

Abstract
Most cellular processes are performed by proteomic units that interact with each other. These units are often stoichiometrically stable complexes comprised of several proteins. To obtain a faithful view of the protein interactome we must view it in terms of these basic units (complexes and proteins) and the interactions between them. This study makes two contributions toward this goal. First, it provides a new algorithm for reconstruction of stable complexes from a variety of heterogeneous biological assays; our approach combines state-of-the-art machine learning methods with a novel hierarchical clustering algorithm that allows clusters to overlap. We demonstrate that our approach constructs over 40% more known complexes than other recent methods and that the complexes it produces are more biologically coherent even compared with the reference set. We provide experimental support for some of our novel predictions, identifying both a new complex involved in nutrient starvation and a new component of the eisosome complex. Second, we provide a high accuracy algorithm for the novel problem of predicting transient interactions involving complexes. We show that our complex level network, which we call ComplexNet, provides novel insights regarding the protein-protein interaction network. In particular, we reinterpret the finding that “hubs” in the network are enriched for being essential, showing instead that essential proteins tend to be clustered together in essential complexes and that these essential complexes tend to be large.
Biological processes exhibit a hierarchical structure in which the basic working units, proteins, physically associate to form stoichiometrically stable complexes. Complexes interact with individual proteins or other complexes to form functional modules and pathways that carry out most cellular processes. Such higher level interactions are more transient than those within complexes and are highly dependent on temporal and spatial context. The function of each protein or complex depends on its interaction partners. Therefore, a faithful reconstruction of the entire set of complexes in the cell is essential to identifying the function of individual proteins and complexes as well as serving as a building block for understanding the higher level organization of the cell, such as the interactions of complexes and proteins within cellular pathways. Here we describe a novel method for reconstruction of complexes from a variety of biological assays and a method for predicting the network of interactions relating these core cellular units (complexes and proteins).
Our reconstruction effort focuses on the yeast Saccharomyces cerevisiae. Yeast serves as the prototypical case study for the reconstruction of protein-protein interaction networks. Moreover the yeast complexes often have conserved orthologs in other organisms, including human, and are of interest in their own right. Several studies (1–4) using a variety of assays have generated high throughput data that directly measure protein-protein interactions. Most notably, two high quality data sets (3, 4) used tandem affinity purification (TAP)1 followed by MS to provide a proteome-wide measurement of protein complexes. These data provide the basis for attempting a comprehensive reconstruction of a large fraction of the protein complexes in this organism. Indeed a number of works (5, 6) have attempted such a reconstruction. Generally speaking, all use the same general procedure: one or more data sources are used to estimate a set of affinities between pairs of proteins, essentially measuring the likelihood of that pair to participate together in a complex. These affinities induce a weighted graph whose nodes are proteins and whose edges encode the affinities. A clustering algorithm is then used to construct complexes, sets of proteins that have high affinity in the graph. Although similar at a high level, the different methods differ significantly on the design choices made for the key steps in the process.
Recent works (since 2006) all focus on processing the proteome-wide TAP-MS data and using the results to define complexes. Gavin et al. (3), Collins et al. (7), and Hart et al. (5) all use probabilistic models that compare the number of interactions observed between proteins in the data versus the number expected in some null model. Collins et al. (7) and Hart et al. (5) both used all three of the available high throughput data sets (2–4) in an attempt to provide a unified interaction network. The two unified networks resulting from these studies were shown to have large overlap and to achieve comparable agreement with the set of co-complex interactions in the MIPS data set (8) that are collated from previous small scale studies. The interaction graphs resulting from the computed affinity scores are then clustered to produce a set of identified complexes. Gavin et al. (3), Hart et al. (5), and Pu et al. (6) all use a Markov clustering (MCL) (9) procedure; Collins et al. (7) use a hierarchical agglomerative clustering (HAC) procedure but do not suggest a computational procedure for using the resulting dendrogram to produce specific complex predictions.
Despite the fairly high quality of these networks and the agreement between them, they still contain many false positives and negatives. False negatives can arise, for example, from the difficulty in detecting interactions involving low abundance proteins or membrane proteins or from cases where the tag added to the bait protein during TAP-MS prevents binding of the bait to its interacting partners. False positives can arise, for example, from complexes that share components or from the contaminants that bind to the bait nonspecifically after cell lysis. Therefore, the set of complexes derived from the protein-protein interaction network alone has limited accuracy. Less than 20% of the MIPS complexes (8), which are derived from reliable small scale experiments, are exactly captured by the predictions of Pu et al. (6) or by those of Hart et al. (5).
In this study, we constructed a method that generates a set of complexes with higher sensitivity and coverage by integrating multiple sources of data, including mRNA gene expression data, cellular localization, and yeast two-hybrid data. The data integration approach was used in some early works on predicting protein-protein interactions (10, 11) and more recently by Qiu and Noble (12), but these studies focus only on predicting pairs of proteins in the same complex and not on reconstructing entire complexes. Many recent studies (13–21) have successfully integrated multiple types of data to predict functional linkage between proteins, constructing a graph whose pairwise affinity score summarizes the information from different sources of data. However, because the data integration is not trained toward predicting complexes, the high affinity pairs contain transient binding partners and even protein pairs that never interact directly but merely function in the same pathways. When these graphs are clustered, the clusters correspond to a variety of cellular entities, including pathways, functional modules, or co-expression clusters. We developed a data integration approach that is aimed directly at the problem of predicting stoichiometrically stable complexes.
We used a two-phase automated procedure that we trained on a new high quality reference set that we generated from annotations in MIPS and SGD and from manual curation of the literature. In the first phase, we used boosting (22), a state-of-the-art machine learning method, to train an affinity function that is specifically aimed at predicting whether two proteins are co-complexed. Unlike most other learning methods, boosting is capable of inducing useful features by combining different aspects of the raw data, making it particularly well suited to a data integration setting. Once we generated the learned affinity graph over pairs of proteins, we predicted complexes by using a novel clustering algorithm called hierarchical agglomerative clustering with overlap (HACO). The HACO algorithm is a simple and elegant extension of HAC that addresses many of its limitations, such as the irreversible commitment to a possibly incorrect clustering decision. HACO can be applied to any setting where HAC is applied; given the enormous usefulness of HAC for the analysis of biological data sets of many different types (e.g. Refs. 7, 23, and 24), we believe that HACO may be applicable in a broad range of other tasks.
To validate our approach, we tested the ability of our methods and other methods to predict reference complexes that were not used in training. By integrating multiple sources of data, we recovered more reference complexes than other state-of-the-art methods (5, 6) when applied to the same set of yeast proteins. We also validated our predicted set of complexes against external data sources that are not used in the training. In all cases, our predictions were shown to be more coherent than other methods and, in many cases, more coherent even than the set of reference complexes.
A detailed examination of our predicted complexes suggests that many of them were previously known but not included in our (comprehensive) reference set, suggesting that our complexes form a valuable new set of reference complexes. In several cases, our predicted complexes were not previously characterized. We experimentally validated two of these predictions: a new component in the recently characterized eisosome complex (25), which marks the site of endocytosis in eukaryotes, and a newly characterized six-protein complex, including four phosphatases, that appears to be involved in the response to nutrient starvation and that we named the nutrient starvation complex (NSC).
The complex-based view provides a new perspective on the analysis and reconstruction of the protein interaction network. In the past, Jeong et al. (26) have suggested that the degree of a protein in an interaction network is positively correlated with its essentiality and have argued that “hubs” in the network are more likely to be essential because they are involved in more interactions. Our analysis presents a complex-based alternative view: essential proteins tend to cluster together in essential complexes (5), and essential complexes tend to be large; thus, the essential hubs in the network are often members in large complexes comprised mostly of essential proteins. We also reformulate the task of reconstructing the protein interaction network. Rather than considering interactions between individual proteins (27–29), a somewhat confusing network that confounds interactions within complexes and interactions between complexes, we tackle the novel task of predicting a comprehensive protein interaction network that involves both individual proteins and larger complexes. We argue that these entities are the right building blocks in reconstructing cellular processes, providing a view of cellular interaction networks that is both easier to interpret than the complex network of interactions between individual proteins and more faithful to biological reality. Moreover a complex, which is a stable collection of many proteins that act together, provides a more robust basis for predicting interactions as we can combine signals for all its constituent proteins, reducing sensitivity to noise.
To accomplish this goal, we constructed a reference set of complex-complex interactions, considering two complexes to interact if they are significantly enriched for reliable interactions between their components. We further augmented this set with a hand-curated list of established complex-complex interactions. We then used a machine learning approach to detect the “signature” of such interactions from a large set of assays that are likely to be indicative. We explored different machine learning methods and showed that a partially supervised naïve Bayes model, where we learned the model from both labeled and unlabeled interactions, provides the best performance. This model was applied both to our predicted complexes and to individual proteins, providing a new, comprehensive reconstruction of the S. cerevisiae interaction network, which can be downloaded from our project Web page.2 We showed that entities that are predicted to interact are more likely to share the same functional categories. A detailed investigation of our new predicted interactions presents many that are established in the literature as well as some that are novel but consistent, presenting plausible hypotheses for further investigation.
EXPERIMENTAL PROCEDURES
Complex Prediction
Constructing a Set of Reference Complexes—
We compiled a reference set of complexes by combining literature-derived results from small scale experiments in MIPS (8) and SGD (31) with a hand-curated list (see our supporting Web site3) that we generated. The MIPS, SGD, and hand-curated sets contain 225, 195, and 164 complexes, respectively (supplemental Fig. S1a). Below we describe our method for establishing correspondence between the three lists and combining them into a high confidence reference set suitable for training our method and for evaluating the accuracy of its predictions.
Our approach consisted of five processing steps. First, we merged similar complexes from the original lists (see below), resulting in a list of 543 complexes. Second, we removed 112 redundant complexes that were proper subsets of other complexes. Third, we removed the five largest complexes: the four ribosomal subunits and the small nucleolar ribonucleoprotein complex; these complexes are so large that they greatly overwhelm the signal both in training the method and in evaluating the results. Fourth, we restricted the complexes to the set of 2195 proteins that have adequate amount of experimental evidence (see below). Finally we removed single protein complexes, arriving at the final list of 340 complexes. With at least two and on average 4.9 proteins per complex, this set of complexes contained 1100 unique proteins and a total of 1661 protein members, showing that the reference complexes contain notable overlap (proteins that are shared by multiple complexes).
In the first step of this merging process, we define each candidate complex from the three curated lists as a node in an undirected graph (or network). Two complexes are connected by an edge if they overlap significantly, i.e. their Jaccard similarity coefficient is greater than 0.7 (see Jaccard coefficient (JC) metric below) with an edge weight equal to the JC value. We found 422 isolated nodes in the graph, corresponding to unique complexes that do not overlap significantly with any other complexes in the list. The task of merging similar complexes is equivalent to that of finding several types of connected components in this graph. A complete subgraph with average edge weight of 1 is equivalent to a group of complexes with identical protein content that appear under multiple names in at least two of the curated lists. We found 66 such groups, which correspond to complexes that we regard as very high confidence because of multiple corroborating evidence. A complete subgraph in the rest of the network with average edge weight less than 1 (but greater than 0.7) is equivalent to a group of complexes whose protein contents are reported differently by the different curated lists. We found 45 such groups and produced a consensus complex for each, resolving conflicts by a majority vote: a protein was included in the resulting complex only if it was found in more than half of the candidate complexes from the conflicted group. The remaining 18 nodes formed four connected components but no complete subgraphs, each component indicating non-transitive overlaps between three or more candidate complexes (e.g. A overlaps with B, and B overlaps with C, but A does not overlap significantly with C). Manual inspection and consultation with experts resulted in 10 unique complexes being added to the reference list. The distribution of complex sizes in our reference set is shown in supplemental Fig. S2.
Constructing Positive and Negative Co-complex Protein Pairs—
The set of positive co-complexed protein pairs consists of all protein pairs that appear in the same complex in the reference set. For the negative set, we first consider all protein pairs (P1, P2) such that P1 is in a reference complex and P2 is outside any version of that complex in any of the three hand-curated sets; we then exclude any pair that is within some other reference complex. The result of this process was 5065 positive pairs and about one million negative pairs.
Features for Predicting Co-complexed Relations—
We constructed features for our protein-protein interaction network using five different data sources: the purification enrichment (PE) score from the consolidated network of Collins et al. (7), a cellular component from a truncated version of the Gene Ontology (GO) (33), transmembrane proteins (31), co-expression (34), and yeast two-hybrid (Y2H) interactions (35, 36).
Our highest coverage source regarding protein-protein interaction comes from high throughput TAP-MS data of the Gavin et al. (3) and Krogan et al. (4) data sets. The recent work of Collins et al. (7) provides a coherent and systematic way of integrating the data from these separate assays into a high quality score that measures the probability of a protein pair to be co-complexed. The recent work of Hart et al. (5) provides a different integration method, but the results are quite similar, providing support for both of these procedures. We derived five features from the PE analysis: the direct score is computed based only on bait-prey information in the purifications; the indirect score is computed based on prey-prey information; the actual PE score is the sum of direct and indirect scores; the scaled score maps the PE score to a value between 0 and 1 to approximate the confidence value that the pair represents a true interaction; finally each protein is represented by a vector of its scaled PE scores with all the other proteins (where we assign its interaction with itself a score of 1), and we define our PE-distance feature as the cosine distance between the vectors of two proteins.
