that in the sample processing for 2DC the SDS-solubilized proteins must first be precipitated before being resolubilized for trypsin digestion (usually in a urea solution) (Fig. 1). To assess possible protein loss during the precipitation and resolubilization steps, equal amounts of purified plasma membranes were processed in three ways. (i) Proteins were directly solubilized in CLB for SDS-PAGE, (ii) CLB-solubilized proteins were precipitated and then resolubilized by urea (as in the 2DC process), and (iii) CLB-solubilized proteins were precipitated and then resolubilized by CLB. The resolubilized samples from ii and iii were centrifuged, and the supernatants were collected for further tests. The proteins in these fractions were separated by SDS-PAGE and electrotransferred to filters for immunoblotting to detect specific proteins (Fig. 7). As expected, the more soluble proteins such as ezrin and calpain-5 were equivalently present regardless of process and showed minimal losses after precipitation and urea resolubilization. However, the signals of IMPs and even lipid-anchored proteins were significantly affected by sample processing. Some proteins were not affected by the precipitation step but did not resolubilize well in urea solution, such as claudin-5 junctional adhesion molecule A, and the glycosylphosphatidylinositol-anchored protein contactin. Their signal was reduced in the urea lane, which typified the procedure used in 2DC. Other proteins showed a dramatic loss in signal from the precipitation step regardless of resolubilization conditions, such as VAP-1, GPCR116, and the lipid-anchored protein Gαq. The sample loss for 2DC was fairly apparent, ranging from 0% for soluble proteins to >90% for many lipid-embedded proteins. A 10-fold or more decrease of a protein in the final analyte can greatly hinder its downstream detection. These results help to explain the challenges encountered by the 2DC method in identifying certain proteins, particularly proteins embedded in the lipid bilayer. They also provide further rationale favoring SDS-PAGE beyond just any prefractionation scheme that simply separates soluble proteins well to reduce sample complexity. The additional power of SDS-PAGE to resolve nearly all IMPs and lipid-anchored proteins with minimal losses clearly contributed substantially to (i) G2DC dominance over 2DC, (ii) increased overall peptide detections and protein identifications by MS, (iii) greater sensitivity, especially relative to 2DC, and (iv) enhanced identification of membrane-embedded proteins, many of which are simply lost during sample processing for 2DC.
Fig. 7.

Protein partitioning during sample processing for 2DC. An immunoblot of plasma membrane proteins treated with different procedures and reagents is shown. Membrane samples were solubilized directly in CLB for SDS-PAGE, or CLB-solubilized proteins were first precipitated and then resolubilized by either 8 m urea as for 2DC or CLB. The resolubilized samples from the precipitations (Precip.) were centrifuged, and the supernatants were collected for SDS-PAGE. The number of putative or known transmembrane helices (TMH) and number of unique peptides detected by each method are shown in the table on the right. 0*, partially membrane-embedded with high hydrophobic region; Lipid-AP, lipid-anchored proteins; GPI-AP, glycosylphosphatidylinositol-anchored proteins. See supplemental Table 1 for information on antibodies and protein names. VE-cad, VE-cadherin; JAM A, junctional molecule A.
DISCUSSION
Although recent improvements in MS instrumentation provide higher sensitivity and resolution, there remains a need for reduced sample complexity and improved detection of lower abundance proteins, especially IMPs. Various strategies to enhance MS-based proteomics analysis have been developed, such as different off-line sample fractionations and different mass spectrometers with different ion sources and different search engines (34, 35). Recent studies on various isolated cell membranes have used different instruments (36), ionization techniques (11), different temperatures of C18 column (37), and protein separation methods (11, 38) to identify 107 to >1306 proteins, many based on one peptide identifications. Organelles isolated from mammalian cells and tissues are rich in lipid membranes and have also been intensively studied by various proteomics approaches, mostly using SDS-PAGE combined with reversed-phase LC/MS/MS. These studies (39–44) identified 67 to >2710 proteins from Golgi, nucleus, mitochondria, endosomes, proteasomes, and microsomes isolated from various tissues based on at least one peptide criterion.
