

Abstract
Despite many efforts in the last decade, the progress in de novo peptide sequencing has been slow with only 30–45% of all peptides correctly reconstructed. We argue that accurate full-length peptide sequencing may be an unattainable goal for some spectra and demonstrate how to accurately sequence gapped peptides instead. We further argue that gapped peptides are nearly as useful as full-length peptides for error-tolerant database searches. Gapped peptides occupy a niche between long but inaccurate full-length reconstructions and short but accurate peptide sequence tags. Our MS-Profile tool uses spectral profiles, a new representation of tandem mass spectra, to generate gapped peptides that are longer and more accurate than peptide sequence tags of length 3 traditionally used to speed up database searches in proteomics. In addition, spectral profiles also enable intuitive visualization of all high scoring de novo reconstructions of tandem mass spectra.
Recent advances in de novo peptide sequencing have enabled tag-based peptide identification tools (e.g. Inspect (1) and Paragon (2)) that are orders of magnitude faster than traditional MS/MS database search approaches (e.g. Sequest (3) and Mascot (4)). However, reliable full-length de novo peptide sequencing remains an elusive goal, and even the most accurate de novo tools correctly reconstruct only 30–45% of peptides (5). We argue that accurate full-length de novo peptide sequencing may be an unattainable goal for many spectra because they do not provide enough information to disambiguate between correct and incorrect reconstructions. Spectra often have variable local quality (along the peptide length) making some regions not amenable to de novo sequencing. For example, spectra of peptides DGEAAENTDAQK and DSVAAENTDAQK are very similar (supplemental Fig. S1) making it nearly impossible to reliably reconstruct these peptides de novo (the combined mass of Gly and Glu is close to the combined mass of Ser and Val). In such cases, it makes more sense to reconstruct a gapped peptide D[186]AAENTDAQK rather than a contiguous peptide (186 Da refers to the rounded combined mass of Gly and Glu or Ser and Val). Although gapped peptides are less informative than full-length peptides, we argue that there is little difference between these two representations. Indeed in most applications, de novo peptide sequencing is not the final goal in analyzing a spectrum but rather a prelude to error-tolerant database searches and other applications like metaproteomics (6–9). We argue that long gapped peptides are nearly as good for such applications as full-length de novo reconstructions. For example, the gapped peptide D[186]AAENTDAQK has 9 continuous amino acids and thus, for all practical applications, is at least as useful as any peptide of length 9 (or length 11 if one counts Asp and [186] as separate “letters”). Because most mass spectrometrists view peptides of length 9 as useful as peptides of length 12, generating sufficiently long gapped peptides is nearly as useful as generating full-length reconstructions (the full length of D[186]AAENTDAQK is 12).
In this study we introduce the notion of a spectral profile (Fig. 1) that enables accurate de novo sequencing of gapped peptides and reveals the variable spectral quality along the peptide length. For example, for peptides of length 11–12, our MS-Profile tool correctly reconstructs 65% of gapped peptides as compared with 46, 28, and 26% correct reconstructions of full or truncated full-length peptides by PepNovo+ (5, 10), MS-Dictionary (11), and PEAKS (12). Gapped peptides occupy a niche between peptide sequence tags (that in most applications are limited to tags of length 3) and full-length reconstructions: they are nearly as accurate as short tags and, at the same time, typically have a unique match in the protein database. For example, for peptides of length 12, the average length of gapped reconstructions is 8.9, typically resulting in a single hit even when searching against the largest databases used in proteomics today.1
Fig. 1.

An example of the spectral profile. Top, an MS/MS spectrum of the peptide STVAGESGSADTVR with b- and y-peaks painted green and blue, respectively. Middle, the spectral profile of the spectrum above. The overall height of each peak represents the probability of the peak being a correct prefix mass. Each peak is represented as a multicolored bar where various colors (subpeaks stacked on top of each other) correspond to various amino acids (amino acids are color-coded). Similarly to the motif profile in supplemental Fig. S2, the height of each colored subpeak (corresponding to an amino acid X) represents the probability of a prefix with terminal amino acid X ending at the given mass position. Bottom, the database match (DBMatch), full-length de novo reconstruction (DeNovo), and gapped peptide (Gapped) of the spectrum in the top panel. The painted rectangles represent the tags of length 1 ending at each position of the de novo reconstruction: the width of each rectangle corresponds to the mass of the amino acid, and the height corresponds to the probability of the length 1 tag being correct. Although the full-length de novo reconstruction is incorrect, the gapped peptide reconstruction (generated using the spectral profile) is correct. The consecutive amino acids Ser and Leu are represented as a 200-Da gap because the value of the spectral profile at the position separating Ser and Leu is low.
A spectral profile is a novel representation of tandem mass spectra with “intensities” of all masses varying from 0 to 1. Every peptide of length n defines n prefix masses representing masses of the first i amino acids (for 1 ≤ i ≤ n). The spectral profile at mass x is the proportion of peptides with prefix mass x among all high scoring interpretations of the spectrum. Thus, the spectral profile compactly represents information about all high scoring de novo reconstructions (spectral dictionary) even if there are billions of such reconstructions (see Ref. 11). Spectral profiles are conceptually similar to the motif profiles (13) (supplemental Fig. S2) that are used in various areas of bioinformatics (e.g. in regulatory genomics). Whereas motif profiles in regulatory genomics compactly represent all known binding sites of a transcription factor, a spectral profile compactly represents all high scoring de novo reconstructions of an MS/MS spectrum. However, whereas motif profiles represent the center of gravity of known motifs, spectral profiles represent the center of gravity of unknown high scoring de novo reconstructions (spectral dictionaries). This makes computing spectral profiles challenging because in many cases spectral dictionaries cannot be explicitly generated (11). This study extends Kim et al. (11) by showing how to compute the spectral profile of any spectrum without explicitly generating its spectral dictionary. We further show how to use spectral profiles for generating reliable gapped peptides.