As the PE score provides most of the signals in predicting complexes (see “Results”), we only kept the 2390 proteins that have at least one scaled PE score above 0.2 with some other protein. Although this set only covers about 40% of the ∼6000 yeast genes, it covers 81% of all protein members in the lists of high quality complexes that comprised our reference set. As noted earlier, we excluded proteins that appear exclusively in the four ribosomal subunits and the small nucleolar ribonucleoprotein complex. This resulted in the final list of 2195 proteins on which we performed our complex prediction.
Yeast two-hybrid assays also provide a direct measurement of protein-protein interactions. We derived these data from the assays of Ito et al. (35) and Uetz et al. (36). Interacting pairs are assigned a feature value of 1. Pairs of proteins that appeared in the assay but were not observed to interact are assigned a feature value of −1. All other pairs have 0 as their feature values.
The GO cellular component hierarchy (33) was downloaded on June 25, 2007. An examination of the hierarchy showed that many of the smaller categories (lower in the hierarchy) refer to particular complexes whose information is derived from the same small scale experiment that informs our reference set. Thus, to achieve a fair evaluation using the reference set, we removed categories of size less than 120 that can potentially contain the answer. The remaining 44 of 564 categories represent high level cellular localization information, much of which is obtained through high throughput experiments (37). Some sample categories include “endoplasmic reticulum part,” “nuclear chromosome part,” “mitochondrial membrane,” and “cytoplasm.”
We derived two pairwise localization features from the GO cellular component. One is the semantic distance measure (38), which is the log size of the smallest category that contains both proteins. However, this feature is a pessimistic assessment regarding the co-localization of the two proteins as lack of annotation of a protein in some category, particularly one that is a subset of its most specific category, does not necessarily mean that it cannot belong to this category. Therefore, we constructed a second feature, which is the log size of the smallest possible group that could contain both proteins (given the current evidence). It is computed in the following way between protein A and protein B whose most specific categories are X and Y, respectively. If X is a subcategory of Y, then the two proteins might belong together to any group if they were to be annotated with enough detail. Therefore, we use log of 120, the size of the smallest category, as our second feature. On the other hand, if X and Y are not subcategories of each other, we denote Z to be the smallest common supercategory of X and Y. We then denote X′ (respectively Y′) to be the category one level down the path from Z to X (respectively Y). Thus, assuming that A and B belong to the two different categories at X′ and Y′, the smallest semantic category that we can form that may contain them both is X ′ ∪ Y′. Thus, our second feature is log(|X ′ ∪ Y′|).
A list of membrane proteins is obtained by parsing the transmembrane annotations in SGD (31). A pair of proteins is considered to be membrane if at least one of the proteins is found in the membrane. The first membrane feature is 1 if the pair is membrane and 0 otherwise. The second and third features are the product of the first feature with the direct and indirect PE score of the two proteins, respectively. This allows our boosting model to take into account the known fact that TAP-MS purifications work differently on membrane proteins from non-membrane proteins.
Microarray data were downloaded from the Stanford Microarray Database (34) on December 5, 2006; it contains a total of 902 experiments for yeast divided into 19 categories. The data were normalized to mean 0 and standard deviation 1. We constructed a feature by computing the mean-centered Pearson correlation coefficient between the expression profiles of two proteins.
A final feature is obtained from small scale physical interactions. We downloaded protein-protein interactions from MIPS (8) and DIP (39) on March 21, 2006. We extracted from MIPS those physical interactions that are non-high throughput yeast two hybrid or affinity chromatography. For DIP, we picked non-genetic interactions that are derived from small scale experiments or verified by multiple experiments. This feature has a value of 1 for observed interactions and feature value 0 for all other pairs. Importantly there is a risk of cyclicity between these small scale interactions and the reference complexes. Therefore, to avoid a positive bias in our results, we omitted this feature in the cross-validation runs, which are evaluated against the reference complexes. For those runs that are trained on the entire set of reference complexes, this cyclicity is not a concern, so this feature was included. There are a total of 12 features for cross-validation runs and 13 features for runs that are trained on the entire reference set.
Integrating Multiple Features Using the LogitBoost—
Boosting (22) is a class of algorithms that iteratively combines weak learners to give a representative ensemble. Each weak learner is a simple classifier, such as a decision stump, that may only weakly correlate with the labels. After a weak learner is trained, we add it to the ensemble with appropriate weight. In the next iteration, the algorithm puts more weights on the data points that are classified incorrectly by the current ensemble, which the next weak learner will focus on. Boosting is able to perform automatic feature selection and has accuracy that is better or comparable with other state-of-the-art classifiers such as support vector machines (40) in many domains. We implemented a version of boosting algorithms called LogitBoost (22) that uses decision stumps as weak learners and the logit function as the loss function. This variant is shown to be more robust to outliers and overfitting than the standard AdaBoost variant (41). Our experiments (data not shown) showed that this method performs well on our data compared with other versions of boosting and other classification algorithms such as logistic regression and support vector machines. The prediction of the learned ensemble classifier on a given protein pair is taken to be the affinity of the pair in the clustering algorithm below.
The HACO Algorithm—
The standard HAC algorithm with average linkage (42) maintains a pool of merging candidate sets where the distance between two non-overlapping sets is as follows.
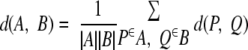 |
(Eq. 1) |
In our setting, we take d(P, Q) as the negative of the affinity between protein P and protein Q. Note that d(A, B) is the average of the edge distance between proteins in A and proteins in B.
In HAC, at each step, we pick the two non-overlapping sets with the closest distance, A and B, and merge them to create a new set, M. M is added to the pool, while the sets A and B are removed. Therefore in later steps, we could only consider the superset M and would never be able to use A or B again to merge with some other set. Assume that there is another set C whose distance to A is only slightly larger than d(A, B). In this case, the decision to merge A with B rather than with C is arbitrary and unstable. When the actual clusters overlap, a more appropriate solution would be to have two overlapping merged candidates: M = A ∪ B and N = A ∪ C. We adapt HAC to accommodate this intuition. We define the divergence between A and M as a measure of the cohesiveness of the set M that is outside of A (supplemental Fig. S3),
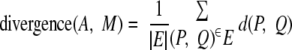 |
(Eq. 2) |
where E is the set of pairs in M but not in A: E = {(P, Q) | (P, Q) ε M × M − A × A, P < Q}. (Here “<” can be any ordering among the proteins, such as alphabetical, to avoid a pair appearing twice in the set E).
If M is not overlapping with C, we have the choice of whether to use A or M to merge with C. If divergence(A, N) − divergence(A, M) is small, it makes sense to merge A and C to create a new set N that is almost as coherent as M. On the other hand, if the difference is large, we would prefer to replace A with its superset M as the merging candidate to C.
In practice, we use d(A, C) to approximate divergence(A, N): we check whether Δ = d(A, C) − divergence(A, M) is small. Divergence(A, N) is the weighted average of d(A, C) and d(C), the distance within C. d(C) tends to be smaller than d(A, C) because pairs within C, which is formed earlier by some merging, are more coherent than pairs between A and C. Therefore, d(A, C) tends to be smaller than divergence(A, N), so keeping Δ small is generally a more stringent requirement for ensuring that N is almost as coherent as M. Moreover by forcing d(A, C) to be small, we make sure the set N is coherent not just because the distance within C is small. With this consideration, we defined the modified distance between A and C (supplemental Fig. S3) as follows.
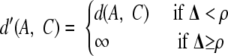 |
(Eq. 3) |
The modified distance d′ is used to pick the two closest sets to merge in the next iteration. If Δ is smaller than a margin, we make d′ equal to d and thus allow A and C to merge. On the other hand, if Δ is large, we make d′ infinity and thus prohibit A and C from merging in favor of merging their supersets. ρ is the margin parameter: the larger the margin ρ, the more likely a set A is to be reused, resulting in more overlapping subsets constructed by the algorithm. If the margin is 0, it reduces to the standard HAC. Therefore, our HACO algorithm is a generalization of the HAC. Note that we can eliminate a set from the merging candidate pool when its modified distances to all other sets are ∞. Of course we can define another modified distance as long as it is larger when Δ is large and close to d(A, C) when Δ is small.
In practice, A might have multiple supersets in the pool. Therefore, we look at all of the supersets of A in the pool that are not overlapping with C and use the set MA, C with smallest divergence from A, i.e. the one that provides the best replacement for A in terms of the proposed merger with C.
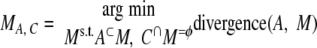 |
(Eq. 4) |
We do the same thing with C for its proposed merger with A.
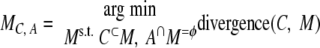 |
(Eq. 5) |
The smaller of divergence(A, MA, C) and divergence(C, MC, A) is used to compute the modified distance.
The algorithm terminates when there are no more non-overlapping sets to merge. The output is a cluster-lattice where the same cluster can be a child of multiple parents in the lattice. The lattice is cut at a certain threshold to generate a set of overlapping clusters. These predicted clusters are the sets that are still in the candidate pool when the distance in the merging process reaches the threshold.
Training and Test Regime—
To evaluate our prediction accuracy against the reference set, we divided the 340 reference complexes into five disjoint subsets, or folds. As there are about a million negative pairs, for computational expediency, we randomly sampled one-tenth of the negative pairs to be used in training while setting each negative pair to have 10 times the weight of the positive pairs.
For each fold in the 5-fold cross-validation, we hide one set and use the remaining four sets to train the affinity function for the protein pairs, the margin ρ for the HACO, and the cutoff threshold for the resulting cluster-lattice. We use the same training set in all steps of our pipeline and evaluate the final predictions on complexes in a separate test set that is hidden during all steps of the training process. We select the cutoff threshold by maximizing the coverage (see below for the definition) on the training set. To pick the margin ρ, we cannot use coverage alone because our model would always prefer a bigger margin that keeps more sets in the pool. Therefore, we choose ρ by maximizing the product of coverage and sensitivity (see below for the definition) on the training set. This approach trades off between the match with the reference set and the number of predicted complexes.
To evaluate our predictions against external data sources, such as biological coherence and essentiality, we augmented our model with a feature constructed from small scale physical interactions and trained it on the entire set of 340 reference complexes. To avoid circularity between features and evaluation, we did not evaluate the predictions from such runs against the reference complexes.
Evaluation Metrics for Matching between Predictions and Reference Complexes—
The overlap between a reference complex R and a predicted complex C can be quantified in several ways (43) (supplemental Fig. S4).
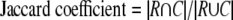 |
(Eq. 6) |
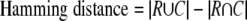 |
(Eq. 7) |
We use both measures because of the size effect. For example, a Hamming distance of 2 between two large complexes, say both of size 5, is a good match. In this case JC = 4/6 = 0.67. On the other hand, a Hamming distance of 2 between two small complexes of size 2 implies an overlap of only one protein, which could arise simply by chance. In this case JC = 1/3 = 0.33.
We define the coverage and sensitivity of a set of predictions so we can systematically evaluate genome-wide predictions. For each reference complex, we find the prediction that has the highest Jaccard coefficient. We define the scaled Jaccard coefficient (SJC) as follows: SJC(R, C) = max{0, 2JC(R, C) − 1}. We truncate the value at 0 because it may represent random overlap. In the above examples, the matching of the two large complexes of size 5 and Hamming distance 2 would have SJC = 0.33, whereas the small ones of size 2 and Hamming distance 2 would have SJC = 0. We define the coverage as the average Jaccard coefficient per reference complex,
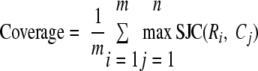 |
(Eq. 8) |
where m is the number of reference complexes and n is the number of predicted complexes.
For sensitivity, we sum the Jaccard coefficients of all the overlapping (reference and prediction) complex pairs and normalize by the total number of predicted complexes.
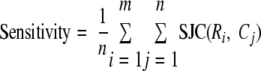 |
(Eq. 9) |
Biological Coherence of Predicted Complexes—
We evaluate biological coherence of the predicted complexes using several metrics. The first is average semantic distance in the GO biological process hierarchy. GO data were downloaded on June 25, 2007. We computed the distance between two proteins as the log size of their smallest common category (38) as for the cellular component hierarchy above.
We downloaded the protein expression data from Ghaemmaghami et al. (44). We used log of measured protein levels in terms of molecules per cell as the protein abundance value.
The growth phenotype data were obtained from Hillenmeyer et al. (45). For each gene, its homozygous deletion strain is grown in 418 experiments with different drug treatments. The log ratio of the deletion strain's growth in no-drug control to its growth with the drug treatment is used to define the growth phenotype in that particular condition. For each pair of genes, we computed the Pearson correlation of the growth phenotypes across all 418 conditions; this is the measure used in the original study.
We downloaded the transcriptional regulation data from MacIsaac et al. (46) and Harbison et al. (47). We used a p value cutoff at 0.001 and required conservation across species to define the transcription factors for each protein. We computed how many transcription factors are shared by any two proteins.