Here we combined four different MS-based proteomics approaches to identify 1,833 proteins using a criterion of two peptides in any single MS measurement. Of the total proteins identified here, almost 30% are embedded in the lipid membrane. In comparison with our past 2DC study (∼15% lipid membrane-embedded proteins) (26), we identified significantly more lipid membrane-embedded proteins in this current work (>30%). Classically plasma membranes are isolated by zonal centrifugation using a density gradient or by two-phase partitioning. They are often processed further by affinity purification and selective solubilizations (10, 18, 45). These techniques are designed to eliminate proteins not well intercalated in the membrane and in so doing to enrich for IMPs. In our plasma membrane preparation, we avoided such treatments to allow the endogenous spectrum of protein types to remain. This allows a more diverse, realistic, and accurate characterization of the membranome, including relative abundance of each protein.
Using the estimated protein quantity, we showed that the three gel-based methods identified proteins over a dynamic range of ∼5 orders of magnitude. 2DC data were excluded from this calculation because of the protein loss during sample processing and the resultant measurement uncertainty and insensitivity (Fig. 7). Lastly it may be noteworthy that eliminating data redundancy based on a genecentric parsimony principle should be the practice but can be very time-consuming. In this study, we indeed removed all redundancy to prevent inflation in the number of protein identifications.
Our past 2DC proteomics mapping of the endothelial plasma membranes identified 450 proteins (26) using a three-peptide identification criterion that counts peptides from any of the 12 MS measurements performed rather than peptides from a single MS measurement. In this study, we used more stringent overall analytical criteria yet still found that the gel-based 3D approach greatly increases the comprehensiveness of the analysis by >4-fold. We detected more proteins including IMPs and known EC marker proteins (>100). Specifically we detected the well known protein-tyrosine kinase receptors (VEGFR-1 and ephrin receptors), semaphorin receptors (plexin D1, plexin B2, and neuropilin-1), and multipass IMPs (apelin receptor, GPCR-124, GPCR-116, γ-aminobutyric acid, type A receptor, vasoactive intestinal polypeptide receptor, latrophilin-2, latrophilin-3, and P2X purinoceptor 7). The advantages of SDS-PAGE prefractionation and 2D LC/MS/MS analysis include enhanced protein coverage, dynamic range, and sensitivity of detection. Perhaps most importantly, this step clearly improves the detection of lipid-embedded proteins.
Although 2DC has been a popular method in the field, more recently SDS-PAGE followed by 1D LC/MS/MS analysis has gained considerable favor. Unfortunately most studies do not perform multiple replicate MS measurements, and even fewer attempt to define the MS analysis statistically, ultimately to achieve a level of 95% analytical completeness. To our knowledge, we are the first to combine SDS-PAGE with continuous 2D LC/MS/MS and repetitive measurements and to compare analyses of four such technologies. We found one report in the literature that identified 544 proteins (one peptide/protein criterion) in rat brain membranes (of which <11% were IMPs) by using SDS-PAGE and trypsin digestion of gel slices and then performing batch elution of peptides from SCX off line. These SCX fractions were individually analyzed by C18 reversed-phase LC/MS/MS (46). Our 3D approach is seamlessly automated to provide continuous analysis necessary for the greater throughput required in large scale applications.