The difficult challenge in de novo spectral interpretations is how to figure out which ion type every peak represents (e.g. how to distinguish b-series peaks from y-series peaks) and how to analyze the widely varying intensities in a single probabilistic framework. The spectral profile collapses all possible ion type interpretations and varying intensities into a single ion type (b-ion) with rigorously defined probability. In contrast to real MS/MS spectra (that contain peaks corresponding to b- and y-ions, various neutral losses, etc.), spectral profiles only represent (putative) b-ions. In some sense, spectral profiles represent a trade-off between (hard-to-interpret but compact) real spectra and (easy-to-interpret but huge) spectral dictionaries. We emphasize that spectral profiles are different from “scored spectra” (e.g. sequence spectra (14, 15) or prefix residue mass spectra (1)) that are commonly used for de novo sequencing and MS/MS database searches. Although profile probabilities are global (i.e. they take into account complex dependences between all peaks in the spectrum), scored spectra take into account only a few local satellite peaks explaining a given mass.
Similar to the diverse applications of motif profiles, spectral profiles have a multitude of applications that we describe below. Fig. 2 illustrates recently implemented alternative approaches to peptide identification: peptide sequence tag approaches (1, 2, 16, 17) and full-length de novo reconstruction approaches (7, 8, 11, 18) (see also lookup peak approach (19)). Although these approaches significantly speed up conventional peptide identification tools, each of them presents certain challenges, leading to deteriorating performance on long (15+-aa2) peptides. Most of these approaches do not automatically adjust to varying spectral qualities or different peptide lengths. For example, InsPecT generates the same number of tags for every spectrum, although a more sensible approach would be to generate a larger number of tags for long peptides (tag generation deteriorates for longer peptides) or for low quality spectra. Although MS-Dictionary (11) generates an adaptive but large number of full-length reconstructions (for both high and low quality spectra), dictionaries of spectra of long peptides may become so large that their generation becomes impractical. To overcome this problem, we show how to quickly construct spectral profiles of even huge dictionaries without explicitly generating them.
Fig. 2.

Various filtering approaches to peptide identifications. The tag-based approach (e.g. InsPecT (1) and Paragon (2)) extracts short (usually length 3) peptide sequence tags and filters databases by considering only peptides that match tags. Full-length de novo approaches either reconstruct a single full-length peptide and find sequence matches (e.g. MS-BLAST (6), OpenSea (7), and SPIDER (8)) or generate multiple full-length reconstructions and find sequence matches to the protein database ((RAId) (18) and MS-Dictionary (11)). Spectral profiles represent an alternative approach to peptide identification generating gapped peptides and matching them to the database.
MS-Profile currently works in two modes (Fig. 3). In the first mode, the input is an MS/MS spectrum and a spectral probability threshold (described below), and the output is a spectral profile. In the second mode, the constructed spectral profile, in addition to a de novo reconstruction, and a MinProbability threshold (described below) serve as an input, and the output is a gapped peptide. MS-Profile in the second mode represents a new de novo peptide sequencing tool that improves accuracy of de novo reconstructions produced by other tools (e.g. PepNovo+, PEAKS, or MS-Dictionary). In particular, it generates gapped peptides that can be used for mutation-tolerant database searches and speed up existing database search tools. MS-Profile is available both as open source software and as a Web server.
Fig. 3.

Overview of the MS-Profile tool. MS-Profile works in two modes: mode 1 is for the spectral profile generation and mode 2 is for the gapped peptide generation.
EXPERIMENTAL PROCEDURES
What Is the Spectral Profile?—
For the sake of simplicity, we will first introduce the notion of a spectral profile under the assumption that amino acid masses are integers.3 Given a peptide P1 … Pn, we define its prefix masses as a series mass(P1), mass(P1) + mass(P2), …, Σj = 1i mass(Pj), …, Σj = 1n mass(Pj) where Σj = 1n mass(Pj) = k is defined as the parent mass. We further represent the peptide P1, … Pn as a k-mer boolean vector P = x1 … xk where xt = 1 if t represents a prefix mass and xt = 0 otherwise (see Refs. 11, 20, and 21) for applications of boolean spectra and peptides). Given a set of boolean peptides Dictionary = {P1, …, Pm}, we define the spectral profile as simply the center of gravity of all peptides (boolean vectors) in the set, i.e. Profile(Dictionary) = 1/m Σj = 1m Pj. This definition assumes that all peptides in the Dictionary are equally likely.
Kim et al. (11) introduced the notion of spectral dictionary and described an MS-Dictionary approach to peptide identification. Given a spectrum Spectrum and a score threshold Threshold, Dictionary(Spectrum, Threshold) is defined as the set of all peptides with scores above the Threshold. We define the spectral profile Profile(Spectrum, Threshold) as Profile(Dictionary(Spectrum, Threshold)).