Complex-Complex and Complex-Protein Interaction Prediction
Constructing a Reference List of Positive and Negative Complex-Complex Interactions—
We derived a reliable set of S. cerevisiae protein-protein interactions from MIPS (8) and DIP (39) downloaded on March 21, 2006. We extracted from MIPS those physical interactions that are non-high throughput yeast two hybrid or affinity chromatography. For DIP, we picked non-genetic interactions that are derived from small scale experiments or verified by multiple experiments. We computed the number of reliable interactions between proteins of two complexes and compared it with what we expect if the reliable interactions are distributed randomly. We define the two complexes to be interacting if the enrichment of reliable interactions is more than 20 standard deviations above the mean. Such strong enrichment is needed because the reliable interactions are very sparse, and the presence of even a very small number would result in a large deviation from the mean (e.g. for two complexes of size 2 and 5 respectively, we only need one reliable interaction of the total 10 pairs to get an enrichment of 10 standard deviations above the mean). We ended up with a list of 82 interactions between the set of 383 complexes we just predicted. To augment this list, we generated a list of 59 additional known interactions between 81 named complexes. To avoid the redundancy between those 81 named complexes and our 383 predicted complexes, we replace a predicted complex by a named complex if they overlap with JC > 0.5. This process gave us a total of 421 complexes with 133 unique interactions between them that are used as our positive reference set. We created a negative reference set of 3173 non-interactions by using all pairs of named complexes that are not in our positive set. The interaction status of all the remaining pairs of complexes, named or predicted, is treated as unknown.
For protein-complex interactions, in addition to the above negative set between complexes, we randomly sampled 6560 protein-complex pairs that are not in the positive set and added them to our negative reference set. The number 6560 was chosen so the ratio of positive to negative pairs for protein-complex interactions is the same as the ratio for complex-complex interactions. All our reference lists are available from our supporting Web site.3
Features for Predicting Interactions—
Because there is no direct measurement of complex-complex or complex-protein interactions, we try to use as much indirect evidence as possible. Besides all data sources used for identifying complexes, we added four additional data sources based on correlation of growth fitness, correlation of transcription factor profile, protein-protein interaction prediction, and condition-specific expression correlation.
The correlation of growth fitness profile (45) is computed as described above under “Biological Coherence of Predicted Complexes.” For each protein, we create a transcription factor (TF) profile vector where each position in the vector represents a TF and its value is 1 if the TF is found to regulate the protein (46) and 0 if it is not. We used the same transcription regulation data as described above under “Biological Coherence of Predicted Complexes.” For any pair of proteins, we compute the mutual information between the profile vectors of the two proteins using the method described Date and Marcotte (48).
There are many works in integrating multiple sources of data to predict protein-protein interactions. In particular, the InSite method (49) integrates protein sequence motifs, evidence for protein-protein interactions, and evidence for motif-motif interactions in a principled probabilistic framework to make high quality predictions of protein-protein interactions. Here we use the InSite method but trained without the reliable interactions between complexes in our positive reference set. We use the predicted probabilities that two proteins interact as one more data source.
Here we processed the expression data in accordance with our intuition that transient interactions occur under specific conditions, and we should only expect expression profiles of interacting proteins to be correlated only when at least one of the pair is active. Specifically we divided our expression data into 76 conditions (50–58), each of which represents a particular time course. In accordance with convention, we quantify the activity of a protein under certain condition according to its maximum deviation from norm, or in other words the maximum absolute expression (assuming norm to be 0). For each condition, we define a protein to be differentially expressed, or active, if its maximum absolute expression is above a cutoff, which we specify to be 1.0. For each pair of proteins, we compute Pearson correlation coefficient (PCC) separately in each condition. If a protein in the pair is inactive under a condition, the PCC value for the condition is assumed to be 0. We use the PCC value, averaged across all conditions under which at least one protein of the pair is active, as our last feature type. Initial investigation showed that this feature is better correlated with the reference complex-complex interactions than the overall PCC across all conditions. We note that, for the task of predicting when two proteins are co-complexed, the simple correlation performed better (data not shown), consistent with the fact that the activity of two members of a stable complex is likely to be similar across a wide range of conditions.
Aggregating Signals between Proteins into Features between Complexes—
All forms of evidence in our analysis involve a pair of proteins. To predict interactions between two complexes, C and D, we aggregate the signals for all protein pairs between C and D and produce the following features,
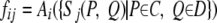 |
(Eq. 10) |
where Ai() is some aggregating function, such as sum, maximum, mean, minimum, decayed maximum, decayed minimum, etc. (See supplemental Table 1 for a complete list of aggregating functions and their definitions.) Sj represents the jth feature type between a pair of proteins. We also use four global features, independent of the data sources: size of the first complex, size of the second complex, number of protein pairs between the two complexes, and number of overlapping proteins between the two complexes. The features for interactions between a protein P and a complex C are identical except that we only need to aggregate the signals over all pairs (P, Q) for Q in C.
The naïve Bayes model that we use assumes all features to be conditionally independent of each other given the status of whether two complexes interact or not. Therefore for each data source, we pick only the best aggregating function to reduce the conditional dependences between the features. To do this, we define rij to be the area under the ROC curve if we use the feature fij alone to predict complex-complex interactions: the greater rij, the stronger the correlation between the feature and the presence of a complex-complex interaction. Therefore, for naïve Bayes, we use, for each feature type j, the feature fj = fij where i gives rise to the maximum value rij. Supplemental Table 2 lists the aggregating function chosen for each feature type.
Learning and Predictions—
We experimented with different machine learning algorithms for making our predictions: 1) a simple naïve Bayes model where the effects of different feature types are assumed to be independent, 2) a discriminative boosting algorithm as we used in predicting co-complexed affinities between protein pairs above, and 3) a naïve Bayes model where the unlabeled complex-complex interactions are taken to be unobserved variables, and the model is trained via the expectation maximization (EM) algorithm. This last approach is based on the fact that the amount of labeled training data is quite limited in this task, but the unlabeled data also provide us with useful information about the behavior of different features in interacting and non-interacting pairs. A variant of this same approach was used with success in the InSite model (49).
More formally, for each pair of complexes, we construct an “interaction variable” whose value is 1 if the two complexes are in the positive reference set of interacting complexes, 0 if they are in the negative reference set, and unobserved otherwise. Each feature of the complex pair is associated with two conditional distributions: one for the case of an interacting pair and the other for the case of a non-interacting pair. These distributions are defined via some parametric class (see supplemental Table 3). The distributions for the different features are taken to be independent of each other within each of the two cases. The model is trained via the following EM procedure. We initialize the model parameters to those that would be obtained from maximum likelihood estimation using the pairs in our reference set alone. We then iteratively repeat the following two steps until convergence. In the E-step, we use our current model to compute the marginal probability of each unobserved interaction variable given the features associated with the pair. We use the computed probability as a soft assignment to the interaction variable. In the M-step, we learn the parameters for the distributions using maximum likelihood estimation based on the inferred soft assignment to all interaction variables; the variables in the reference set are always fixed to their known value. We use the model obtained at convergence to predict, for each pair of complexes not in our reference set, the probability with which the pair interacts.
We used the same naïve Bayes + EM procedure when making predictions using only one of the features (PE score or InSite probability), which we used as a comparison base line. In these comparisons, we used the same aggregator selected for the model using all the features.
When training using the LogitBoost model, we are not making independence assumptions between the different features. Hence there we include all features fij instead of just picking the best aggregating function for each feature type. We used the same naïve Bayes + EM procedure for the protein-complex interaction predictions, although the best aggregating functions picked and the set of parametric classes used for the feature distributions were a little different. (See supplemental Tables 2 and 4.)
Functional Coherence of Complexes Predicted to Interact—
We evaluate whether two interacting complexes are more likely to share the same functional category. We used functional categories from MIPS (8), which has 18 functional categories with an average of 684 proteins per category. A complex is assigned to a particular functional category if more than half of its components belong to the functional category. We only perform our evaluation on complex pairs where both complexes are assigned to some MIPS functional category.
Experimental Validation
TAP Purification—
Two liters of yeast culture expressing Pil1-TAP was grown to A600 = 0.8 and subsequently harvested. The resulting pellet was resuspended in 8 ml of buffer A (150 mm potassium acetate, 20 mm HEPES, pH = 7.4, 2 mm magnesium acetate, 5% glycerol) and frozen in liquid nitrogen. Total proteins were extracted by bead milling of the frozen pellet followed by addition of Triton X-100 to 1% (w/v) final concentration. Solubilized extracts were cleared by two centrifugations of 4 min at 4000 × g and incubated with IgG-Sepharose for 2 h. Beads were washed six times with 50 ml of buffer B (150 mm potassium acetate, 20 mm HEPES, pH = 7.4, 2 mm magnesium acetate, 5% glycerol, 1% Triton X-100). Proteins were eluted by tobacco etch virus protease cleavage in 200 μl of buffer for 2 h and analyzed by SDS-PAGE by Coomassie staining. Bands were cut and digested with trypsin, and peptides were extracted and analyzed by LC-MS/MS as described previously (59).
EMAP Experiments—
EMAP experiments and subsequent data analysis were done as described previously (60, 61). Data from these experiments are presented on our supporting Web site.3
RESULTS
Method Overview
We compiled a reference set of complexes by combining literature-derived results from small scale experiments in MIPS (8) and SGD (31) with a hand-curated list (see our supporting Web site3) that we generated. The MIPS, SGD, and hand-curated sets contain 225, 195, and 164 complexes, respectively (supplemental Fig. S1a). We established correspondence between the three lists and combined them into a high confidence reference set suitable for training our method and for evaluating the accuracy of its predictions (see “Experimental Procedures”). This curated set was compiled prior to the development of our method and was not subsequently revised.
We then formulated the task of predicting whether two proteins were members of the same complex as a machine learning task. We used our reference set to construct a high quality set of positive and negative examples. We constructed features that are useful for predicting this relationship from five different data sources: the PE score from the consolidated network of Collins et al. (7), a cellular component from a truncated version of the GO (33), transmembrane proteins (31), co-expression (34), and Y2H interactions (35, 36) (see “Experimental Procedures”). We then applied the boosting algorithm (22) for training the predictor. Boosting was selected because of its high accuracy, robustness to outliers, and ability to perform automatic feature selection. The prediction of the boosting classifier on a given protein pair is taken to be the affinity of the pair in the clustering algorithm below.
Our initial experiments showed that HAC, which progressively merges sets of proteins with strongest affinity, produces the best results for complex reconstruction if trained to optimize for that task. However, HAC has several significant limitations. First, it does not allow clusters to overlap, whereas actual complexes do share subunits. Second, it uses a single cutoff to decide the granularity of the complexes constructed. A cluster near the cutoff in the dendrogram can be formed even if it is the result of merging two relatively weakly connected subclusters A and B. Such a cluster, although of lower confidence, still excludes both A and B from being predicted as a complex; this occurs even if A and B are strong candidates for being a complex. Finally once a set of proteins is merged with another set, it cannot merge with anything else even if the affinity is only slightly lower. Therefore an incorrect decision cannot be fixed later in the process.
To address these limitations, we constructed a novel clustering algorithm called HACO that allows a set of proteins to be merged with multiple other sets with which it has comparably strong affinity (see “Experimental Procedures”). HACO addresses all of the limitations above. First, it produces clusters that can overlap. Second, when merging A and B into a single cluster C, it also has the option of leaving A and/or B as candidate complexes, avoiding a wrong decision because of an arbitrary cutoff. Finally as it allows the same cluster to be used in multiple places, it avoids many mistakes that arise from an almost arbitrary breaking of near ties. Both our boosting algorithm and the HACO code are freely available on our project Web page,3 allowing them to be used for predicting complexes with other forms of data.
Complex Predictions
Coverage and Sensitivity of Predicted Complexes—
We compiled a reference set of complexes from MIPS (8), SGD (31), and hand-curation (see our supporting Web site3) that is more comprehensive than previous studies (5, 6). Although it still contains noise and bias, it provides us with the ultimate evaluation of our predictions. There are 340 complexes in our reference set with an average of 4.9 proteins per complex (supplemental Fig. S1b).
To predict complexes, we first trained our model to predict pairwise co-complex interactions and then used our HACO algorithm to cluster the resulting pairwise affinity network into complexes. We constructed features for our protein-protein interaction network using five different data sources: the PE score from the consolidated network of Collins et al. (7), a cellular component from a truncated version of the GO (33), transmembrane proteins (31), co-expression (34), and Y2H interactions (35, 36). We tested our approach using a standard 5-fold cross-validation regime, training on 80% of the complexes and testing on the remaining 20%; the test set was not used in any aspect of the training of the model. For each fold in the 5-fold cross-validation, we applied HACO to the affinity measure learned using the boosting model on the training data. We evaluated the resulting clusters on the hidden test set. We predicted 417.8 complexes per fold with at least two proteins for each complex. Each complex contains 4.30 proteins on average (supplemental Fig. S1).
We define a complex to be well predicted if it is within Hamming distance (see “Experimental Procedures”) of 2 to some predicted complex. However, two small complexes can be quite different even if their Hamming distance is 2. Therefore we also require the Jaccard coefficient (see “Experimental Procedures”), which takes into account the size of the complexes, to be above 0.5. We also measure the coverage and sensitivity of the set of predictions (see “Experimental Procedures”): coverage measures how well the reference set is covered by our predictions, and sensitivity measures how well each predicted complex overlaps with the reference set, a measure that takes into consideration the number of predicted complexes.