Here we used SDS-PAGE to prefractionate the membrane proteins according to size before 2D LC/MS/MS analysis. The SDS-PAGE obviates the problem of trying to digest IMPs in solution as depicted in Fig. 7. The IMPs do not tend to stay in solution without the presence of detergents and thus are not readily amenable to trypsin digestions in solution. The comparisons between 2DC and G2DC revealed the enormous benefit of prefractionation by SDS-PAGE over in-solution trypsin digestion to generate peptides for detection by conventional 2D LC/MS/MS. The gain in part comes from the reduced sample complexity afforded by any prefractionation with sufficient protein resolving and separating power. More importantly as illustrated in part in Fig. 7, the improved solubilization and resolution for IMPs and lipid-anchored proteins by SDS-PAGE significantly enhances MS identification and thus provides an additional remarkable advantage over other protein separation techniques not so well suited for IMPs.
G2DC is obviously the superior gel-based method among the three gel-based approaches in this study, indicating the importance of the second dimension in the nano-HPLC separation of complex peptides in the MS analysis. This extra separation can greatly improve sensitivity by diminishing peptide co-elution and thus enhancing identification of less abundant peptides not previously detected. Using the same mass spectrometer and same prefractionation, G2DC clearly outperformed GRPC even though GRPC individually analyzes ∼50 gel slices of equivalent sample, whereas G2DC uses only seven such groupings. We appear to gain more from the second dimension of peptide separation by nano-HPLC than from the 7-fold decrease in sample complexity. This advantage is neither obvious nor completely predictable a priori to these results. This benefit appears rather considerable when comparing G2DC (using LCQ) versus GRPT (using LTQ). GRPT not only uses a more powerful and sensitive mass spectrometer but also individually analyzes ∼70 gel slices, ultimately reducing sample complexity 10-fold over G2DC. As summarized in Table III, when each gradient step used to elute the peptides from the columns before MS/MS analysis is considered, 119 MS-analyzed steps were performed by the combination of SDS-PAGE and 2D nano-HPLC in G2DC. Thus, G2DC intrinsically reduced sample complexity beyond that of 2DC, GRPC, and GRPT methods (3D versus 2D). The 2D nano-HPLC peptide separation scheme continues to be an important foundation to enhance protein identifications, sensitivity, and dynamic range of detection.
Table III.
Summary of sample fractions applied per MS measurement
The number of fractions generated by protein prefractionation (column 2), type of chromatography used in peptide separation (column 3), HPLC elution steps (column 4), total number of in-line HPLC elution steps per measurement (column 5), and number of peptides from the proteins identified by each MS step (column 6) are listed by method.
| Method | Pre-MS fraction | Nano-HPLC dimension | Peptide elution step | Total MS steps | No. of peptides/step |
|---|---|---|---|---|---|
| 2DC | 1 | 2 (SCX-RP) | 17 | 17 | 171 |
| GRPC | 43–50 | 1 (RP) | 1 | 43–50 | 106 |
| GRPT | 70–80 | 1 (RP) | 1 | 70–80 | 253 |
| G2DC | 7 | 2 (SCX-RP) | 17 | 119 | 308 |
All large scale proteomics investigations must balance high throughput with data quality. Large sample analyses on a daily basis require rapid sample turnover so that the speed of the instrumentation and methodology will be a major priority. In contrast, if the goal is greater protein coverage or protein identifications, more detailed and comprehensive studies will be essential. Taking 95% analytical completeness as the desired end point, 2DC requires at least seven replicate MS measurements, each using 2 days of instrument time to yield 14 total measurement days and identifies an average of ∼17 new proteins per day. Similarly gel-based methods require no more than five replicate measurements to reach 95% analytical completeness. Thus, GRPC needs 50 total measurement days and identifies 10 proteins per day. GRPT needs 35 total measurement days and identifies 40 proteins per day, whereas G2DC needs 70 total measurement days and identifies ∼20 proteins per day. The autosampler being used with the LTQ is likely to reduce overall performance but probably enhances overall efficiency and throughput. In our hands, GRPT can generate twice the number of identified proteins as GRPC, but the overall number of peptides identified was less than with GRPC (Figs. 3f and 4h). Furthermore our analysis of method combinations showed that G2DC plus GRPT identifies ≥95% of the total proteins identified by all four methods. Although this is obviously the most efficient combination, the gain of nearly 400 proteins beyond G2DC alone requires 35 total measurement days (via GRPT) to yield about 10 proteins not detected by G2DC per total measurement days.