When the Dictionary is explicitly given, computing Profile(Dictionary) amounts to computing the center of gravity of k-dimensional boolean vectors from Dictionary. Although MS-Dictionary (11) is capable of quickly generating spectral dictionaries for short peptides (less than 15 aa), the spectral dictionaries of spectra of long peptides are so large (even for sensible choices of Threshold) that MS-Dictionary becomes impractical. For example, for a typical spectrum of a 15-aa long peptide, the spectral dictionary consists of ≈4·109 high scoring peptides that would typically result in statistically significant database hits (11). Below we show how to quickly generate spectral profiles of such huge dictionaries without explicitly generating the dictionary. MS-Profile takes only ≈0.2 s to generate the spectral profiles even for spectra of long peptides. Thus MS-Profile bypasses the need to explicitly generate large spectral dictionaries that limited applications of MS-Dictionary in the case of long peptides.
Computing Spectral Profiles—
The transformation of spectra into spectral profiles can be done efficiently by the forward-backward dynamic programming algorithm (22). For the sake of simplicity, we first represent a spectrum with parent mass k as a boolean spectrum S = s1 … sk where si = 1 if there is a peak at mass i in the spectrum and si = 0 otherwise. This representation assumes that spectra are discretized and that all masses are integers. Below we use the term mass of peptide/spectra to refer to the dimension of the corresponding vectors (parent mass k). The score (denoted as Score(P, S)) between a boolean peptide P = p1 … pk and a boolean spectrum S = s1 … sk (of the same mass) is defined as Σj = 1k pj·sj. When peptide P and spectrum S differ in mass, we define Score(P, S) as −∞.
We define Siprefix as s1 … si and Sisuffix as sk-i + 1 … sk. Given a spectrum S = s1 … sk, we define ρprefix(i, t) as the set of all boolean peptides P ε ρprefix(i, t) with length i and Score(P, Siprefix) = t. Let xfwd(i, t) be the size of ρprefix(i, t). As shown in Ref. 23, xfwd(i, t) can be computed using the forward dynamic programming as follows.
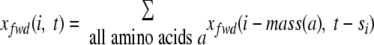 |
(1) |
We initialize xfwd(0,0) = 1, xfwd(0, t) = 0 for t > 0, and xfwd(i, t) = 0 for negative i.
Given a spectrum S = s1 … sk, we define ρsuffix(i, t) as the set of all boolean peptides P ε ρsuffix(i, t) with length k − i and Score(P, Sk − isuffix) = t. Let xbwd(i, t) be the size of ρsuffix(i, t). The variable xbwd(i, t) can be computed using the reverse dynamic programming as follows.
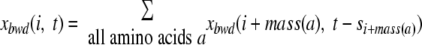 |
(2) |
We initialize xbwd(k, 0) = 1, xbwd(k, t) = 0 for t > 0, and xbwd(i, t) = 0 for i > k.
Given a score threshold Threshold to generate a Dictionary, it is easy to see that the size of the Dictionary can be computed as follows.
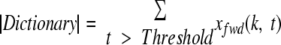 |
(3) |
Below we demonstrate that Profile(Spectrum, Threshold) = f1 … fk can be computed using the forward-backward algorithm.
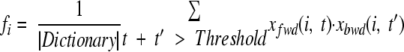 |
(4) |
Indeed Equation 5 follows.
 |
(5) |
Every peptide in Dictionary with xi = 1 can be decomposed into Pref = x1 … xi and Suff = xi + 1 … xk peptides. Because all peptides in the Dictionary score above Threshold, Score(Pref, s1 … si) + Score(Suff, si + 1 … sk) > Threshold. Thus, the number of such peptides is given by Σt + t′ > Threshold xfwd(i, t)·xbwd(i, t′). Conversely concatenation of two arbitrary peptides Pref = x1 … xi and Suff = xi + 1 … xk contributes to the Dictionary as long as Score(Pref, s1 … si) + Score(Suff, si + 1 … sk) > Threshold. Because the number of such concatenations with xi = 1 is given by Σt + t′ > Threshold xfwd(i, t)·xbwd(i, t′), fi can be computed by the forward-backward algorithm as described above.
Fig. 4 illustrates computation of a spectral profile. In practice, we compute spectral profiles for a fixed spectral probability (23) (rather than for a fixed score threshold). The spectral probability of a Peptide-Spectrum Match (PSM) is defined as the total probability of all peptides with scores exceeding the score of the PSM.4 One can also define a spectral probability depending on a score Threshold as the total probability of all peptides with scores above Threshold (the total probability of all peptides in the corresponding spectral dictionary). Given a spectral probability p, one can approximate the expectation value as p·DatabaseSize. See Refs. 11 and 23 for the background on spectral probabilities and spectral dictionaries. For each spectrum, MS-Profile dynamically sets Threshold as the minimum score s such that the spectral probability of the reconstructions with scores above s does not exceed a predefined spectral probability (e.g. 10−8) and computes the spectral profile. For example, the spectral profile in Fig. 1 was computed for spectral probability 10−8. Supplemental Fig. S3 illustrates that the spectral profile remains rather stable for a range of spectral probabilities.
Fig. 4.