We compared our results with those of Bader and Hogue (62), Gavin et al. (3), Krogan et al. (4), Hart et al. (5), and Pu et al. (6). As we discussed, each method made different decisions for defining the affinity function and for clustering it. Bader and Hogue (62) used a novel clustering algorithm called molecular complex detection (MCODE) to detect densely connected regions in the protein-protein interaction network. Gavin et al. (3) computed a socioaffinity score between each pair of proteins that compares the number of times the two proteins are observed together in some purifications relative to what is expected by chance. The pairwise network of socioaffinity scores is then subjected to a procedure that produces overlapping clusters. Complexes are composed of a “core” that appears in most runs of the clustering algorithm and “attachments” that appear only in some. Most of the recent methods appear to have converged on using the MCL algorithm (9) albeit on different affinity functions. Krogan et al. (4) used a machine learning approach, trained on MIPS reference complexes, to predict the confidence that a pair of proteins is in the same complex. Hart et al. (5) defined a p value by comparing observed relative to expected number of interactions applied to three sets of purifications (2–4). Pu et al. (6) applied MCL directly to the PE score of Collins et al. (7). All of these MCL-based methods produce non-overlapping clusters, although the method of Pu et al. (6) used a postprocessing phase to identify proteins that are likely to be recruited by multiple complexes.
Fig. 1a shows the accuracy of our method in reconstructing the reference complexes as compared with the best of these other approaches. As we can see, our affinity score achieves significantly better results than any of these methods; the results are better even when we use simple HAC for the clustering and improve further when we use HACO. We note that Hart et al. (5) and Pu et al. (6) are the state of the art in complex predictions and have been extensively compared with other complex prediction methods. In particular, Pu et al. (6) applied MCL to the same set of PE scores (7) as we used. HACO was able to perfectly recover 42 and 46% more reference complexes compared with Hart et al. (5) and Pu et al. (6), respectively (p values <0.01). The corresponding increase in sensitivity is 6 and 29%, respectively, and increase in coverage is 28 and 33%, respectively. The results suggest that these improvements are a consequence of our use of data integration with state-of-the-art machine learning. In particular, the Pu et al. (6) method and the Hart et al. (5) method, both of which used MCL applied to different affinities obtained from the TAP-MS data, performed very similarly. Interestingly HAC applied to the PE score performed slightly better than MCL applied to the PE score (HAC PE versus Pu et al. (6)). These three methods performed better than those of Bader and Hogue (62), Gavin et al. (3), and Krogan et al. (4) likely because of the fact that these earlier methods used only a single set of purifications. These results demonstrate the importance of combining data from multiple data sources integrated appropriately. We note that MIPS complexes are used, albeit in a very limited way, in generating the PE score. To avoid any risk of circular reasoning, we ran the same experiments using the SGD complexes alone as an independent reference set; the results (supplemental Fig. S2a) show that the improvement of our method over others remains consistent in this reference set as well.
Fig. 1.

Accuracy in reconstructing reference complexes. A comparison of predicted complexes to other state-of-the-art methods in the ability to accurately reconstruct reference complexes is shown. a, the number of reference complexes well matched by our predictions (y axis) and for the different methods we compared (x axis). The prediction quality is shown as bars: black, perfect prediction; dark gray, predictions that differ by a single protein (one extra or one fewer); light gray, predictions that differ by two proteins. Hart et al. (5) and Pu et al. (6) are state-of-the-art methods that outperform Gavin et al. (3) and Krogan et al. (4). The method of Bader and Hogue (62) has even lower accuracy (data not shown). Applying HAC to PE score (HAC PE) performed slightly better than Hart et al. (5) and Pu et al. (6), which use MCL. Our model, which uses LogitBoost and clustering, is able to achieve significantly better results than any other method by integrating multiple sources of data. The results are better even when we use simple HAC (HAC all; 88 perfect matches) for the clustering and improve further when we use HACO (95 perfect matches). This improvement is consistent over all five folds in our cross-validation process: over the five folds, HAC PE recovers 15, 11, 16, 22, and nine of the complexes; HAC all recovers 21, 13, 21, 23, and 10; and HACO recovers 24, 13, 23, 23, and 12. This consistency over folds demonstrates the robustness in the improvement we obtain using our method. In “Train all,” we trained on all data and tested on the same data; this method achieves only slightly higher accuracy, which indicates little overfitting to the training data and supports evaluating biological coherence of our predictions on this set. b, the x axis is the sensitivity of our predictions, which quantifies how likely a prediction is to match some reference complexes; the y axis is the coverage of our predictions, which quantifies how many reference complexes are matched by our predictions (see “Experimental Procedures”). Our approach has higher sensitivity and coverage than other methods. HACO has the highest product of sensitivity and coverage except for Train all, which trains and tests on the same data and thus provides an unachievable upper bound on performance.
The HACO algorithm helps address several of the limitations of the HAC approach. First it reduces the sensitivity of the complex definitions to a single universal threshold in the hierarchy. One such example involves the 15-protein SAGA complex. Here HAC predicts a 24-protein superset of the SAGA complex. This cluster is a much weaker cluster than SAGA itself: the average affinity between the SAGA proteins is 0.35 as compared with the average affinity of −1.19 for pairs within the 23 proteins excluding pairs of SAGA proteins. By comparison, HACO, by keeping multiple hypotheses relative to the cutoff, predicted a 23-protein cluster (similar to the HAC prediction) but also predicted the subcluster that corresponds perfectly to the SAGA complex. The second limitation addressed by HACO is that it avoids an early commitment to incorrect outcomes. For example, the affinity between Rad23 and Png1 is slightly higher than that between Rad23 and Rad4. HAC incorrectly merges Rad23 and Png1 and now cannot reuse Rad23 in any other complex. HACO can reuse Rad23, merging it with Rad4 to create a complex that perfectly matches the NEF2 (nucleotide excision repair factor 2) complex in the reference set.
Contribution of Each Data Source for Predicting Complexes—
Given the importance of data integration, it is useful to see which data sources play the most important role in our results. We first considered the contribution of each feature to our learned affinity function. Our approach uses LogitBoost (22), which defines the affinity function as the weighted sum of many weak learners, each of which is a decision stump on one of the features. The top weak learners involve features that are deemed to be most predictive. The top features in the order of their importance are: correlation of PE score (weight, 3.84); semantic distance in the truncated GO cellular component categories (−2.2); direct PE score, which is based only on direct bait-prey interactions (0.58); small scale physical interactions (0.55); and co-expression (0.16). It is interesting to note that the correlation of the PE score is deemed more informative than the PE score itself. One explanation is that the pairwise PE score between proteins P and Q is still a noisy measure for co-complexness, but if P and Q are truly co-complexed, they are likely to have similar interactions with other proteins.
As another metric for assessing the importance of each data source to the quality of our predictions, we successively applied our pipeline with HAC to the data source alone and to all data sources except that data source (Fig. 2 and supplemental Fig. S2). The PE score plays the dominant role and by itself predicts most of the complexes. Importantly our method here combines different variations of PE score (direct, indirect, scaled, total, and correlation) using boosting, generating an affinity score that is quite a bit better at predicting complexes than the original scaled PE score (73/54/16 perfect matches/one away/two away for HAC PE in Fig. 1a versus 81/50/19 for the PE-based features alone in Fig. 2). This result demonstrates the value of applying machine learning methods specifically optimized for the problem of complex identification. Nevertheless we still get a significant improvement by integrating in other data sources.
Fig. 2.
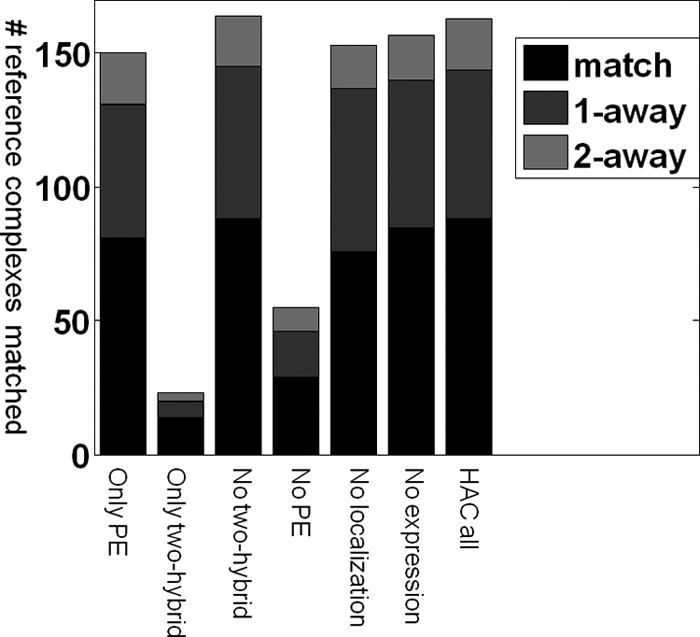
Contribution of each data source. To assess the contribution of each data source, we successively applied our pipeline with HAC to each data source alone and to all data sources except one; shown are the interesting cases (see also supplemental Fig. S2b) using the same format as in Fig. 1. The PE score by itself predicts most of the complexes, but we still get a significant improvement by integrating other data sources. Localization or expression are nonspecific and by themselves do not predict any complexes at all, but removing them decreases the accuracy, suggesting that they help clarify ambiguities in the TAP-MS data. Conversely the yeast two-hybrid feature by itself predicts a reasonable number of complexes, but removing it does not decrease accuracy at all, suggesting that it is redundant with the TAP-MS data.
Localization and expression have a similar effect. By itself, neither predicts any complexes at all; this is not surprising, as both are features with low precision. However, removing each of them decreases the accuracy, suggesting that they provide a signal that is independent of the PE score, and can help resolve some of its ambiguities and errors. The yeast two-hybrid feature has the opposite behavior: in isolation, it predicts a reasonable number of complexes; however, removing it does not decrease accuracy at all. This behavior can be explained by the hypothesis that yeast two-hybrid data largely correlate with PE score; thus, although the feature is predictive, it does not add much given the PE score data. This last hypothesis is further verified by the fact that localization and expression features appear within the top five weak learners, whereas yeast two-hybrid feature does not.
Biological Coherence of Predicted Complexes—
Having tested the ability of our method to reconstruct reference complexes, we produced a final set of predictions from our method. Here we train on all reference complexes and introduce an additional feature relating to interaction in small scale experiments; this feature was not used in the comparison with reference complexes to avoid potential circularity between this feature and the definition of the reference complexes. Overall this process resulted in 383 predicted complexes, which can be found on our supporting Web site.3 We evaluated the validity of these complexes by comparing with external data sources not used in the training and not directly related to reference complexes. For all biological coherence validations, we compute the coherence for each complex as the average of the coherence measure for all pairs in the complex. Then we take the average across all complexes predicted. We compare with the methods of Hart et al. (5) and Pu et al. (6), which consistently out-performed all previous methods. As a different benchmark, we also compare with the coherence for the highest affinity protein pairs (those that are most likely to belong to the same complex).
We validate our predictions by looking at various measures of biological coherence (Fig. 3): similarity of GO biological process, similarity in the level of protein abundance for different complex components, correlation of growth defect profiles across a broad range of conditions, and co-regulation as measured by sharing of transcription factors. For all measures, HACO with our affinity function considerably outperformed all other approaches with the method of Hart et al. (5) being the closest competitor. Most striking were the improvements in correlation of growth phenotypes across multiple conditions and in coherence of the transcriptional regulation program. To specifically test our complex formation process, we also compared pairs of co-complexed proteins with pairs that have high affinity (as computed by our boosting algorithm). The results were largely comparable with the notable exception of protein abundance where our complexes are 12% less coherent than the top affinity pairs; this suggests that proteins with lower affinity scores can be members of the complex but also play other roles in the cell, reducing their correlation with other proteins in the same complex. The comparison with the reference complexes is also interesting. Our complexes are considerably more coherent than the reference complexes on regulator overlap and perform similarly on correlation of abundance and growth phenotype. Conversely our complexes are significantly less coherent than the reference complexes on GO biological process annotations; this is not surprising as the reference complexes and GO annotations are derived (at least in part) from similar data sources, such as literature and small scale experiments. Overall when comparing with data sources that were not used in constructing the reference complexes, our predictions seem to perform as well or better than the reference set, suggesting that our predictions provide a strong set of complexes that can be used as a new reference.
Fig. 3.

Coherence of our predicted complexes. We computed the functional coherence between proteins in the same complex against external data sources that are not used in training. More coherent complexes have a smaller difference in protein abundance, have a smaller semantic distance in GO biological process, share more transcriptional regulators, and have a higher growth fitness correlation. The y axis shows the values for these metrics of functional coherence; also shown is the performance of random pairs (thick horizontal line). Our predicted set of complexes significantly outperforms other state-of-the-art methods. For GO biological processes, our complexes have a semantic distance 8 and 17% lower than the methods of Hart et al. (5) and Pu et al. (6), respectively. For protein abundance, the improvement over Hart et al. (5) and Pu et al. (6) is 5 and 10%, respectively; conversely our complexes are 12% less coherent than the top affinity pairs, suggesting that proteins with lower affinity scores can be members of the complex but also play other roles in the cell, reducing their correlation with other proteins in the same complex. For the correlation of growth phenotypes across different conditions, our predicted complexes are 19 and 31% more coherent, respectively, a very significant improvement. Finally protein pairs within our complexes on average share 30 and 59%, respectively, more transcription factors than those of Hart et al. (5) and Pu et al. (6). The comparison with the reference complexes shows that our complexes are considerably more coherent on regulator overlap and perform similarly on correlation of abundance and growth phenotype. Conversely our complexes are 21% less coherent than the reference complexes on GO biological process annotations; this is not surprising as the reference complexes and GO annotations are derived (at least in part) from similar data sources, such as literature and small scale experiments.