The methods developed and described in this study may have great importance for disease biomarker and target discovery efforts. Just because a protein is not detected in one or two MS measurements does not mean it is not present in the sample. A single MS measurement of a complex protein mixture likely provides only a small fraction of the total protein space. For example, if 10% of each space is analyzed then any protein appearing distinct between samples is very likely to be in the remaining 90% not yet identified. Hence comparison of distinct samples by single MS measurements would seem fruitless and may explain the paucity of candidate biomarkers that have been validated to date. Moreover using a statistically defined end point not only helps eliminate the inherent variance between single MS measurements but also increases the replicability of findings between different laboratories as well as improves the utility of future MS-based analyses (47).
It is clear that higher stringencies, including reaching 95% analytical completeness, are not optional but actually required for identifying meaningful protein distinctions between different biological samples; however, doing so requires 5–10 MS measurements of each distinct sample. The choice of balancing machine times and methods to accomplish analytical comprehensiveness will be a personal one. For our laboratory, we plan to adhere to a criterion of 95% analytical completion. The extra MS measurement days may well be time well spent to avoid a long list of candidates that may be temporarily satisfying yet ultimately overwhelming with few actual “true positives.” Furthermore up until now, membrane-embedded proteins have been notoriously difficult to detect by past traditional MS methods. Although throughput and efficiency were more modest when using G2DC (Table II), the significantly higher number of peptides and proteins identified, especially IMPs, and the enhanced quality of the data (i.e. coverage) may well be worth the extra time invested (Figs. 2–5 and Table III).
The results described here constitute the most comprehensive characterization to date of plasma membranes, specifically luminal EC plasma membranes isolated from rat lung. By combining data from multiple gel-based MS studies and a gel-free MS analysis, we expanded the EC plasma membrane proteome from a few hundred proteins to nearly 2,000 including >100 known EC markers and >500 lipid-embedded proteins. Systematic, comparative analyses of the different MS methods used here revealed that a 3D approach combining SDS-PAGE prefractionation with 2D-LC/MS/MS analysis enhances protein identification, especially of IMPs, improves overall protein coverage, reduces the number of replicates required, allows the use of less starting material, and increases detection sensitivity and dynamic range. Notably these gains were made despite the higher stringency imposed (tryptic peptide only, two-peptide criterion within a single measurement, better mass tolerances, and Sequest scores) than in our past work (26) and thus increase our confidence in the expanded data set. Mapping the cell surface membranomes, especially lipid-embedded proteins on the luminal surface of in vivo EC plasma membranes, is an essential prelude to the discovery of tissue- and disease-specific markers. The unique comparison of 2D and 3D strategies, the repeated measurements within each technique until 95% completeness was achieved statistically, and the label-free protein quantification described here may help establish new standards for analytical comprehensiveness in membranome mapping while assisting future biomarker discovery efforts.
Acknowledgments
We thank Kerri Massey for critical reading of the manuscript and fruitful discussion. We thank Dr. John Yates (The Scripps Research Institute) for kindly providing the DTASelect software. We thank Dr. J. Abe (Department of Biological Sciences, Tokyo Institute of Technology, Tokyo, Japan) for the GPCR116 antibody, Dr. Z. Chen (Department of Neuropharmacology, The Scripps Research Institute) for the Claudin-5 antibody, and Dr. M. Kinoshita (Department of Cell Biology, Harvard Medical School) for the Septin-2 antibody.