An example of the dynamic programming algorithm for computing the spectral profile of a “toy” boolean spectrum 011010100 with four peaks at masses 2, 3, 5, and 7 (parent mass, 9). The MS-Profile algorithm is illustrated with the help of a toy amino acid model (only two amino acids with masses 2 and 3 Da) and a simplified discretized spectrum. The scoring function Score(Peptide, Spectrum) used for this illustration is the number of matching peaks between boolean peptide and boolean spectrum. There are only five peptides with parent mass 9 Da: 3222 (score 3), 2322 (score 3), 2232 (score 2), 2223, and 333 (both score 1). These five peptides correspond to nine-dimensional boolean vectors: 001010101, 010010101, 010100101, 010101001, and 001001001. If one considers all peptides with scores 1 and above, the spectral profile is a nine-dimensional vector (0, ⅗, ⅖, ⅖, ⅖, ⅖, ⅗, 0, 1) representing the center of gravity of these five vectors. However, if one considers the dictionary of all peptides with scores 2 and above then the spectral profile (0, ⅔, ⅓, ⅓, ⅔, 0, 1, 0, 1) is the center of gravity of three peptides 001010101, 010010101, and 010100101. The forward-backward dynamic programming generates the spectral profile without explicitly generating any of the peptides in the dictionary. For the threshold 1 (peptides of scores 2 and above are considered), the size of the spectral dictionary is 3, and the spectral profile of the dictionary is (0, ⅔, ⅓, ⅓, ⅔, 0, 1, 0, 1). Numbers in bold and dashed (mass 2) and solid (mass 3) arrows represent paths to reach the dictionary with the threshold 1.
Note that the simple boolean model for scoring peptide-spectrum matches can easily be extended to more complicated models without any algorithmic changes. Indeed MS-Profile uses the scoring model of MS-Dictionary (23) that considers various features such as ion types, peak intensities, and mass errors.
RESULTS
Data Set—
We used the Standard Protein Mix database consisting of 1.1 million spectra generated from 18 proteins using eight different mass spectrometers (24). For this study, we considered only the charge 2 spectra generated by Thermo Electron LTQ where 1388 peptides of length between 7 and 20 are reliably identified with false discovery rate 2.5% using Sequest (3) and PeptideProphet (25) in the search against the Haemophilus influenzae database appended with sequences of the 18 proteins (567,460 residues). Although this study focuses on doubly charged spectra, MS-Profile can also be applied to MS/MS spectra of higher charges as long as an additive scoring model for highly charged MS/MS spectra is available.
For each peptide, we randomly selected one representative spectrum and formed a data set of 1388 PSMs grouped by the length of their peptide identifications. To avoid computational artifacts introduced by errors in the parent mass, the parent masses of the spectra are corrected according to the Sequest identifications. Below we refer to this data set as the Standard data set. Throughout the paper we measure accuracy of a de novo sequencing tool as the percentage of spectra with error-free reconstructions among all spectra in the Standard data set.
Fig. 5 shows the distribution of spectral probabilities (false positive rates) of the Standard data set. Most PSMs (91%) have spectral probabilities lower than 10−8. We used 10−8 as the spectral probability threshold to generate spectral profiles.
Fig. 5.

Distribution of spectral probabilities for PSM in the standard data set. A bar at position i represents the portion of the spectra with spectral probabilities varying from 10−i − 0.5 to 10−i + 0.5.
Table I shows the results of de novo peptide sequencing of the Standard data set with PEAKS, PepNovo+, and MS-Dictionary. PEAKS and MS-Dictionary correctly reconstructed peptides for less than 30% of the spectra, and the accuracy of both tools greatly deteriorates as the peptide length increases. PepNovo+ reported shorter de novo reconstructions (especially for spectra of long peptides) by allowing gaps in the start and the end of the peptides, resulting in accuracy better than that of the other tools. Below we show that MS-Profile improves the accuracy of these tools at the cost of a small reduction in the length of reconstructed peptides.
Table I.
Accuracy and average length of PEAKS, PepNovo+, and MS-Dictionary for the Standard data set
PEAKS (online 2.0), PepNovo+ (released July 24, 2008), and MS-Dictionary (released November 7, 2007) were run with parent mass and fragment mass tolerances of 0.5 Da, fixed modification of Cys + 57, no optional modifications, and without any enzyme preference. In contrast to PEAKS and MS-Dictionary, PepNovo+ allows gaps at the start/end of peptides thus giving PepNovo+ significant leverage when it comes to the reported accuracy of reconstruction. Although MS-Dictionary is designed for generating spectral dictionaries (rather than ensuring that the correct reconstruction has the top score), it can be used in de novo mode as well (it has slightly higher accuracy than PEAKS while generating slightly longer peptides). PEAKS and MS-Dictionary have a tendency to output de novo reconstructions that are longer than the correct peptides (e.g. for peptides of length 11–12, the average length of PEAKS and MS-Dictionary reconstructions is 12.1 and 12.2, respectively). Accuracy of each tool is defined as the percentage of the error-free reconstructions among all reconstructions for the Standard data set.