In-depth Study of Predicted Complexes—
We also did a systematic, manual evaluation of many of our predicted complexes. We first considered the complexes that were one away from the reference set, that is a protein P and a complex A where P was either added to A or removed from A in contradiction to the reference set. Most of these cases represented situations where it is unclear whether P really did belong in A or not, and different biologists often have different opinions. For example, the Torpedo complex, which is involved in transcriptional termination by RNA polymerase II (63), was reported to be comprised of three subunits: the exonuclease Rat1, Rai1, and Rtt103. We predicted that Rtt103 was not a component of this complex, consistent with the weaker stoichiometric association of Rtt103 with the two other tightly associated members of the complex (63). In another example, we predicted that Csn12 was not a component of the COP9 signalosome, which is involved in deneddylation (27). Consistent with this, Maytal-Kivity et al. (64) demonstrated that Csn12 is the only component of this complex that is not required for the deneddylation activity. Furthermore we found that Csn12, but not other signalosome subunits, is required for efficient mRNA splicing at a number of genes in budding yeast,4 suggesting that Csn12 plays multiple cellular roles and may not be an integral member of the complex. Finally we predicted that Ski7 is part of the exosome complex, which harbors 3′-to-5′ exonuclease activity and acts on many different types of RNA. Evidence suggests that Ski7 acts as an adaptor to target the exosome to mRNAs lacking stop codons (65).
In other cases, however, the predictions made by our algorithm were interesting and worthy of further investigation. One such example is the eisosome, previously described to be primarily comprised of two subunits (Pil1 and Lsp1) (25); however, we predicted that the complex contains another, previously undescribed component, Mrp8. Consistent with this prediction, TAP purification of Mrp8 reveals that it is indeed a stoichiometrically stable member of the eisosome complex (see Fig. 4a). Further work will be required to determine the role that this novel subunit plays in eisosome function.
Fig. 4.

Validation of novel biological findings. a, Pil1, Lsp1, and Mrp8 form a stable complex. TAP-tagged Pil1 was affinity-purified from yeast. Highly enriched fractions were run on SDS-PAGE, and co-purifying proteins were identified by LC-MS/MS as Lsp1 and Mrp8, indicated on the left; protein sizes are shown in kDa on the right. The result supports our prediction that Mrp8 is a component of the eisosome complex. b and c, support for newly uncovered NSC complex comprised of six genes (YCR095C, YHL029C, YNL032W, YNL056W, YNL099C/OCA1, and YDR067C), four of which are phosphatases. Five of these components were predicted by HACO to be a stoichiometrically stable complex; based on other data (shown in this figure) we conjecture that the sixth (YDR067C) may also be a member of this complex. b, support in chemical genomics data of Hillenmeyer et al. (45), which measured the fitness profiles of all non-essential homozygous yeast mutants under 418 conditions. Left, the fitness profiles of the six predicted NSC members cluster tightly together. Right, shown are the conditions in which at least one of the six components had a fitness defect with p < 1e−10; the consistently strong sensitivity to rapamycin, lysine dropout, and synthetic complete medium suggests the involvement of these proteins in response to nutrient starvation. c, support in new EMAP data, which measured quantitative genetic interaction profiles with ∼500 genes involved in signaling. Left, the genetic interaction profiles of the six components cluster tightly together. Right, the components have positive genetic interactions among them and exhibit significant interactions with genes involved in nutrient starvation response, including Sch9 and Gcn2. CBP, calmodulin-binding peptide.
We also studied the novel complex predictions, those that did not match any of the reference complexes above our match threshold. A number of these turned out to be well characterized complexes that, for some reason, had not (yet) been included into any of our three reference sets. For example, we identified the Sit4/Sap185 heterodimer phosphatase complex (66); a complex comprised of Yos9, Hrd3, Usa1, and Hrd1 that is involved in endoplasmic reticulum-associated degradation (67, 68); and the U3-processome complex (complex 1129) involved in the generation and regulation of the small ribosomal particle (69). Many others comprised plausible complexes that, to our knowledge, have not yet been characterized and are worthy candidates for further investigation.
One such example is a complex (complex 1014) comprised of five components (YNL099C/OCA1, YNL056W/OCA2, YNL032W/SIW14/OCA3, YCR095C/OCA4, and YHL029C/OCA5), four of which are putative phosphatases. One of the proteins (Oca1) has been previously shown to be required for cell cycle arrest in response to exposure to a lipid peroxide (70). We note that the individual pairwise connections between these proteins were observed before and that various forms of evidence support their shared function (14), including a shared phenotype of oxidant-induced cell cycle arrest, which underlies the current name of many of these genes in SGD. However, this group was not previously identified as a complex nor was its function characterized. Further supporting our prediction of this group as a complex is the fact that the chemical-genetic interaction profiles of the five genes were tightly clustered in a recent high throughput study (45) (Fig. 4b). Mutations in the components of the complex resulted in significant sensitivity to a number of conditions, including several that are related to nutrient starvation, including exposure to rapamycin, lysine dropout, and synthetic complete medium. To further characterize the functions of these factors, we subjected the mutants to quantitative genetic interaction profiling using an EMAP (60, 61, 71) focused on genes implicated in signaling, including protein and small molecule kinases and phosphatases (see our supporting Web site3). Again we found that the components of the complex had strong positive genetic interactions between them and clustered tightly together within the set of ∼500 genes included in the EMAP, both factors that indicate a strong functional connection (60). Specifically we found that all components have strong negative genetic interactions with Sch9, the yeast homolog of S6 kinase and a central node in nutrient signaling (72, 73). Conversely we found strong positive genetic interactions with GCN2, a protein kinase that phosphorylates the α subunit of translation initiation factor eIF2 (Sui2) in response to nutrient starvation (74). Collectively these data suggest an involvement of these proteins in response to nutrient starvation. Interestingly both the chemogenomic profiling and the genetic interactions suggested a functional connection to another uncharacterized gene, YDR067C, which may form a sixth member of the complex. Based on the coherence of this complex and its strong links to nutrient starvation, we propose to name this six-protein complex NSC.
The predictions made by our algorithm also contained a number of mistakes, which fell into two main categories. The first comprised subsets of known complexes, such as subsets of the pre-60 S ribosomal particle (complexes 1088 and 1106). These may represent functionally distinct submodules within larger complexes and therefore may provide useful insight about complex structure. Consistent with this notion, we identified the deubiquitination unit of SAGA (Ubp8/Sgf11) (75–77). The other category of error involved pairs of complexes that either interact or share subunits and were merged by the HACO procedure into a single complex. For example, complex 1125 is comprised of two chromatin-remodeling complexes, INO80-C and SWR-C, which have shared components, including Rvb1 and Rvb2, members of the RuvB family of helicases (78). These two error modes illustrate the difficulty in selecting the appropriate granularity for making complex predictions where some complexes occur fairly low in the clustergram so that they have very high affinity with components outside the complex, whereas others occur very high in the clustergram so that they contain components that have low affinity among themselves. This difficulty is perhaps one of the biggest challenges in accurately determining complexes. We note, however, that in some cases (such as the SAGA complex described above), the correct complexes themselves (or a slight variant) were sometimes also members in our set of predictions, a situation possible because of the ability of HACO to make predictions at multiple levels of granularity. Thus, HACO is occasionally able to circumvent this challenge by trading off coverage for precision.
A Comprehensive Interaction Network
Complexes together with individual proteins comprise the basic units of the interaction network of the cell. So far, most of the work (27–29) has focused on predictions of interactions between pairs of individual proteins. However, the view of the network in terms of pairwise interactions loses much of its structure. Many interactions arise from co-complexness so that a single large complex can give rise to a very dense (almost complete) subgraph in the network. Other pairwise interactions are representatives of interactions between larger complexes. We therefore set out to construct a comprehensive network of interactions between all basic units in the proteome, both complexes and proteins.
We compiled a reference set of CCIs and protein-complex interactions from reliable protein-protein interactions and hand-curation (see “Experimental Procedures”). Importantly to avoid circular reasoning, any interactions that we used in the construction of the gold standard CCIs and protein-complex interactions were not given as features to the prediction algorithm. We used 10-fold cross-validation to evaluate the ability of our model in accurately predicting CCIs. We randomly divide our reference interactions into 10 sets. In each fold, we hide one set and train on the remaining nine sets. We then make predictions on the held-out set using the learned model. We compare three methods (see “Experimental Procedures”): simple naïve Bayes, a discriminative boosting method, and naïve Bayes with EM (NB + EM) that also makes use of the data for pairs that are not in our reference set. As we can see in Fig. 5a, NB + EM performs better than both other methods, achieving very high performance: 44 of the top 50 predictions (88%) are in the positive reference set. We also compared these results with two state-of-the-art methods for predicting protein-protein interactions: the PE score and the InSite probabilities. As we can see, by integrating multiple sources of data, we are able to improve the accuracy to 0.88 (area under the ROC curve) from 0.85 and 0.79 for PE score and InSite probabilities, respectively.
Fig. 5.

Verification of complex-based interaction network. a, verification of our complex-complex interaction predictions relative to our reference set. Complex pairs in the hidden set of a 10-fold cross-validation are ranked based on their predicted interaction probabilities. Blue, green, and red curves are for the three models we tried. Light blue and pink curves are for the predictions using only PE score or InSite probabilities, respectively. Each point on the curve corresponds to a different threshold, giving rise to a different number of predicted interactions. The value on the x axis is the number of pairs not in the reference set but predicted to interact. The value on the y axis is the number of reference interactions that are predicted to interact. The bars in the right bottom corner are the areas under the ROC curves. Our naïve Bayes model with EM achieves the highest accuracy. The prediction made by PE score alone is slightly worse than our integrated models. b, functional coherence of interacting complexes measured by joint membership in the same MIPS functional category, a feature not used in training. We only consider those interacting complexes if both of them are assigned to some MIPS category. We picked the top 500 predictions from our NB + EM model and the top 500 obtained from the PE score alone. We compared them with complex pairs in our reference set and randomly selected pairs. The y axis shows the proportion of interacting complexes that are assigned to the same MIPS category. As we can see, 59.2% of our predicted interacting complexes share the same MIPS category, whereas only 35.2 and 45.5% share the same category for random complex pairs and for those predicted by PE score alone, respectively. The reference complexes are the most coherent, a fact that is not surprising as the functional classification of reference complexes is sometimes derived from the same literature sources as the interactions between those complexes. c, verification against a reference set of our unified predictions of protein-complex and complex-complex interaction set. Complex pairs in the hidden set of a 10-fold cross-validation are ranked based on their predicted interaction probabilities. The blue curve is for our naïve Bayes model with EM. The light blue curve is for the predictions using only PE score. The pink curve is for the prediction using only InSite probability. Each point on the curve corresponds to a different threshold, giving rise to a different number of predicted interactions. The value on the x axis is the number of pairs not in the reference set but predicted to interact. The value on the y axis is the number of reference interactions that are predicted to interact. The areas under the curves are shown by the bars in the bottom right corner.
The PE score provides the strongest signal and provides, by itself, accuracy on our reference set that is only somewhat lower than that of our integrated model. However, when evaluated on other metrics, our data integration provides more significant benefits. We expect interacting complexes to be involved in similar biological processes. Therefore, we measured the coherence of our CCI predictions relative to GO biological process annotations and MIPS functional categories (neither of which were used in training). Here our predictions are considerably better than those obtained from the PE score alone (Fig. 5b), suggesting that our set of reference complexes is perhaps somewhat biased toward areas that are well covered by the TAP-MS assays.
We also apply our model to predict a unified network involving both proteins and complexes, a network that we call ComplexNet. In ComplexNet, we have both the interactions between two complexes and the interactions between a protein and a complex. As we can see from Fig. 5c, by integrating multiple data sources, our naïve Bayes model with EM is able to achieve higher accuracy than using either PE score or InSite probability alone. We generated predictions for all protein-complex pairs and complex-complex pairs by training on the entire reference set (see our supporting Web site3 for the complete list of the predictions). Overall our predictions provide a comprehensive network of all of the interactions involving complexes. It can be combined with a set of high quality protein-protein interactions (such as Ref. 49) to provide a complete set of predictions for the S. cerevisiae protein interaction network. Fig. 6 presents a fragment of the network.
Fig. 6.

A complex-level interaction network. a, a fragment of our ComplexNet, comprising a subset of the interactions between the largest complexes. Shown are the 500 highest confidence predictions plus the reference interactions restricted to interactions between complexes of size ≥3. The color of each complex indicates the fraction of essential components, demonstrating the enrichment of essential proteins in larger complexes. The complexes are placed in regions based on their cellular localization determined by majority vote based on the data of Huh et al. (37). The inset shows all interactions that involve the kinetochore complex. b, a list of the complexes associated with the numbers in the figure. Complexes are associated with a name of a known complex when they overlap with that complex with F-score >0.5. Otherwise the number associated with a complex is a unique identifier used in our supporting Web site.3 ER, endoplasmic reticulum.