Footnotes
Published, MCP Papers in Press, January 19, 2009, DOI 10.1074/mcp.M800215-MCP200
The abbreviations used are: IMP, integral membrane protein; LCQ, LCQ DecaXP mass spectrometer; LTQ, LTQ mass spectrometer; 2D, two-dimensional; 1D, one-dimensional; 3D, three-dimensional; 2DC, gel-free 2D LC/MS/MS on line with the LCQ DecaXP mass spectrometer; G2DC, gel-based 2D LC/MS/MS SDS-PAGE on line with the LCQ DecaXP mass spectrometer; GRPC, gel-based 1D (reversed-phase) LC/MS/MS on line with the LCQ DecaXP mass spectrometer; GRPT, gel-based 1D (reversed-phase) LC/MS/MS on line with the LTQ mass spectrometer; EC, endothelial cell; TMH, transmembrane helices; STM, single transmembrane helix; MTM, multiple transmembrane helices; CLB, cell lysis buffer; SCX, strong cation exchange; SI, spectral index; RPQ, relative protein quantity; RP, reversed-phase; CV, coefficient of variation.
This work was supported, in whole or in part, by National Institutes of Health Grants R24CA95893, R01HL58216, R01CA83989, R01HL074063, R33CA118602, and PO1CA164898 (to J. E. S.).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Josic, D., and Clifton, J. G. ( 2007) Mammalian plasma membrane proteomics. Proteomics 7, 3010–3029 [DOI] [PubMed] [Google Scholar]
- 2.Chen, E. I., Hewel, J., Felding-Habermann, B., and Yates, J. R., III ( 2006) Large scale protein profiling by combination of protein fractionation and multidimensional protein identification technology (MudPIT). Mol. Cell. Proteomics 5, 53–56 [DOI] [PubMed] [Google Scholar]
- 3.Koch, H. B., Zhang, R., Verdoodt, B., Bailey, A., Zhang, C. D., Yates, J. R., III, Menssen, A., and Hermeking, H. ( 2007) Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6, 205–217 [DOI] [PubMed] [Google Scholar]
- 4.Wang, Y., Rudnick, P. A., Evans, E. L., Li, J., Zhuang, Z., Devoe, D. L., Lee, C. S., and Balgley, B. M. ( 2005) Proteome analysis of microdissected tumor tissue using a capillary isoelectric focusing-based multidimensional separation platform coupled with ESI-tandem MS. Anal. Chem. 77, 6549–6556 [DOI] [PubMed] [Google Scholar]
- 5.Wasinger, V. C., Locke, V. L., Raftery, M. J., Larance, M., Rothemund, D., Liew, A., Bate, I., and Guilhaus, M. ( 2005) Two-dimensional liquid chromatography/tandem mass spectrometry analysis of Gradiflow fractionated native human plasma. Proteomics 5, 3397–3401 [DOI] [PubMed] [Google Scholar]
- 6.Wetterhall, M., Palmblad, M., Hakansson, P., Markides, K. E., and Bergquist, J. ( 2002) Rapid analysis of tryptically digested cerebrospinal fluid using capillary electrophoresis-electrospray ionization-Fourier transform ion cyclotron resonance-mass spectrometry. J. Proteome Res. 1, 361–366 [DOI] [PubMed] [Google Scholar]
- 7.Breci, L., Hattrup, E., Keeler, M., Letarte, J., Johnson, R., and Haynes, P. A. ( 2005) Comprehensive proteomics in yeast using chromatographic fractionation, gas phase fractionation, protein gel electrophoresis, and isoelectric focusing. Proteomics 5, 2018–2028 [DOI] [PubMed] [Google Scholar]
- 8.Shin, J. H., Krapfenbauer, K., and Lubec, G. ( 2006) Large-scale identification of cytosolic mouse brain proteins by chromatographic prefractionation. Electrophoresis 27, 2799–2813 [DOI] [PubMed] [Google Scholar]