| Peptide length | PEAKS
|
PepNovo+
|
MS-Dictionary
|
|||
|---|---|---|---|---|---|---|
| Accuracy | Length | Accuracy | Length | Accuracy | Length | |
| 7–8 | 0.59 | 8.1 | 0.65 | 7.8 | 0.54 | 7.8 |
| 9–10 | 0.33 | 10.0 | 0.54 | 9.3 | 0.40 | 9.9 |
| 11–12 | 0.27 | 12.1 | 0.47 | 10.4 | 0.31 | 12.2 |
| 13–14 | 0.12 | 14.1 | 0.36 | 12.0 | 0.14 | 14.2 |
| 15–16 | 0.10 | 16.0 | 0.34 | 13.2 | 0.12 | 16.1 |
| 17–18 | 0.07 | 16.9 | 0.29 | 14.1 | 0.02 | 18.3 |
| 19–20 | 0.05 | 19.0 | 0.38 | 14.0 | 0.04 | 20.8 |
| Total | 0.26 | 12.6 | 0.46 | 11.0 | 0.28 | 12.8 |
De Novo Sequencing of Gapped Peptides—
De novo peptide sequencing algorithms usually correctly recover some amino acids within a peptide and misinterpret others. The key challenge is to figure out which portions of the peptide are reconstructed incorrectly and to limit reconstructions to highly accurate portions. Gapped peptide reconstruction addresses this challenge by reporting only reliably reconstructed regions of the peptide.
Given a Peptide = x1 … xk, a Profile = f1 …, fk, and a parameter MinProbability, we define GappedPeptide(Peptide, Profile, MinProbability) = g1 … gk as gi = xi if fi ≥ MinProbability and gi = 0 otherwise. Fig. 1 shows a spectral profile for the spectrum of peptide STVAGESGSADTVR and (incorrect) de novo reconstruction SSLAGESGSADTVR. One can notice that although profile values for most prefix masses in STVAGESGSADTVR are relatively high (0.207, 0.084, 0.475, 0.518, 0.310, 0.522, 0.791, 0.718, 0.730, 0.709, 0.323, 0.149, and 0.353) the profile value for one prefix mass falls below 0.1. This low profile value points to an unreliable portion of the reconstruction. Converting peptide STVAGESGSADTVR into a gapped peptide (with MinProbability = 0.1) results in a (correct) gapped peptide S[200]AGESGSADTVR. Increasing MinProbability to 0.2 results in a shorter gapped peptide S[200]AGESGSAD[200]R.
MS-Profile generates gapped peptides as follows. For each spectrum, it first constructs the spectral profile and generates optimal de novo reconstructions by backtracking its forward matrix. Indeed because MS-Profile uses the MS-Dictionary scoring (11), the reconstructions are the same as reconstructions generated by MS-Dictionary. Both PEAKS and MS-Dictionary may generate (a small number of) multiple optimal de novo reconstructions, and we first convert them into a single consensus reconstruction. For example, the set of reconstructions YWAGELTR, YWASVLTR, YWAVSLTR, and YWAEGLTR will be converted into a single consensus reconstruction YWA[186]LTR by retaining only the prefix masses present in all reconstructions. Next MS-Profile discards all prefix masses in the consensus reconstruction whose corresponding profile values are below MinProbability as described above. The remaining prefix masses represent the gapped peptide generated by applying MS-Profile to MS-Dictionary (referred to as MS-Profile(MS-Dictionary)). Fig. 6 compares the accuracy of de novo reconstructions generated by MS-Dictionary and the gapped peptide generated by MS-Profile(MS-Dictionary). Applying MS-Profile increases the percentage of correct reconstructions from 28 to 42% while decreasing the average length of reconstructions from 12.8 to 9.1 amino acids when MinProbability = 0.1. We remark that the Standard data set contains some low quality spectra that are nearly impossible to reconstruct in de novo fashion. Supplemental Fig. S4 illustrates the performance of MS-Profile when the Standard data set is restricted to high quality spectra. Supplemental Fig. S5 illustrates similar comparisons for the different MinProbability thresholds. One can increase the accuracy by increasing the MinProbability threshold. For example, when MinProbability = 0.2, the accuracy increases to 50% while the average length of gapped peptide decreases to 7.9. When MinProbability = 0.3, the accuracy increases to 54% while the average length of gapped peptide becomes 7.2.
Fig. 6.

a, accuracy of best scoring reconstructions generated by MS-Dictionary and the gapped peptide generated by MS-Profile (MS-Dictionary) for different peptide lengths (MinProbability = 0.1). If the length of a gapped peptide is less than 5 (5.6% of the spectra in the Standard data set), we counted it as incorrect (even if the gapped peptide is correct) to penalize very short gapped reconstructions. b, average length of reconstructions generated by MS-Dictionary and the gapped peptides generated by MS-Profile (MS-Dictionary) for various peptide lengths.
PepNovo+ and PEAKS represent some of the most accurate de novo peptide sequencing tools. MS-Profile can be used to convert PepNovo+ and PEAKS reconstructions into gapped peptides resulting in MS-Profile(PepNovo+) and MS-Profile(PEAKS) tools. Applying MS-Profile to PepNovo+ increases the percentage of correct reconstructions from 46 to 65% while decreasing the average length of reconstructions from 11.0 to 8.9 amino acids (MinProbability = 0.1). Applying MS-Profile to PEAKS increases the percentage of correct reconstructions from 26 to 48% while decreasing the average length of reconstructions from 12.6 to 9.2 amino acids (MinProbability = 0.1). Although gapped peptides generated by MS-Profile(PepNovo+) and MS-Profile(PEAKS) are shorter than PepNovo+ and PEAKS reconstructions, they are still long enough to uniquely identify most peptides even in large protein databases. Fig. 7 compares the accuracy and lengths of PepNovo+, PEAKS, MS-Profile(PepNovo+), and MS-Profile(PEAKS) reconstructions.
