

Abstract
(±) SKF83959, like many other arylbenzazepines, elicits powerful neuroprotection in vitro and in vivo. The neuroprotective action of the compound was found to partially depend on its D1-like dopamine receptor agonistic activity. The precise mechanism for the (±) SKF83959-mediated neuroprotection remains elusive. We report here that (±) SKF83959 is a potent blocker for delayed rectifier K+ channel. (±) SKF83959 inhibited the delayed rectifier K+ current (I K) dose-dependently in rat hippocampal neurons. The IC 50 value for inhibition of I K was 41.9±2.3 µM (Hill coefficient = 1.81±0.13, n = 6), whereas that for inhibition of I A was 307.9±38.5 µM (Hill coefficient = 1.37±0.08, n = 6). Thus, (±) SKF83959 is 7.3-fold more potent in suppressing I K than I A. Moreover, the inhibition of I K by (±) SKF83959 was voltage-dependent and not related to dopamine receptors. The rapidly onset of inhibition and recovery suggests that the inhibition resulted from a direct interaction of (±) SKF83959 with the K+ channel. The intracellular application of (±) SKF83959 had no effects of on I K, indicating that the compound most likely acts at the outer mouth of the pore of K+ channel. We also tested the enantiomers of (±) SKF83959, R-(+) SKF83959 (MCL-201), and S-(−) SKF83959 (MCL-202), as well as SKF38393; all these compounds inhibited I K. However, (±) SKF83959, at either 0.1 or 1 mM, exhibited the strongest inhibition on the currents among all tested drug. The present findings not only revealed a new potent blocker of I K , but also provided a novel mechanism for the neuroprotective action of arylbenzazepines such as (±) SKF83959.
Introduction
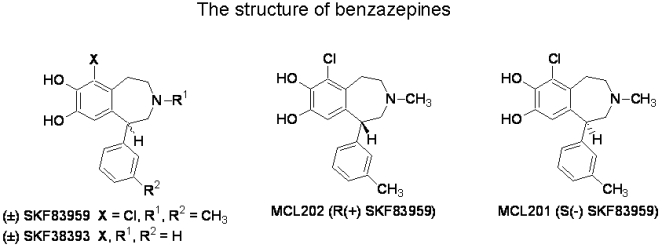
Atypical D1 receptor agonist (±) SKF83959, (±) 3-methyl-6-chloro-7,8-hydroxy-1-(3-methylphenyl)-2,3,4,5-tetrahydro-1H-3-benzazepine (Fig. 1), possesses a unique pharmacological property. (±) SKF83959 does not stimulate cAMP formation in brain tissues but activates the phosphatidylinositol (PI)-linked pathway via a D1-like dopamine receptors (D1DAR) [1]–[5], although it produces D1 agonist-mediated behavioral responses in animals [6]. This compound was shown to have anti-parkinsonian effect in an experimental primate model as well as in the unilateral lesioned rodent model for Parkinson's disease [7]–[11]. Earlier studies with (±) SKF83959 and its isomers, showed that such D1-like agonists increase eye blinking in monkeys [12] and rats [13] and that the magnitude of this effect may be related to agonistic efficacy. Although there is a recent report by employing a different approach depicting that (±) SKF83959 is able to stimulate native brain tissues coupling to both Gs and Gq protein[14], the antiparkinsonian mechanism of the agent, however, is known not to relate to the cAMP pathway and may associate with the (±) SKF83959-mediated PI-linked D1DAR activation [3], [6], [15]–[17]. In agreement with a previous report in a model of primate Parkinson's disease, in which (±) SKF83959 not only reduced the dyskinesias but also reduced the occurrence of motor fluctuation and wearing-off dyskinesia [15], we have recently demonstrated that chronic treatment of (±) SKF83959 resulted in a significantly lower dyskinesia while eliciting its potent anti-parkinsonian action in a model of 6-hydroxydopamine (6-OHDA) unilaterally -lesioned rats. Moreover, a chronic administration of (±) SKF83959 also significantly reduced levo-DOPA-induced dyskinesias in PD rats [18]. The underlying mechanism for the anti-dyskinesia action of (±) SKF83959 is believed to associate with the drug's powerful neuroprotective action [19]. Since it was suggested that occurrence of motor fluctuation and wearing-off dyskinesia are associated with anti-PD drug treatment-induced further loss of dopaminergic neurons [20], [21]. We recently demonstrated that in both cultured neurons and in HEK293 cells, (±) SKF83959 inhibited GSK3β activity via a D1-like receptor-dependent mechanism. However, blockage of D1-like receptor activation which blunted SKF83959-mediated inhibition on GSK3β,was found to attenuated partially the neuroprotective effect of (±) SKF83959 [19], indicating that D1-like receptor-independent mechanism may be also involved in the neuroprotection of the drug.