- 9.Tang, H. Y., Ali-Khan, N., Echan, L. A., Levenkova, N., Rux, J. J., and Speicher, D. W. ( 2005) A novel four-dimensional strategy combining protein and peptide separation methods enables detection of low-abundance proteins in human plasma and serum proteomes. Proteomics 5, 3329–3342 [DOI] [PubMed] [Google Scholar]
- 10.Blonder, J., Conrads, T. P., Yu, L. R., Terunuma, A., Janini, G. M., Issaq, H. J., Vogel, J. C., and Veenstra, T. D. ( 2004) A detergent- and cyanogen bromide-free method for integral membrane proteomics: application to Halobacterium purple membranes and the human epidermal membrane proteome. Proteomics 4, 31–45 [DOI] [PubMed] [Google Scholar]
- 11.Burré, J., Beckhaus, T., Schägger, H., Corvey, C., Hofmann, S., Karas, M., Zimmermann, H., and Volknandt, W. ( 2006) Analysis of the synaptic vesicle proteome using three gel-based protein separation techniques. Proteomics 6, 6250–6262 [DOI] [PubMed] [Google Scholar]
- 12.Chen, P., Li, X., Sun, Y., Liu, Z., Cao, R., He, Q., Wang, M., Xiong, J., Xie, J., Wang, X., and Liang, S. ( 2006) Proteomic analysis of rat hippocampal plasma membrane: characterization of potential neuronal-specific plasma membrane proteins. J. Neurochem. 98, 1126–1140 [DOI] [PubMed] [Google Scholar]
- 13.Coughenour, H. D., Spaulding, R. S., and Thompson, C. M. ( 2004) The synaptic vesicle proteome: a comparative study in membrane protein identification. Proteomics 4, 3141–3155 [DOI] [PubMed] [Google Scholar]
- 14.Everberg, H., Leiding, T., Schioth, A., Tjerneld, F., and Gustavsson, N. ( 2006) Efficient and non-denaturing membrane solubilization combined with enrichment of membrane protein complexes by detergent/polymer aqueous two-phase partitioning for proteome analysis. J. Chromatogr. A 1122, 35–46 [DOI] [PubMed] [Google Scholar]
- 15.Gauthier, D. J., Gibbs, B. F., Rabah, N., and Lazure, C. ( 2004) Utilization of a new biotinylation reagent in the development of a nondiscriminatory investigative approach for the study of cell surface proteins. Proteomics 4, 3783–3790 [DOI] [PubMed] [Google Scholar]
- 16.Ihling, C., Berger, K., Hofliger, M. M., Fuhrer, D., Beck-Sickinger, A. G., and Sinz, A. ( 2003) Nano-high-performance liquid chromatography in combination with nano-electrospray ionization Fourier transform ion-cyclotron resonance mass spectrometry for proteome analysis. Rapid Commun. Mass Spectrom. 17, 1240–1246 [DOI] [PubMed] [Google Scholar]
- 17.Schindler, J., Jung, S., Niedner-Schatteburg, G., Friauf, E., and Nothwang, H. G. ( 2006) Enrichment of integral membrane proteins from small amounts of brain tissue. J. Neural Transm. 113, 995–1013 [DOI] [PubMed] [Google Scholar]
- 18.Schindler, J., Lewandrowski, U., Sickmann, A., Friauf, E., and Nothwang, H. G. ( 2006) Proteomic analysis of brain plasma membranes isolated by affinity two-phase partitioning. Mol. Cell. Proteomics 5, 390–400 [DOI] [PubMed] [Google Scholar]
- 19.Shin, B. K., Wang, H., Yim, A. M., Le Naour, F., Brichory, F., Jang, J. H., Zhao, R., Puravs, E., Tra, J., Michael, C. W., Misek, D. E., and Hanash, S. M. ( 2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 278, 7607–7616 [DOI] [PubMed] [Google Scholar]