We identified many CCIs that were expected and well characterized but not in the reference set, such as interactions between histones and several chromatin-modifying complexes including the ISW1 complex, the HAT1 complex, and RSC. ComplexNet also suggests novel hypotheses, several of which have support in the literature. For example, we predicted an interaction between complex 1035, which consists of the poorly characterized proteins Yer071c and Yir003w/Aim21, with the yeast actin-capping protein (a Cap1-Cap2 heterodimer). Consistent with this prediction, high throughput fluorescence microscopy found that Yir003w co-localizes with components of the actin cytoskeleton (37), and two-hybrid data has connected Yir003w to the actin-binding protein Abp1 (79). Additionally like deletion of CAP1 or CAP2, deletion of YER071C or YIR003W results in strong sensitivity to the actin-depolymerizing agent latrunculin (45). Our observation suggests a more specific placement of this complex among the actin regulatory machinery. We also found several interesting interactions involving the centromere-localized kinetochore complex (Fig. 6), some of which have independent support. Our prediction of an interaction between the kinetochore and the proteasome is supported by a recent report that levels of Cse4, a centromere-localized histone, are regulated by ubiquitin-proteasome-mediated proteolysis (80). Our predicted link between the kinetochore and the spliceosome is consistent with evidence of a functional connection between these two factors (81). The remaining connections we observed with the kinetochore (pre-60 S ribosomal particle and RNA polymerase III) are intriguing, but more work will be required to determine the validity and functional significance of these predicted relationships. We can also learn from the false positive predictions of CCIs. Our algorithm does make some apparently false positive predictions, and many of them fall into two main categories. Pairs of complexes that share a substantial number of common components, such as the SWR complex and NuA4, are sometimes identified as interacting. Additionally pairs of complexes that do not interact directly but are one link away in the interaction network are sometimes identified. Along these lines, we identified an interaction between the NuA4 histone acetylase complex and the opposing RPD3(L) deacetylase complex. Both complexes have subunits with specificity for binding Lys4-trimethylated histone H3 (82) and have been found to be regulated by binding to 14-3-3 proteins (83). Thus, even such a false positive may still provide interesting biological insights.
Essentiality and Complex Size
Much discussion has occurred regarding the relationship between essentiality and the structure of the protein-protein interaction network. Early work of Jeong et al. (26) and Han et al. (84) found that hub proteins in a protein-protein interaction network are more likely to be encoded by essential genes. More recent work (85) suggests that highly connected proteins are simply more likely to participate in essential protein-protein interactions and are therefore more likely to be essential. However, a deeper insight on the relationship between the protein network and essentiality can be obtained by considering the network at the level of complexes rather than pairwise interactions. Such an analysis was recently performed by Hart et al. (5), who showed that essential proteins are concentrated in certain complexes, resulting in a dichotomy of essential and non-essential complexes. This phenomenon was also found in our predicted complexes (Fig. 7a). However, that finding does not explain why hubs in the network are more likely to be essential. We therefore looked into the distribution of essential proteins in complexes of different sizes and found that the fraction of essential components in a complex tends to increase with complex size (Fig. 7b). Moreover when we aggregate over all complexes of a given size, larger complexes tend to have a far greater proportion of essential proteins among their components (Fig. 7b). Components in a large complex are naturally highly connected in the protein interaction network and therefore often form hubs. Thus, the finding regarding the essentiality of hubs very likely arises from the fact that large complexes are more likely to have a much higher ratio of essential genes. Our finding is consistent with the recent work of Zotenko et al. (86), who argue that essential hubs are often members of a densely connected set of proteins performing an essential cellular function. However, this analysis is still performed on the pairwise protein network and hence is unable to identify the strong dependence between the size of a complex and its essentiality.
Fig. 7.

Relationship between complex size and essentiality. a, fraction of complexes with different essentiality fractions. Each complex is represented by its size and the fraction of essential components. The different colors represent different ratios of essentiality in a complex discretized into five bins. The x axis represents the complex size, and the y axis represents the fraction of complexes of that size that have this particular essentiality ratio. We can see that the large majority of complexes of size 2 have essentiality ratio in the range 0–0.2, whereas larger complexes tend to have a larger essentiality ratio. Also shown on the x axis, in parentheses, is the number of complexes in each category (e.g. there are 54 complexes of size 3). b, the relationship between complex size and the proportion of essential proteins in complexes of that size. The x axis is the size bin of the complexes. The y axis is the proportion of essential proteins in all complexes within the size bin. As we can see, larger complexes tend to have a higher proportion of essential proteins. c, evaluation of different metrics as predictive of essentiality: size of the largest enclosing complex versus degree in the protein-protein interaction network (hubness). For the red and light blue curves, we rank each protein based on the size of the largest complex to which is belongs; the red curve uses predicted complexes, and the light blue curve uses the reference complexes. For the blue curve and green curve, we use the hubness, the degree of protein in a protein-protein interaction network; the blue curve uses the yeast two-hybrid protein-protein interaction network, and the green curve uses a network where pairs are connected if they have a scaled PE score >0.5. The x axis is the number of essential proteins in the K top ranked proteins (for different values of K), and the y axis is the number of non-essential proteins. Complex size in our predicted complexes (red) is the best predictor for essentiality. The hubness based on PE score (green) performs better than the other metrics presumably because it also correlates directly with co-membership in a complex. The reference complexes (light blue) perform slightly worse but considerably better than interactions in the Y2H data.
To test whether our finding truly explains the phenomenon of essential hubs, we tested whether essentiality is better explained by complex size or by hubness. We rank every protein based on the size of the largest complex to which it belongs and for the K top-ranked proteins (for different values of K) plot the number of essential versus non-essential proteins (Fig. 7c). We plotted a similar curve by using the hubness of the protein, the degree in the yeast two-hybrid protein-protein interaction network (35, 36). As we can see, complex size is a much better predictor for essentiality than hubness. We note that if we use the scaled PE score (at threshold >0.5) to define a protein-interaction network the hubness becomes a strong predictor of protein essentiality. However, PE score is more related to co-complexness than interaction, and thus this metric of hubness is directly related to complex size. Nevertheless using complex size directly is still better than using scaled PE score. Interestingly if we use the size of the largest enclosing reference complex to rank each protein, the result is slightly less predictive than using our predicted complexes or even the scaled PE score directly.
DISCUSSION
Identifying a comprehensive set of protein complexes in yeast is an important but challenging task. The high quality and high throughput TAP-MS data, which directly measure co-complexness, provide a starting point for accurately reconstructing these complexes. Indeed two recent studies (5, 6) used the TAP-MS data to produce a set of complexes with state-of-the-art performances. Both methods applied a simple clustering algorithm to a score derived directly from the TAP-MS data. In this study, we are able to significantly improve the accuracy of the complex reconstruction in three ways. First, we carefully constructed a large set of reference complexes and trained our model so it specifically predicts co-membership in stoichiometrically stable complexes. Second, we integrated multiple sources of heterogeneous data so our predictions are more robust to noise and incomplete coverage in the TAP-MS data. Finally we extended the highly effective HAC algorithm to allow reconstruction of clusters with overlap, a flexibility that allows it to circumvent many of the limitations of the standard HAC algorithm. We show that the resulting set of predicted complexes (available from our Web site3) has significantly higher accuracy and is more biologically coherent than that of other recent methods. In many cases, it is even more coherent than the reference set, indicating it is of high quality and can be used as a new reference set. When combined with our comprehensive, hand-curated reference set (also available from our Web site3), our work provides a significant new resource to the research community.
With our high quality set of complexes, we are able to take a higher level perspective on the protein-protein interaction network, viewing it in terms of interactions between atomic units (whether individual proteins or stable complexes). There has been much work on predicting protein-protein interactions. However, these pairwise interactions are often induced by higher level relationships: those within a complex and those between complexes. Interactions within a complex give rise to densely connected subgraphs in the interaction network; interactions between complexes can give rise to a network of interconnections involving different members of the two complexes. Viewing the network in terms of its atomic units can help clarify its structure and its basic properties. We therefore defined the novel problem of predicting interactions between complexes and other complexes or proteins and constructed a new, high accuracy method for making such predictions. The result of our analysis is ComplexNet, a unified interaction network involving both proteins and complexes. We can now analyze the properties of this network, which better captures the true interactions underlying cellular processes. In particular, this network provides a new perspective on the previously observed relationship between the “hubness” of a protein in the network and its essentiality, demonstrating that larger complexes are more likely to be essential and comprise a large fraction of essential proteins. It would also be of interest to study other properties of this network, such as its connectivity or hierarchical structure.
To find a coherent set of proteins that form a complex, we have the choice of many different clustering algorithms. Brohee and van Helden (87) showed that MCL works well on a protein-protein interaction network by comparing it with three other clustering algorithms in the literature. So not surprisingly, Pu et al. (6) and Hart et al. (5) applied MCL to the TAP-MS network; MCL is confirmed by our results to be better than other existing methods in terms of reconstructing reference complexes and biological coherence. On the other hand, we found that HAC achieves about the same accuracy as MCL. Therefore, we focus on the best proven method and try to further improve it by addressing some of its limitations. One of the significant advantages of our HACO algorithm, which extends the HAC, is its ability to create overlapping complexes. Indeed the inability of traditional HAC to generate overlapping clusters is one of its major deficiencies in other types of data as well. Interestingly in our results, there were relatively few cases where two “correct” complexes shared subcomponents. Most of the benefit of HACO arose from avoiding mistakes arising from the greedy decisions of HAC and from allowing predictions at different levels of granularity (e.g. a complex and one of its subunits). Nevertheless the lack of extensive sharing of components between complexes was surprising given that such sharing is present in the reference set. To some extent, this phenomenon is due to the trade-off in HACO parameters between increasing the amount of component sharing and errors arising from merging of distinct complexes. However, HACO applied to other data sets (data not shown) did give rise to much more extensive sharing among different clusters. Thus, a complementary hypothesis is that some of the sharing of components between complexes arises when a protein plays roles in different complexes in different conditions. Our data, having been acquired almost entirely in YPD, would not reveal this condition-specific pleiotropy. It would be of great interest to acquire TAP-MS data in different conditions and study the extent to which complex structure is condition-specific.
We note that there are other clustering algorithms (88, 89) that also generate overlapping complexes. However, both of them are applicable only to a binary interaction network so an application to our task would require that we discretize the continuous affinities between protein pairs into two values (interacting and non-interacting) using some fixed threshold. Our analysis of the affinities for reference complexes suggested strongly that proteins that are co-complexed often exhibit affinities over a very broad range so that such a discretization would result in an unacceptable loss of useful information. On the other hand, HACO uses the continuous valued affinities directly, allowing the finer resolution of the computed affinities to be used by the algorithm. We also note that we devised several other novel methods that attempt to construct overlapping clusters. For example, one method directly learns an affinity function to predict the likelihood that a set of proteins forms a complex, aiming to take advantage of features involving more than two proteins. HACO significantly outperformed all of our other proposed methods, and so we omit details.
There are still many reference complexes that are not matched by our predicted complexes. Many of them fall into roughly two categories. In the first category, proteins in the reference complex have high affinities with each other and are grouped as a set during the HACO procedure. However, they are not selected in our predictions because they are not at the granularity where we cut our HACO cluster-lattice. They then become subsets or supersets of some predicted complexes. In fact, if we use all the sets generated during our HACO procedure as predicted complexes, 136 reference complexes would be perfectly predicted and 243 would be well matched by some predicted complexes in comparison with 95 perfect matches and 189 good matches in our current predictions. However, this approach would result in far too many predictions (3478), greatly reducing sensitivity. This fact highlights the limitations in defining a universal level of affinity at which one determines that a group of proteins forms a stable complex and suggests that a more flexible technique may be a useful direction for future work. In the second category, the proteins in the reference complex do not have high affinities with each other. This situation arises when the signal in the data is not sufficiently strong to indicate that two proteins are likely to interact. As most of our signal comes from the TAP-MS data, such “blind spots” can arise from limitations of this assay, such as complexes of low abundance or that are membrane-bound. In particular, we note that the TAP-MS data were all acquired in a single condition (rich media), and some complexes may simply not be present in the cell in that condition. Our inability to recover such complexes arises not from computational limitation, but from limitations in the data. New experimental assays are needed before these complexes can be reconstructed.
Like other previous approaches, our method was developed in the context of S. cerevisiae where we have the most data relevant to protein-protein interactions. Having a high quality set of predicted complexes is of significant value even in yeast as yeast provides an excellent model for many core biological processes. Moreover many key complexes are conserved from yeast to human, making our complex predictions valuable also to analysis of higher level organisms. Finally our method is general purpose and can easily be applied more broadly. Its ability to integrate multiple sets of diverse data makes it suitable for other organisms where we may not have the same type of data available as in yeast. With the increasing amount of high throughput protein-protein interaction data, both from TAP-MS (90) and other assays (30, 32), we should soon be able to provide a high quality reconstruction of protein complexes in other organisms, including human.
Our work takes a step toward a more hierarchical view of the protein-protein interaction network, moving up from individual proteins to complexes as the basic interacting units. The next level of the hierarchy is the pathways that comprise cellular pathways. Although the notion of a “pathway” is not as well defined, it would nevertheless be very useful to reconstruct pathways that are comprised of interacting complexes and proteins. This type of analysis will give us a unified perspective on the underlying hierarchical organization of the cell and provide significant insight.