Fig. 7.

a, accuracy of best scoring reconstructions generated by PEAKS, PepNovo+, MS-Profile(PEAKS), and MS-Profile(PepNovo+) for different peptide lengths. The reconstructions are converted into gapped peptides using MS-Profile with MinProbability = 0.1. If the length of a gapped peptide is less than 5, we consider it incorrect. b, average length of best scoring reconstructions and gapped peptides for different peptide lengths. The length of PepNovo+ reconstructions is not proportional to the peptide length because PepNovo+ allows variable length gaps at the start and end of the peptide.
PepNovo+ allows users to generate up to 2000 reconstructions per spectrum. When multiple reconstructions are generated, the probability of at least one of them being correct increases. For each reconstruction, we generate a gapped peptide using MS-Profile(PepNovo+). Because different PepNovo+ reconstructions may correspond to the same gapped peptide, the number of gapped peptides generated by MS-Profile(PepNovo+) is typically smaller than the original number of PepNovo+ reconstructions. Supplemental Table S1 illustrates that although the number of gapped peptides generated by MS-Profile(PepNovo+) is 3–15 times smaller than the number of PepNovo+ reconstructions the length of the reconstructed gapped peptides is typically sufficient to ensure a unique database hit. Fig. 8 compares accuracy and length of peptides and gapped peptides generated by PepNovo+ and MS-Profile(PepNovo+) for the top 100 and the top 1000 reconstructions. Again MS-Profile(PepNovo+) outperforms PepNovo+ while generating much smaller numbers of gapped peptides.
Fig. 8.

Accuracy (a) and length (b) of top 100 PepNovo+ reconstructions (PepNovo+(#Recs = 100)), top 1000 PepNovo+ reconstructions (PepNovo+(#Recs = 1000)), MS-Profile gapped peptides converted from top 100 PepNovo+ reconstructions (MS-Profile(PepNovo+ #Recs = 100)), and MS-Profile gapped peptides converted from top 1000 PepNovo+ reconstructions (MS-Profile(PepNovo+ #Recs = 1000)). #Recs, number of reconstructions.
The improved performance of MS-Profile(PepNovo+) in generating gapped peptides suggests that it can be used for database filtration in the same way as peptide sequence tags are used in InsPecT (1). For the Standard data set, we ran InsPecT to generate 1, 10, and 25 tags of lengths 3 and 4 and measured for how many spectra InsPecT generates at least one correct tag (Fig. 9). The same number of gapped peptides are also generated by MS-Profile(PepNovo+). It turned out that the best gapped peptide is longer and more accurate than the best tag of length 3 (the gapped peptide is correct for 65% of spectra, whereas the best 3-aa long tag is correct for 44% of spectra). Also top 10 and 25 gapped peptides are roughly as accurate as the same number of tags of length 3. For 83% of spectra, at least one of the top 10 gapped peptides is correct, whereas for 80% of spectra, at least one of the top 10 tags of 3 aa is correct. For 86% of spectra, at least one of the top 25 gapped peptides is correct, whereas for 88% of spectra, at least one of the top 25 tags of 3 aa is correct. This is surprising because gapped peptides generated by MS-Profile(PepNovo+) represent a much better filter for database search than InsPecT tags. To test the filtering efficiency, we matched the top gapped peptide of each spectrum and its top 3-aa tag against the Swiss-Prot database (release 56.4, 145 million residues) counting the number of false matches to the database. Although 90% of gapped peptides have no false matches, only 29% of tags have no false match. The average number of false matches is 1.6 for gapped peptides, 50 times smaller than 80.3 false tag matches on average. The average number of false matches is a key parameter in filtration-based MS/MS searches because it is roughly proportional to the time required for peptide identification (1). Therefore, a 50-fold reduction in the number of false matches can potentially translate into a 50-fold speedup as compared with (already fast) InsPecT. The contrast between gapped peptides and tags is particularly pronounced in searches against very large databases like proteogenomic six-frame translation searches of the repeat-masked human genome of size 2.7 billion residues (11). Gapped peptides longer than 8 aa (63% of spectra in the Standard data set) are expected to have only 0.24 false matches in this database, whereas 3-aa tags are expected to have 1400 false matches on average.
Fig. 9.

Comparison of accuracy of InsPecT tags and gapped peptides generated by MS-Profile. InsPecT (released April 4, 2008) was run with parent mass and fragment mass tolerances of 0.5 Da, fixed modification of Cys + 57, no optional modifications, and without any enzyme preference. The same number of InsPecT tags of length 3, InsPecT tags of length 4, and MS-Profile(PepNovo+) gapped peptides are generated, and their accuracies are shown. a, accuracy of one tag or gapped peptide. b, accuracy of 10 tags or gapped peptides. c, accuracy of 25 tags or gapped peptides.
This comparison suggests that MS-Profile can significantly improve on previous filtration approaches to MS/MS database searches. In contrast to peptide sequence tags (that typically have many false hits in a database), gapped peptides typically have few false hits (if any) thus speeding up the database searches. We comment that use of gapped seeds in traditional BLAST-like genomics searches is well studied (26).