- 20.Zhang, L. J., Wang, X. E., Peng, X., Wei, Y. J., Cao, R., Liu, Z., Xiong, J. X., Yin, X. F., Ping, C., and Liang, S. ( 2006) Proteomic analysis of low-abundant integral plasma membrane proteins based on gels. CMLS Cell. Mol. Life Sci. 63, 1790–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, W., Zhou, G., Zhao, Y., and White, M. A. ( 2003) Affinity enrichment of plasma membrane for proteomics analysis. Electrophoresis 24, 2855–2863 [DOI] [PubMed] [Google Scholar]
- 22.Zhao, Y., Zhang, W., Kho, Y., and Zhao, Y. ( 2004) Proteomic analysis of integral plasma membrane proteins. Anal. Chem. 76, 1817–1823 [DOI] [PubMed] [Google Scholar]
- 23.Schnitzer, J. E. ( 1998) Vascular targeting as a strategy for cancer therapy. N. Engl. J. Med. 339, 472–474 [DOI] [PubMed] [Google Scholar]
- 24.Oh, P., Li, Y., Yu, J., Durr, E., Krasinska, K. M., Carver, L. A., Testa, J. E., and Schnitzer, J. E. ( 2004) Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature 429, 629–635 [DOI] [PubMed] [Google Scholar]
- 25.Oh, P., Borgstrom, P., Witkiewicz, H., Li, Y., Borgstrom, B. J., Chrastina, A., Iwata, K., Zinn, K. R., Baldwin, R., Testa, J. E., and Schnitzer, J. E. ( 2007) Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat. Biotechnol. 25, 327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durr, E., Yu, J., Krasinska, K. M., Carver, L. A., Yates, J. R. I., Testa, J. E., Oh, P., and Schnitzer, J. E. ( 2004) Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat. Biotechnol. 22, 985–992 [DOI] [PubMed] [Google Scholar]
- 27.Schnitzer, J. E., McIntosh, D. P., Dvorak, A. M., Liu, J., and Oh, P. ( 1995) Separation of caveolae from associated microdomains of GPI-anchored proteins. Science 269, 1435–1439 [DOI] [PubMed] [Google Scholar]
- 28.Oh, P., Caver, L., and Schnitzer, J. E. ( 2006) Isolation and subfractionation of plasma membranes to purify caveolae separately from lipid rafts, in Cell Biology: a Laboratory Handbook (Celis, J. E., ed) 3rd Ed., pp. 11–26, Elsevier, Amsterdam
- 29.Oh, P., and Schnitzer, J. E. ( 2001) Segregation of heterotrimeric G proteins in cell surface microdomains. Gq binds caveolin to concentrate in caveolae, whereas Gi and Gs target lipid rafts by default. Mol. Biol. Cell 12, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilm, M., Shevchenko, A., Houthaeve, T., Breit, S., Schweigerer, L., Fotsis, T., and Mann, M. ( 1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469 [DOI] [PubMed] [Google Scholar]
- 31.Washburn, M. P., Wolters, D., and Yates, J. R., III ( 2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 32.Tabb, D. L., McDonald, W. H., and Yates, J. R., III ( 2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallin, E., and von Heijne, G. ( 1998) Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kapp, E. A., Schutz, F., Connolly, L. M., Chakel, J. A., Meza, J. E., Miller, C. A., Fenyo, D., Eng, J. K., Adkins, J. N., Omenn, G. S., and Simpson, R. J. ( 2005) An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics 5, 3475–3490 [DOI] [PubMed] [Google Scholar]