Acknowledgments
We thank Maureen Hillenmeyer for useful discussions and for early access to the chemical genomics data.
Footnotes
Published, MCP Papers in Press, January 27, 2009, DOI 10.1074/mcp.M800490-MCP200
The abbreviations used are: TAP, tandem affinity purification; EMAP, epistatic miniarray profile; EM, expectation maximization; GO, Gene Ontology; HAC, hierarchical agglomerative clustering; HACO, hierarchical agglomerative clustering with overlap; JC, Jaccard coefficient; PCC, Pearson correlation coefficient; PE, purification enrichment; TF, transcription factor; Y2H, yeast two-hybrid; MIPS, Munich Information Center for Protein Sequences; MCL, Markov clustering; SGD, Saccharomyces Genome Database; NSC, nutrient starvation complex; DIP, Database of Interacting Proteins; SJC, scaled Jaccard coefficient; ROC, receiver operating characteristic; CCI, complex-complex interaction; NB, naïve Bayes.
Complex-complex interactions (CCI) (dags.stanford.edu/CCI/).
Complex (dags.stanford.edu/Complex/).
Wilmes, G. M., Bergkessel, M., Bandyopadhyay, S., Shales, M., Braberg, H., Cagney, G., Collins, S. R., Whitworth, G. B., Kress, T. L., Weissman, J. S., Ideker, T., Guthrie, C., and Krogan, N. J. (2008) A genetic interaction map of RNA-processing factors reveals links between sem1/Dss1-containing complexes and mRNA export and splicing Mol. Cell 32, 735–746
This work was supported by the National Science Foundation and the Defense Advanced Research Projects Agency under the program Cognitive Assistant that Learns and Organizes (to H. W., B. K., and D. K.), the Howard Hughes Medical Institute (to S. R. C. and K. M. S.), an Ernst Schering postdoctoral fellowship (to D. F.), and Sandler Family funding (to N. J. K.).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J. M., Michon, A. M., Cruciat, C. M., Remor, M., Hofert, C., Schelder, M., Brajenovic, M., Ruffner, H., Merino, A., Klein, K., Hudak, M., Dickson, D., Rudi, T., Gnau, V., Bauch, A., Bastuck, S., Huhse, B., Leutwein, C., Heurtier, M. A., Copley, R. R., Edelmann, A., Querfurth, E., Rybin, V., Drewes, G., Raida, M., Bouwmeester, T., Bork, P., Seraphin, B., Kuster, B., Neubauer, G., and Superti-Furga, G. ( 2002) Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147 [DOI] [PubMed] [Google Scholar]
- 2.Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., Yang, L., Wolting, C., Donaldson, I., Schandorff, S., Shewnarane, J., Vo, M., Taggart, J., Goudreault, M., Muskat, B., Alfarano, C., Dewar, D., Lin, Z., Michalickova, K., Willems, A. R., Sassi, H., Nielsen, P. A., Rasmussen, K. J., Andersen, J. R., Johansen, L. E., Hansen, L. H., Jespersen, H., Podtelejnikov, A., Nielsen, E., Crawford, J., Poulsen, V., Sorensen, B. D., Matthiesen, J., Hendrickson, R. C., Gleeson, F., Pawson, T., Moran, M. F., Durocher, D., Mann, M., Hogue, C. W., Figeys, D., and Tyers, M. ( 2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183 [DOI] [PubMed] [Google Scholar]
- 3.Gavin, A. C., Aloy, P., Grandi, P., Krause, R., Boesche, M., Marzioch, M., Rau, C., Jensen, L. J., Bastuck, S., Dumpelfeld, B., Edelmann, A., Heurtier, M. A., Hoffman, V., Hoefert, C., Klein, K., Hudak, M., Michon, A. M., Schelder, M., Schirle, M., Remor, M., Rudi, T., Hooper, S., Bauer, A., Bouwmeester, T., Casari, G., Drewes, G., Neubauer, G., Rick, J. M., Kuster, B., Bork, P., Russell, R. B., and Superti-Furga, G. ( 2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636 [DOI] [PubMed] [Google Scholar]
- 4.Krogan, N. J., Cagney, G., Yu, H., Zhong, G., Guo, X., Ignatchenko, A., Li, J., Pu, S., Datta, N., Tikuisis, A. P., Punna, T., Peregrin-Alvarez, J. M., Shales, M., Zhang, X., Davey, M., Robinson, M. D., Paccanaro, A., Bray, J. E., Sheung, A., Beattie, B., Richards, D. P., Canadien, V., Lalev, A., Mena, F., Wong, P., Starostine, A., Canete, M. M., Vlasblom, J., Wu, S., Orsi, C., Collins, S. R., Chandran, S., Haw, R., Rilstone, J. J., Gandi, K., Thompson, N. J., Musso, G., St Onge, P., Ghanny, S., Lam, M. H., Butland, G., Altaf-Ul, A. M., Kanaya, S., Shilatifard, A., O'Shea, E., Weissman, J. S., Ingles, C. J., Hughes, T. R., Parkinson, J., Gerstein, M., Wodak, S. J., Emili, A., and Greenblatt, J. F. ( 2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440, 637–643 [DOI] [PubMed] [Google Scholar]
- 5.Hart, G. T., Lee, I., and Marcotte, E. R. ( 2007) A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pu, S., Vlasblom, J., Emili, A., Greenblatt, J., and Wodak, S. J. ( 2007) Identifying functional modules in the physical interactome of Saccharomyces cerevisiae. Proteomics 7, 944–960 [DOI] [PubMed] [Google Scholar]
- 7.Collins, S. R., Kemmeren, P., Zhao, X. C., Greenblatt, J. F., Spencer, F., Holstege, F. C., Weissman, J. S., and Krogan, N. J. ( 2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol. Cell. Proteomics 6, 439–450 [DOI] [PubMed] [Google Scholar]
- 8.Mewes, H. W., Frishman, D., Mayer, K. F., Munsterkotter, M., Noubibou, O., Pagel, P., Rattei, T., Oesterheld, M., Ruepp, A., and Stumpflen, V. ( 2006) MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res. 34, D169–D172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enright, A. J., Van Dongen, S., and Ouzounis, C. A. ( 2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jansen, R., Yu, H., Greenbaum, D., Kluger, Y., Krogan, N. J., Chung, S., Emili, A., Snyder, M., Greenblatt, J. F., and Gerstein, M. ( 2003) A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302, 449–453 [DOI] [PubMed] [Google Scholar]
- 11.Zhang, L. V., Wong, S. L., King, O. D., and Roth, F. P. ( 2004) Predicting co-complexed protein pairs using genomic and proteomic data integration. BMC Bioinformatics 5, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu, J., and Noble, W. S. ( 2008) Predicting co-complexed protein pairs from heterogeneous data. PLoS Comput. Biol. 4, e1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, J., and Yuan, B. ( 2006) Detecting functional modules in the yeast protein-protein interaction network. Bioinformatics 22, 2283–2290 [DOI] [PubMed] [Google Scholar]
- 14.Lee, I., Li, Z., and Marcotte, E. M. ( 2007) An improved, bias-reduced probabilistic functional gene network of baker's yeast, Saccharomyces cerevisiae. PLoS ONE 2, e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marcotte, E. M., Pellegrini, M., Thompson, M. J., Yeates, T. O., and Eisenberg, D. ( 1999) A combined algorithm for genome-wide prediction of protein function. Nature 402, 83–86 [DOI] [PubMed] [Google Scholar]
- 16.Schlitt, T., Palin, K., Rung, J., Dietmann, S., Lappe, M., Ukkonen, E., and Brazma, A. ( 2003) From gene networks to gene function. Genome Res. 13, 2568–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strong, M., Mallick, P., Pellegrini, M., Thompson, M. J., and Eisenberg, D. ( 2003) Inference of protein function and protein linkages in Mycobacterium tuberculosis based on prokaryotic genome organization: a combined computational approach. Genome Biol. 4, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Mering, C., Zdobnov, E. M., Tsoka, S., Ciccarelli, F. D., Pereira-Leal, J. B., Ouzounis, C. A., and Bork, P. ( 2003) Genome evolution reveals biochemical networks and functional modules. Proc. Natl. Acad. Sci. U. S. A. 100, 15428–15433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. ( 2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yanai, I., and DeLisi, C. ( 2002) The society of genes: networks of functional links between genes from comparative genomics. Genome Biol. 3, research0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yellaboina, S., Goyal, K., and Mande, S. C. ( 2007) Inferring genome-wide functional linkages in E. coli by combining improved genome context methods: comparison with high-throughput experimental data. Genome Res. 17, 527–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins, M., Schapire, R., and Singer, Y. ( 2002) Logistic regression, AdaBoost and Bregman distances. Mach. Learn. 48, 253–285 [Google Scholar]
- 23.Eisen, M. B., Spellman, P. T., Brown, P. O., and Botstein, D. ( 1998) Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95, 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins, S. R., Miller, K. M., Maas, N. L., Roguev, A., Fillingham, J., Chu, C. S., Schuldiner, M., Gebbia, M., Recht, J., Shales, M., Ding, H., Xu, H., Han, J., Ingvarsdottir, K., Cheng, B., Andrews, B., Boone, C., Berger, S. L., Hieter, P., Zhang, Z., Brown, G. W., Ingles, C. J., Emili, A., Allis, C. D., Toczyski, D. P., Weissman, J. S., Greenblatt, J. F., and Krogan, N. J. ( 2007) Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446, 806–810 [DOI] [PubMed] [Google Scholar]
- 25.Walther, T. C., Brickner, J. H., Aguilar, P. S., Bernales, S., Pantoja, C., and Walter, P. ( 2006) Eisosomes mark static sites of endocytosis. Nature 439, 998–1003 [DOI] [PubMed] [Google Scholar]
- 26.Jeong, H., Mason, S. P., Barabasi, A. L., and Oltvai, Z. N. ( 2001) Lethality and centrality in protein networks. Nature 411, 41–42 [DOI] [PubMed] [Google Scholar]
- 27.Wei, N., and Deng, X. W. ( 2003) The COP9 signalosome. Annu. Rev. Cell Dev. Biol. 19, 261–286 [DOI] [PubMed] [Google Scholar]
- 28.Liu, Y., Liu, N., and Zhao, H. ( 2005) Inferring protein-protein interactions through high-throughput interaction data from diverse organisms. Bioinformatics 21, 3279–3285 [DOI] [PubMed] [Google Scholar]
- 29.Bock, J. R., and Gough, D. A. ( 2001) Predicting protein-protein interactions from primary structure. Bioinformatics 17, 455–460 [DOI] [PubMed] [Google Scholar]
- 30.Stelzl, U., Worm, U., Lalowski, M., Haenig, C., Brembeck, F. H., Goehler, H., Stroedicke, M., Zenkner, M., Schoenherr, A., Koeppen, S., Timm, J., Mintzlaff, S., Abraham, C., Bock, N., Kietzmann, S., Goedde, A., Toksoz, E., Droege, A., Krobitsch, S., Korn, B., Birchmeier, W., Lehrach, H., and Wanker, E. E. ( 2005) A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 [DOI] [PubMed] [Google Scholar]
- 31.Cherry, J. M., Adler, C., Ball, C., Chervitz, S. A., Dwight, S. S., Hester, E. T., Jia, Y., Juvik, G., Roe, T., Schroeder, M., Weng, S., and Botstein, D. ( 1998) SGD: Saccharomyces Genome Database. Nucleic Acids Res. 26, 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peri, S., Navarro, J. D., Amanchy, R., Kristiansen, T. Z., Jonnalagadda, C. K., Surendranath, V., Niranjan, V., Muthusamy, B., Gandhi, T. K., Gronborg, M., Ibarrola, N., Deshpande, N., Shanker, K., Shivashankar, H. N., Rashmi, B. P., Ramya, M. A., Zhao, Z., Chandrika, K. N., Padma, N., Harsha, H. C., Yatish, A. J., Kavitha, M. P., Menezes, M., Choudhury, D. R., Suresh, S., Ghosh, N., Saravana, R., Chandran, S., Krishna, S., Joy, M., Anand, S. K., Madavan, V., Joseph, A., Wong, G. W., Schiemann, W. P., Constantinescu, S. N., Huang, L., Khosravi-Far, R., Steen, H., Tewari, M., Ghaffari, S., Blobe, G. C., Dang, C. V., Garcia, J. G., Pevsner, J., Jensen, O. N., Roepstorff, P., Deshpande, K. S., Chinnaiyan, A. M., Hamosh, A., Chakravarti, A., and Pandey, A. ( 2003) Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 13, 2363–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. ( 2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gollub, J., Ball, C. A., Binkley, G., Demeter, J., Finkelstein, D. B., Hebert, J. M., Hernandez-Boussard, T., Jin, H., Kaloper, M., Matese, J. C., Schroeder, M., Brown, P. O., Botstein, D., and Sherlock, G. ( 2003) The Stanford Microarray Database: data access and quality assessment tools. Nucleic Acids Res. 31, 94–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., and Sakaki, Y. ( 2001) A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U. S. A. 98, 4569–4574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., Qureshi-Emili, A., Li, Y., Godwin, B., Conover, D., Kalbfleisch, T., Vijayadamodar, G., Yang, M., Johnston, M., Fields, S., and Rothberg, J. M. ( 2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 [DOI] [PubMed] [Google Scholar]
- 37.Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., and O'Shea, E. K. ( 2003) Global analysis of protein localization in budding yeast. Nature 425, 686–691 [DOI] [PubMed] [Google Scholar]
- 38.Lord, P. W., Stevens, R. D., Brass, A., and Goble, C. A. ( 2003) Investigating semantic similarity measures across the Gene Ontology: the relationship between sequence and annotation. Bioinformatics 19, 1275–1283 [DOI] [PubMed] [Google Scholar]