Evaluating Spectral Profile Probabilities—
Some de novo sequencing programs output the reliability of predicted amino acids. For example, PepNovo+ defines features that reflect the reliability of each predicted amino acid and converts the feature vectors into probabilities (27). PEAKS recently added a similar function that computes the reliability of an amino acid a by locally permuting the reconstruction around a, computing the score difference between the original and permuted reconstructions and using the prelearned distribution of the difference to assign the reliability of a (28). MS-Profile differs from these tools because instead of learning it rigorously computes a probability that a prefix mass is present in a high scoring de novo peptide reconstruction.
We show that the spectral profile probabilities approximate the empirical accuracy of the prefix mass (represented by the profile peak) being correct. To compute the accuracy of the profile value p (for p = 0.1, 0.2, … 0.9, and 1.0), we bundled all the profile peaks with values between p − 0.05 and p + 0.05 and measured the fraction of correct peaks among them. If the empirically computed fraction of correct peaks of the profile value p is close to p then our estimate of profile probabilities is unbiased. Fig. 10 shows that it is indeed the case: the empirical accuracy of the profile peaks with probability p is slightly above p. The slightly higher empirical accuracy (as compared with profile values) is likely a consequence of using the same spectral probability threshold of 10−8 for all spectra, whereas in reality most PSMs have much lower spectral probabilities (Fig. 5).
Fig. 10.

a, the distribution of the average number of profile peaks per spectrum for different profile values generated by MS-Profile. The number of correct peaks is represented by black bars; the number of incorrect peaks is represented by gray bars stacked on the black bars. A peak at position p corresponds to the profile values between p − 0.05 and p + 0.05. b, the empirical accuracy (the number of correct profile peaks divided by the number of total profile peaks) of profile peaks for different profile values. The diagonal line is shown for reference.
DISCUSSION
Although peptide sequence tags were first proposed in 1994 (29), it took 10 years for this idea to become an integral part of the new generation of fast MS/MS database search tools (1, 2). It took such a long time because a seemingly simple problem of generating accurate sequence tags turned out to be more difficult than originally thought. We demonstrated that gapped peptides occupy an important niche between long but inaccurate full-length peptide reconstructions and short but more accurate peptide sequence tags. This niche provides certain advantages because gapped peptides represent a more stringent filter that may enable very fast MS/MS database searches that in many cases will amount to a simple lookup in a database. Spectral profiles reveal poor quality spectra (or poor quality regions within long peptides) that other methods have difficulties analyzing. MS-Profile follows a different route to error-tolerant peptide identifications than OpenSea (7) and SPIDER (8). Instead of trying to generate (unreliable) full-length reconstructions and approximately matching them against the database, MS-Profile generates reliable gapped peptides and matches them against the database exactly.
Some de novo sequencing tools such as Lutefisk (30), PEAKS (12), and PepNovo+ (10) can generate gapped peptides typically trimming the full-length peptides in the beginning/end. Even when internal gaps are allowed (Lutefisk and PEAKS), they are limited to gaps of 2 aa or shorter. For long peptides where multiple consecutive peaks are missing, it is hard to generate correct gapped peptides when only short gaps are allowed. On the other hand, PepNovo+ improves on these tools by allowing long gaps in the start/end of the peptides. As a result, PepNovo+ has a tendency to generate incorrect solutions when it tries to reconstruct all amino acids in the middle. To the best of our knowledge, MS-Profile is the only program that allows both short and long gaps regardless of the position. Also MS-Profile can convert any de novo reconstructions into gapped peptides thus making it a useful addition to various de novo peptide sequencing tools.
Acknowledgments
We thank Vineet Bafna, Ari Frank, and Eunok Paek for useful discussions.
Footnotes
Published, MCP Papers in Press, March 2, 2009, DOI 10.1074/mcp.M800535-MCP200
We define the length of the gapped peptide as the number of masses and amino acids describing the peptide. For example, the length of the gapped peptide [186]DK[246]FK is 6, whereas the length of a 3-aa long peptide sequence tag [307]GTP[421] is 5.
The abbreviations used are: aa, amino acid(s); PSM, peptide-spectrum match; BLAST, basic local alignment search tool.
One can always adjust the “granularity” of mass measurements (e.g. by multiplying all masses by 1000 in the case of accurate mass measurements) and safely assume that the masses of amino acids become integers after this transformation.
The probability of a peptide is defined as the product of probabilities of its amino acids. Amino acid probabilities are predefined depending on the frequencies of amino acids in a protein database (23).