Figure 1. The structures of benzazepines.
Delayed rectifier K+ channel plays an important role in neuroprotection [22]–[24]. It has been proposed that decreased intracellular K+ due to activation of the K+ channel contributes to the neuroapoptosis induced by various insults such as Aβ-amyloid [22], [25] and serum deprivation [23]. Tetraethylammonium (TEA), a classical blocker of delayed rectifier K+ channel, was found to exert potent neuroprotective effects in vivo [26], [27] and in vitro [23]. Considering the importance of delayed rectifier K+ channel in neuroprotection, we decided to explore the potential effect of (±) SKF83959 on delayed rectifier K+ channel as part of our efforts in understanding the mechanism for the D1-receptor independent neuroprotection of the drug. We here demonstrate that (±) SKF83959 and related benzazepines are potent inhibitors of delayed rectifier K+ channel. The potency of (±) SKF83959 is almost 25-fold higher than that of TEA. Our findings not only revealed a new category of potent blockers of delayed rectifier K+ channel but also provided a novel mechanism for the neuroprotective action of arylylbenzazepines analogs.
Materials and Methods
Materials
(±) SKF38393, (±)-SCH23390 hydrochloride, prazosin and mesulergine were purchased from Sigma (St Louis, MO, USA)/RBI (Natick, MA, USA). R-(+) SKF38393 (MCL-201) and S-(−) SKF38393 (MCL-202) (Fig. 1) were synthesized in the Alcohol and Drug Abuse Research Center at McLean Hospital (Belmont, MA, USA). (±) SKF83959 was synthesized in the Synthetic Organic & Medicinal Chemistry Laboratory, Shanghai Institute of Materia Medica, Chinese Academy of Sciences. Other chemicals were purchased from Sigma-Aldrich China Inc.
Preparation of dissociated hippocampal neurons
All procedures were in compliance with the Guidelines for the Care and Use of Laboratory Animals (National Research Council, People's Republic of China, 1996). Dissociated hippocampal neurons were prepared from newborn (5–9 day) Sprague-Dawley rats as described previously [26]. Briefly, hippocampal slices (500 µm) were cut in oxygenated ice-cold dissociation solution consisted of the following (in mM): 82 Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES, and 10 glucose, pH = 7.3 adjusted with NaOH. The slices were incubated in the dissociation solution containing protease XXIII (3 g/L) at 32°C for 8 min, and then placed in dissociation solution containing trypsin inhibitor type II-S (1 g/L) and bovine serum albumin (1 g/L) at 24–25°C under an oxygen atmosphere. Before recording, the CA1 region of several slices was dissected, and triturated using a series of fire-polished Pasteur pipettes with decreasing tip diameters. Dissociated neurons were placed in a recording dish and superfused with an external solution consisted of the following (in mM): 135 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, 0.001 tetrodotoxin , pH = 7.4 adjusted with NaOH..
Whole-cell voltage clamp recoding
Voltage-activated K+ currents were recorded in large pyramidal-shaped neurons using an Axopatch 200A amplifier (Axon Instruments, USA) at 24–25°C [26]. Voltage protocols were controlled by pClamp 9.0 software via a DigiData-1322A interface (Axon Instruments, USA). Electrodes (a tip resistance of 3–5 MΩ) were pulled from borosilicate grass pipettes (Sutter Instruments, USA) and filled with a pipette solution consisted of the following (in mM): 140 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 EGTA (pH 7.4 with KOH). The neurons were held at −50 mV. Unless otherwise mentioned, the total K+ current (I total) was elicited with 400-ms depolarizing steps to +40 mV following a 600-ms hyperpolarizing prepulse to −110 mV, delivered every 10 s. The delayed rectifier K+ current (I K) was elicited by using a similar protocol, but a 50-ms interval at −50 mV was inserted after the prepulse to inactivate the fast transient K+ current (I A). I A is the subtraction of I K from I total. Signals were filtered at 2–10 KHz and sampled at frequencies of 10–40 KHz. Series resistance was compensated by 75%–85%. Linear leak and residual capacitance currents were subtracted online using a P/4 protocol.