- 39.Xenarios, I., Salwinski, L., Duan, X. Q. J., Higney, P., Kim, S. M., and Eisenberg, D. ( 2002) DIP; the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 30, 303–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vapnik, V. ( 1999) The Nature of Statistical Learning Theory, 2nd Ed., Springer-Verlag, New York
- 41.Freund, Y., and Schapire, R. ( 1997) A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 55, 119–139 [Google Scholar]
- 42.Sokal, R., and Michener, C. ( 1958) A statistical method for evaluating systematic relationships. Univ. Kans. Sci. Bull. 38, 1409–1438 [Google Scholar]
- 43.Krause, R., von Mering, C., and Bork, P. ( 2003) A comprehensive set of protein complexes in yeast: mining large scale protein-protein interaction screens. Bioinformatics 19, 1901–1908 [DOI] [PubMed] [Google Scholar]
- 44.Ghaemmaghami, S., Huh, W. K., Bower, K., Howson, R. W., Belle, A., Dephoure, N., O'Shea, E. K., and Weissman, J. S. ( 2003) Global analysis of protein expression in yeast. Nature 425, 737–741 [DOI] [PubMed] [Google Scholar]
- 45.Hillenmeyer, M. E., Fung, E., Wildenhain, J., Pierce, S. E., Hoon, S., Lee, W., Proctor, M., St Onge, R. P., Tyers, M., Koller, D., Altman, R. B., Davis, R. W., Nislow, C., and Giaever, G. ( 2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320, 362–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacIsaac, K. D., Wang, T., Gordon, D. B., Gifford, D. K., Stormo, G. D., and Fraenkel, E. ( 2006) An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harbison, C. T., Gordon, D. B., Lee, T. I., Rinaldi, N. J., Macisaac, K. D., Danford, T. W., Hannett, N. M., Tagne, J. B., Reynolds, D. B., Yoo, J., Jennings, E. G., Zeitlinger, J., Pokholok, D. K., Kellis, M., Rolfe, P. A., Takusagawa, K. T., Lander, E. S., Gifford, D. K., Fraenkel, E., and Young, R. A. ( 2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431, 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Date, S. V., and Marcotte, E. M. ( 2003) Discovery of uncharacterized cellular systems by genome-wide analysis of functional linkages. Nat. Biotechnol. 21, 1055–1062 [DOI] [PubMed] [Google Scholar]
- 49.Wang, H., Segal, E., Ben-Hur, A., Li, Q. R., Vidal, M., and Koller, D. ( 2007) InSite: a computational method for identifying protein-protein interaction binding sites on a proteome-wide scale. Genome Biol. 8, R192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zakrzewska, A., Boorsma, A., Brul, S., Hellingwerf, K. J., and Klis, F. M. ( 2005) Transcriptional response of Saccharomyces cerevisiae to the plasma membrane-perturbing compound chitosan. Eukaryot. Cell 4, 703–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mercier, G., Berthault, N., Touleimat, N., Kepes, F., Fourel, G., Gilson, E., and Dutreix, M. ( 2005) A haploid-specific transcriptional response to irradiation in Saccharomyces cerevisiae. Nucleic Acids Res. 33, 6635–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Causton, H. C., Ren, B., Koh, S. S., Harbison, C. T., Kanin, E., Jennings, E. G., Lee, T. I., True, H. L., Lander, E. S., and Young, R. A. ( 2001) Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12, 323–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lai, L. C., Kosorukoff, A. L., Burke, P. V., and Kwast, K. E. ( 2005) Dynamical remodeling of the transcriptome during short-term anaerobiosis in Saccharomyces cerevisiae: differential response and role of Msn2 and/or Msn4 and other factors in galactose and glucose media. Mol. Cell. Biol. 25, 4075–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Rourke, S. M., and Herskowitz, I. ( 2002) A third osmosensing branch in Saccharomyces cerevisiae requires the Msb2 protein and functions in parallel with the Sho1 branch. Mol. Cell. Biol. 22, 4739–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gasch, A. P., Spellman, P. T., Kao, C. M., Carmel-Harel, O., Eisen, M. B., Storz, G., Botstein, D., and Brown, P. O. ( 2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11, 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gasch, A. P., Huang, M., Metzner, S., Botstein, D., Elledge, S. J., and Brown, P. O. ( 2001) Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 12, 2987–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeRisi, J. L., Iyer, V. R., and Brown, P. O. ( 1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 [DOI] [PubMed] [Google Scholar]
- 58.Kitagawa, E., Akama, K., and Iwahashi, H. ( 2005) Effects of iodine on global gene expression in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 69, 2285–2293 [DOI] [PubMed] [Google Scholar]
- 59.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. ( 1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 [DOI] [PubMed] [Google Scholar]
- 60.Collins, S. R., Schuldiner, M., Krogan, N. J., and Weissman, J. S. ( 2006) A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 7, R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuldiner, M., Collins, S. R., Thompson, N. J., Denic, V., Bhamidipati, A., Punna, T., Ihmels, J., Andrews, B., Boone, C., Greenblatt, J. F., Weissman, J. S., and Krogan, N. J. ( 2005) Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123, 507–519 [DOI] [PubMed] [Google Scholar]
- 62.Bader, G. D., and Hogue, C. W. ( 2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim, M., Krogan, N. J., Vasiljeva, L., Rando, O. J., Nedea, E., Greenblatt, J. F., and Buratowski, S. ( 2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522 [DOI] [PubMed] [Google Scholar]
- 64.Maytal-Kivity, V., Piran, R., Pick, E., Hofmann, K., and Glickman, M. H. ( 2002) COP9 signalosome components play a role in the mating pheromone response of S. cerevisiae. EMBO Rep. 3, 1215–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van Hoof, A., Staples, R. R., Baker, R. E., and Parker, R. ( 2000) Function of the ski4p (Csl4p) and Ski7p proteins in 3′-to-5′ degradation of mRNA. Mol. Cell. Biol. 20, 8230–8243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luke, M. M., Della Seta, F., Di Como, C. J., Sugimoto, H., Kobayashi, R., and Arndt, K. T. ( 1996) The SAP, a new family of proteins, associate and function positively with the SIT4 phosphatase. Mol. Cell. Biol. 16, 2744–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Denic, V., Quan, E. M., and Weissman, J. S. ( 2006) A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359 [DOI] [PubMed] [Google Scholar]
- 68.Carvalho, P., Goder, V., and Rapoport, T. A. ( 2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 69.Dragon, F., Gallagher, J. E., Compagnone-Post, P. A., Mitchell, B. M., Porwancher, K. A., Wehner, K. A., Wormsley, S., Settlage, R. E., Shabanowitz, J., Osheim, Y., Beyer, A. L., Hunt, D. F., and Baserga, S. J. ( 2002) A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417, 967–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alic, N., Higgins, V. J., and Dawes, I. W. ( 2001) Identification of a Saccharomyces cerevisiae gene that is required for G1 arrest in response to the lipid oxidation product linoleic acid hydroperoxide. Mol. Biol. Cell 12, 1801–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fiedler, D., Braberg, H., Mehta, M., Chechik, G., Cagney, G., Mukherjee, P., Silva, A. C., Shales, M., Collins, S. R., van Wageningen, S., Kemmeren, P., Holstege, F. C. P., Weissman, J. S., Christopher-Keogh, M., Koller, D., Shokat, K. M., and Krogan, N. J. ( 2009) Functional organization of the S. cerevisiae phosphorylation network. Cell 136, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jorgensen, P., Rupes, I., Sharom, J. R., Schneper, L., Broach, J. R., and Tyers, M. ( 2004) A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 18, 2491–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Urban, J., Soulard, A., Huber, A., Lippman, S., Mukhopadhyay, D., Deloche, O., Wanke, V., Anrather, D., Ammerer, G., Riezman, H., Broach, J. R., De Virgilio, C., Hall, M. N., and Loewith, R. ( 2007) Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 26, 663–674 [DOI] [PubMed] [Google Scholar]
- 74.Garcia-Barrio, M., Dong, J., Ufano, S., and Hinnebusch, A. G. ( 2000) Association of GCN1-GCN20 regulatory complex with the N-terminus of eIF2alpha kinase GCN2 is required for GCN2 activation. EMBO J. 19, 1887–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Powell, D. W., Weaver, C. M., Jennings, J. L., McAfee, K. J., He, Y., Weil, P. A., and Link, A. J. ( 2004) Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol. Cell. Biol. 24, 7249–7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ingvarsdottir, K., Krogan, N. J., Emre, N. C., Wyce, A., Thompson, N. J., Emili, A., Hughes, T. R., Greenblatt, J. F., and Berger, S. L. ( 2005) H2B ubiquitin protease Ubp8 and Sgf11 constitute a discrete functional module within the Saccharomyces cerevisiae SAGA complex. Mol. Cell. Biol. 25, 1162–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee, K. K., Florens, L., Swanson, S. K., Washburn, M. P., and Workman, J. L. ( 2005) The deubiquitylation activity of Ubp8 is dependent upon Sgf11 and its association with the SAGA complex. Mol. Cell. Biol. 25, 1173–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caruthers, J. M., and McKay, D. B. ( 2002) Helicase structure and mechanism. Curr. Opin. Struct. Biol. 12, 123–133 [DOI] [PubMed] [Google Scholar]
- 79.Fazi, B., Cope, M., Douangamath, A., Ferracuti, S., Schirwitz, K., Zucconi, A., Drubin, D., Wilmanns, M., Cesareni, G., and Castagnoli, L. ( 2002) Unusual binding properties of the SH3 domain of the yeast actin-binding protein Abp1: structural and functional analysis. J. Biol. Chem. 277, 5290–5298 [DOI] [PubMed] [Google Scholar]
- 80.Collins, K. A., Furuyama, S., and Biggins, S. ( 2004) Proteolysis contributes to the exclusive centromere localization of the yeast Cse4/CENP-A histone H3 variant. Curr. Biol. 14, 1968–1972 [DOI] [PubMed] [Google Scholar]
- 81.Bialkowska, A., and Kurlandzka, A. ( 2002) Proteins interacting with Lin 1p, a putative link between chromosome segregation, mRNA splicing and DNA replication in Saccharomyces cerevisiae. Yeast 19, 1323–1333 [DOI] [PubMed] [Google Scholar]
- 82.Shi, X., Kachirskaia, I., Walter, K. L., Kuo, J. H., Lake, A., Davrazou, F., Chan, S. M., Martin, D. G., Fingerman, I. M., Briggs, S. D., Howe, L., Utz, P. J., Kutateladze, T. G., Lugovskoy, A. A., Bedford, M. T., and Gozani, O. ( 2007) Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 282, 2450–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lottersberger, F., Panza, A., Lucchini, G., and Longhese, M. P. ( 2007) Functional and physical interactions between yeast 14-3-3 proteins, acetyltransferases, and deacetylases in response to DNA replication perturbations. Mol. Cell. Biol. 27, 3266–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Han, J. D., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., Zhang, L. V., Dupuy, D., Walhout, A. J., Cusick, M. E., Roth, F. P., and Vidal, M. ( 2004) Evidence for dynamically organized modularity in the yeast protein-protein interaction network. Nature 430, 88–93 [DOI] [PubMed] [Google Scholar]
- 85.He, X., and Zhang, J. ( 2006) Why do hubs tend to be essential in protein networks? PLoS Genet. 2, e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zotenko, E., Mestre, J., O'Leary, D. P., and Przytycka, T. M. ( 2008) Why do hubs in the yeast protein interaction network tend to be essential: reexamining the connection between the network topology and essentiality. PLoS Comput. Biol. 4, e1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brohee, S., and van Helden, J. ( 2006) Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Palla, G., Derenyi, I., Farkas, I., and Vicsek, T. ( 2005) Uncovering the overlapping community structure of complex networks in nature and society. Nature 435, 814–818 [DOI] [PubMed] [Google Scholar]
- 89.Yu, H., Paccanaro, A., Trifonov, V., and Gerstein, M. ( 2006) Predicting interactions in protein networks by completing defective cliques. Bioinformatics 22, 823–829 [DOI] [PubMed] [Google Scholar]
- 90.Ewing, R. M., Chu, P., Elisma, F., Li, H., Taylor, P., Climie, S., McBroom-Cerajewski, L., Robinson, M. D., O'Connor, L., Li, M., Taylor, R., Dharsee, M., Ho, Y., Heilbut, A., Moore, L., Zhang, S., Ornatsky, O., Bukhman, Y. V., Ethier, M., Sheng, Y., Vasilescu, J., Abu-Farha, M., Lambert, J. P., Duewel, H. S., Stewart, I. I., Kuehl, B., Hogue, K., Colwill, K., Gladwish, K., Muskat, B., Kinach, R., Adams, S. L., Moran, M. F., Morin, G. B., Topaloglou, T., and Figeys, D. ( 2007) Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]