This work was supported, in whole or in part, by National Institutes of Health Grant 1-P41-RR024851 from the National Center for Research Resources.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Tanner, S., Shu, H., Frank, A., Wang, L., Zandi, E., Mumby, M., Pevzner, P. A., and Bafna, V. ( 2005) InsPecT: identification of posttranslationally modified peptides from tandem mass spectra. Anal. Chem. 77, 4626–4639 [DOI] [PubMed] [Google Scholar]
- 2.Shilov, I. V., Seymour, S. L., Patel, A. A., Loboda, A., Tang, W. H., Keating, S. P., Hunter, C. L., Nuwaysir, L. M., and Schaeffer, D. A. ( 2007) The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 3.Eng, J., McCormack, A., and Yates, J. ( 1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 4.Perkins, D., Pappin, D., Creasy, D., and Cottrell, J. ( 1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 5.Frank, A., and Pevzner, P. A. ( 2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 6.Shevchenko, A., Sunyaev, S., Loboda, A., Shevchenko, A., Bork, P., Ens, W., and Standing, K. G. ( 2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and blast homology searching. Anal. Chem. 73, 1917–1926 [DOI] [PubMed] [Google Scholar]
- 7.Searle, B. C., Dasari, S., Turner, M., Reddy, A. P., Choi, D., Wilmarth, P. A., McCormack, A. L., David, L. L., and Nagalla, S. R. ( 2004) High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for ms/ms de novo sequencing results. Anal. Chem. 76, 2220–2230 [DOI] [PubMed] [Google Scholar]
- 8.Han, Y., Ma, B., and Zhang, K. ( 2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 9.Denef, V. J., Shah, M. B., Verberkmoes, N. C., Hettich, R. L., and Banfield, J. F. ( 2007) Implications of strain- and species-level sequence divergence for community and isolate shotgun proteomic analysis. J. Proteome Res. 6, 3152–3161 [DOI] [PubMed] [Google Scholar]
- 10.Frank, A. ( 2009) A ranking-based scoring function for peptide-spectrum matches. J. Proteome Res., in press [DOI] [PMC free article] [PubMed]
- 11.Kim, S., Gupta, N., Bandeira, N., and Pevzner, P. ( 2009) Spectral dictionaries: integrating de novo peptide sequencing with database search of tandem mass spectra. Mol. Cell. Proteomics 8, 53–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma, B., Zhang, K., Hendrie, C., Liang, C., Li, M., Doherty-Kirby, A., and Lajoie, G. ( 2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 13.Gribskov, M., McLachlan, A. D., and Eisenberg, D. ( 1987) Profile analysis: detection of distantly related proteins. Proc. Natl. Acad. Sci. U. S. A. 84, 4355–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartels, C. ( 1990) Fast algorithm for peptide sequencing by mass spectroscopy. Biomed. Environ. Mass Spectrom. 19, 363–368 [DOI] [PubMed] [Google Scholar]
- 15.Taylor, J. A., and Johnson, R. S. ( 1997) Sequence database searches via de novo peptide sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 11, 1067–1075 [DOI] [PubMed] [Google Scholar]
- 16.Tabb, D. L., Saraf, A., and Yates, J. R. ( 2003) GutenTag: high-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 75, 6415–6421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sunyaev, S., Liska, A., Golod, A., and Shevchenko, A. ( 2003) MultiTag: multiple error-tolerant sequence tag search for the sequence-similarity identification of proteins by mass spectrometry. Anal. Chem. 75, 1307–1315 [DOI] [PubMed] [Google Scholar]
- 18.Alves, G., and Yu, Y. ( 2005) Robust accurate identification of peptides (RAId): deciphering MS2 data using a structured library search with de novo based statistics. Bioinformatics 21, 3726–3732 [DOI] [PubMed] [Google Scholar]
- 19.Bern, M., Cai, Y., and Goldberg, D. ( 2007) Lookup peaks: a hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 99, 1393–1400 [DOI] [PubMed] [Google Scholar]
- 20.Tsur, D., Tanner, S., Zandi, E., Bafna, V., and Pevzner, P. A. ( 2005) Identification of post-translational modifications by blind search of mass spectra. Nat. Biotechnol. 23, 1562–1567 [DOI] [PubMed] [Google Scholar]
- 21.Bandeira, N., Olsen, J. V., Mann, J. V., Mann, M., and Pevzner, P. A. ( 2008) Multi-spectra peptide sequencing and its applications to multistage mass spectrometry. Bioinformatics 24, i416–i423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durbin, R., Eddy, S. R., Krogh, A., and Mitchison, G. ( 1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids, Cambridge University Press, Cambridge, UK
- 23.Kim, S., Gupta, N., and Pevzner, P. A. ( 2008) Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res. 7, 3354–3363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klimek, J., Eddes, J. S., Hohmann, L., Jackson, J., Peterson, A., Letarte, S., Gafken, P. R., Katz, J. E., Mallick, P., Lee, H., Schmidt, A., Ossola, R., Eng, J. K., Aebersold, R., and Martin, D. B. ( 2008) The standard protein mix database: a diverse data set to assist in the production of improved peptide and protein identification software tools. J. Proteome Res. 7, 96–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller, A., Nesvizhskii, A., Kolker, E., and Aebersold, R. ( 2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 26.Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. ( 1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frank, A., Tanner, S., Bafna, V., and Pevzner, P. A. ( 2005) Peptide sequence tags for fast database search in mass spectrometry. J. Proteome Res. 4, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 28.Xin, L., Lajoie, G., and Ma, B. ( 2008) New method for the validation of de novo sequencing results, in the 56th ASMS Conference on Mass Spectrometry, Denver, June 1–5, 2008, Abstr. WP645, American Society for Mass Spectrometry (ASMS), Santa Fe, NM
- 29.Mann, M., and Wilm, M. ( 1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 30.Taylor, J., and Johnson, R. ( 2001) Implementation and uses of automated de novo peptide sequencing by tandem mass spectrometry. Anal. Chem. 73, 2594–2604 [DOI] [PubMed] [Google Scholar]