Drug application
(±) SKF83959 and other drugs were dissolved in dimethylsulfoxide (DMSO) to prepare a stock solution of 10 mM and stored at −20°C. Before use, the stock solutions were diluted to desired concentrations. For extracellular application, the drug-containing external solution was delivered to the neuron using RSC-100 rapid solution changer with a 9-tube head (BioLogic Co., France). DMSO (less than 0.1% in the final dilution) had no observed effect on the voltage-activated K+ currents (DMSO: 0.598±0.11 nA; control: 0.660±0.095 nA, n = 4; p>0.05). For intracellular dialysis, (±) SKF83959 contained in the pipette solution was diffused into the recorded neuron immediately after patch membrane ruptured [29].
Data analysis
The peak amplitude of I A was measured, whereas the steady-state amplitude of I K was measured at 300 ms after the initiation of each voltage step. The decay time constants (τ) of the currents were obtained by fitting the decay time course with a mono-exponential function. The concentrations of (±) SKF83959 to yield 50% block of the K+ currents (IC 50) were obtained by fitting normalized concentration–inhibition relationships to the equation: I/I 0 = 1/{1+([C]/IC50)n}, where I 0 and I are the current amplitudes measured in control and in the presence of (±) SKF83959, [C] is the concentration of (±) SKF83959 in the external solution and n is the Hill coefficient. The ratio of inhibition was calculated by using the equation: Inhibition = (1−I/I 0)*100%, where I 0 and I are the current amplitude in control and in the presence of (±) SKF83959, respectively. For analyzing the voltage-dependence of steady-state activation or inactivation of I K, normalized conductance or current was plotted against the membrane potential, and fitted to the Boltzmann equation: Y = 1/{1+exp[(V−V 1/2)/k]}, where Y is the normalized conductance or current, V is the test potential; V 1/2 is the voltage at half-maximal activation or inactivation of I K; k is the slope factor. The time course of recovery of I K from inactivation was fitted with a mono-exponential function: I/I max = A*{1–exp[−Δt/τ]}, where I max is the maximal current amplitude; I is the current after a recovery period of Δt; τ is the time constant; A is the amplitude coefficient. Data are presented as mean±S.E.M. Statistical significance was assessed using a Student's t-test except for the voltage dependence of inhibition of I K by (±) SKF83959 where one-way ANOVA test was used, and P<0.05 was considered significant. All analyses were performed using the software “GraphPad Prism 4” and “Excel 2000”.
Results
Inhibition of voltage-activated K+ currents by (±) SKF83959 in hippoacampal neurons
Superfusion with (±) SKF83959 (10–1000 µM) inhibited both I K and I A in concentration-dependent manner. However, the compound preferentially inhibited I K to I A. As shown in Fig. 2A, (±) SKF83959 (100 µM) markedly suppressed I K, whereas a moderate reduction in the amplitude of I A was observed. The inhibition of (±) SKF83959 on both K+ currents had a rapid onset, as it reached steady-state levels within 10 s. Moreover, the K+ currents were partially recovered upon washing out the compound (Fig. 2B, 2C). The partial recovery may be because of the run-down of the currents with recording, or due to the incompletely drug washing- out.
Figure 2. Inhibition of voltage-activated K+ currents by (±) SKF83959 in rat hippocampal neurons.

(A) Upper and lower are the respective representative traces of the delayed rectifier K+ current (I K) and fast transient K+ current (I A) recorded prior to and during superfusion with (±) SKF83959 (100 µM) and after 10 s of washout. The neuron was held at −50 mV. Upper inset shows the pulse protocol to elicit I K, whereas lower inset shows the protocol to elicit the total K+ current. I A is the subtraction of I K from the total K+ current. (B) and (C) Time courses of the inhibition of I K and I A by (±) SKF83959 (100 µM, n = 5 for each). The bar denotes the surpufusion with SKF83959. A number of symbols in (B) and (C) have error bars smaller than their size.
Analyzing the concentration-inhibition relationships of (±) SKF83959 on K+ currents revealed that the IC 50 value for inhibition of I K was 41.9±2.3 µM (Hill coefficient = 1.81±0.13, n = 6), while that for inhibition of I A was 307.9±38.5 µM (Hill coefficient = 1.37±0.08, n = 6) (Fig. 3A). Because (±) SKF83959 is 7.3-fold more potent in suppressing I K than I A, we thus focused on characterizing the inhibition on I K.
Figure 3. Concentration-dependent inhibition of (±) SKF83959 on the voltage-activated K+ currents.