- 35.Li, X., Gong, Y., Wang, Y., Wu, S., Cai, Y., He, P., Lu, Z., Ying, W., Zhang, Y., Jiao, L., He, H., Zhang, Z., He, F., Zhao, X., and Qian, X. ( 2005) Comparison of alternative analytical techniques for the characterisation of the human serum proteome in HUPO Plasma Proteome Project. Proteomics 5, 3423–3441 [DOI] [PubMed] [Google Scholar]
- 36.Stevens, S. M., Jr., Zharikova, A. D., and Prokai, L. ( 2003) Proteomic analysis of the synaptic plasma membrane fraction isolated from rat forebrain. Brain Res. Mol. Brain Res. 117, 116–128 [DOI] [PubMed] [Google Scholar]
- 37.Speers, A. E., Blackler, A. R., and Wu, C. C. ( 2007) Shotgun analysis of integral membrane proteins facilitated by elevated temperature. Anal. Chem. 79, 4613–4620 [DOI] [PubMed] [Google Scholar]
- 38.Blonder, J., Terunuma, A., Conrads, T. P., Chan, K. C., Yee, C., Lucas, D. A., Schaefer, C. F., Yu, L. R., Issaq, H. J., Veenstra, T. D., and Vogel, J. C. ( 2004) A proteomic characterization of the plasma membrane of human epidermis by high-throughput mass spectrometry. J. Investig. Dermatol. 123, 691–699 [DOI] [PubMed] [Google Scholar]
- 39.Foster, L. J., de Hoog, C. L., Zhang, Y., Zhang, Y., Xie, X., Mootha, V. K., and Mann, M. ( 2006) A mammalian organelle map by protein correlation profiling. Cell 125, 187–199 [DOI] [PubMed] [Google Scholar]
- 40.Mootha, V. K., Bunkenborg, J., Olsen, J. V., Hjerrild, M., Wisniewski, J. R., Stahl, E., Bolouri, M. S., Ray, H. N., Sihag, S., Kamal, M., Patterson, N., Lander, E. S., and Mann, M. ( 2003) Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 115, 629–640 [DOI] [PubMed] [Google Scholar]
- 41.Johnson, D. T., Harris, R. A., French, S., Blair, P. V., You, J., Bemis, K. G., Wang, M., and Balaban, R. S. ( 2007) Tissue heterogeneity of the mammalian mitochondrial proteome. Am. J. Physiol. 292, C689–C697 [DOI] [PubMed] [Google Scholar]
- 42.Kislinger, T., Cox, B., Kannan, A., Chung, C., Hu, P., Ignatchenko, A., Scott, M. S., Gramolini, A. O., Morris, Q., Hallett, M. T., Rossant, J., Hughes, T. R., Frey, B., and Emili, A. ( 2006) Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell 125, 173–186 [DOI] [PubMed] [Google Scholar]
- 43.Forner, F., Foster, L. J., Campanaro, S., Valle, G., and Mann, M. ( 2006) Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteomics 5, 608–619 [DOI] [PubMed] [Google Scholar]
- 44.Gilchrist, A., Au, C. E., Hiding, J., Bell, A. W., Fernandez-Rodriguez, J., Lesimple, S., Nagaya, H., Roy, L., Gosline, S. J., Hallett, M., Paiement, J., Kearney, R. E., Nilsson, T., and Bergeron, J. J. ( 2006) Quantitative proteomics analysis of the secretory pathway. Cell 127, 1265–1281 [DOI] [PubMed] [Google Scholar]
- 45.Cao, R., Li, X., Liu, Z., Peng, X., Hu, W., Wang, X., Chen, P., Xie, J., and Liang, S. ( 2006) Integration of a two-phase partition method into proteomics research on rat liver plasma membrane proteins. J. Proteome Res. 5, 634–642 [DOI] [PubMed] [Google Scholar]
- 46.Lohaus, C., Nolte, A., Bluggel, M., Scheer, C., Klose, J., Gobom, J., Schuler, A., Wiebringhaus, T., Meyer, H. E., and Marcus, K. ( 2007) Multidimensional chromatography: a powerful tool for the analysis of membrane proteins in mouse brain. J. Proteome Res. 6, 105–113 [DOI] [PubMed] [Google Scholar]
- 47.Service, R. F. ( 2008) Proteomics. Proteomics ponders prime time. Science 321, 1758–1761 [DOI] [PubMed] [Google Scholar]