(A) Concentration-inhibition curves of (±) SKF83959 on the delayed rectifier K+ current (I K) and fast transient K+ current (I A)(n = 6 for I K and I A). A number of symbols have error bars smaller than their size. (B) Comparison of the substituted phenylbenzazepine, MCL-201, MCL-202, SKF38393 and (±) SKF83959 in inhibition of I K.
We next examined whether the other substituted phenylbenzazepines such as R-(+) SKF83959 (MCL-201), S-(−) SKF83959 (MCL-202) and SKF38393 affect I K. The results are shown in Fig. 3B. At the concentration of 100 µM, the inhibition of I K by MCL-201, MCL-202 and SKF38393 was 36.7±4.1% (n = 8), 9.7±1.7% (n = 8) and 24.5±1.1% (n = 7), respectively, whereas (±) SKF83959 (100 µM) induced much greater inhibition on I K (76.0±2.7%, n = 6). At the concentration of 1 mM, the inhibition of I K by MCL-201, MCL-202 and SKF38393 was 77.1±4.0% (n = 3), 56.1±2.7% (n = 4) and 62.0±2.7% (n = 5), respectively, whereas (±) SKF83959 (1 mM) almost completely suppress the K+ current (92.0±2.5%, n = 3). Thus, (±) SKF83959 produced the most potent inhibition of I K among the four substituted benzazepines at tested concentrations.
Mechanisms underlying the inhibition of the delayed rectifier K+ current by (±) SKF83959
(±) SKF83959 is an atypical agonist of D1-like receptor. We first examined whether the inhibition of I K by (±) SKF83959 was mediated through activation of D1-like receptor. Pretreatment with the D1 receptor antagonist SCH23390 did not significantly alter (±) SKF83959-induced inhibition on I K (Fig. 4A). The inhibition of I K by (±) SKF83959 (100 µM) in the presence of SCH23390 (20 µM) was 60.1±2.7% (n = 6), while that by (±±) SKF83959 alone was 70.3±4.1% (n = 7, P = 0.074, t = 1.97, df = 11).
Figure 4. Block of the delayed rectifier K+ channel by (±) SKF83959 in rat hippocampal neurons.

(A) Inhibition of the delayed rectifier K+ current (I K) by (±) SKF83959 (100 µM) in the presence of D1 receptor antagonist SCH23390 (20 µM, n = 6), or α1-adrenoceptor antagonist prazosin (10 µM, n = 6), or 5-HT receptor antagonist mesulergine (10 µM, n = 6). (B) Lacking effect of intracellular dialysis of (±) SKF83959 on I K (n = 7). The recording pipettes were filled with pipette solution containing (±) SKF83959 (300 µM). The downward arrow indicates the time when the patch membrane was ruptured.
In addition to D1-like receptor, (±) SKF83959 also exhibits moderate affinities to D2 receptors, α1-adrenoceptors and 5-HT2A receptors [5]. However, neither the α1-adrenoceptors antagonist prazosin, nor the 5-HT receptor antagonist mesulergine affected (±) SKF83959-induced inhibition of I K (Fig. 4A). The inhibition of I K by (±) SKF83959 (100 µM) in the presence of prazosin (10 µM) or mesulergine (10 µM) was 76.6±2.1% (n = 6) and 71.4±4.6% (n = 6), respectively, which was almost identical with that of (±) SKF83959 alone treatment (70.3±4.1%, n = 7, P = 0.23, t = 1.29, df = 11 vs. prazosin and P = 0.86, t = 0.18, df = 11 vs. mesulergine). Taken together, (±) SKF83959 is a potent blocker of the delayed rectifier K+ channel and the inhibitory action is independent of either dopamine or serotonin receptors.
To determine the acting site of the agent on the K+ channel, we investigated the effect of intracellular dialysis of (±) SKF83959 on I K. The concentration for intracellular dialysis was 300 µM, which inhibited I K by nearly 90% and is close to the value of IC50 for I A, when applied externally (Fig. 3A). Throughout the 10-min recording period, the relative amplitudes of I K in the neurons dialyzed with (±) SKF83959 were almost identical with that of in the control group (Fig. 4B), so was the I K (data not shown). At the end of 10-min recording, the relative amplitude of I K in control and in the neurons dialyzed with (±) SKF83959 was 85.4±6.0% (n = 5) and 83.4±5.3% (n = 7), respectively (P = 0.81, t = 0.25, df = 10). The results suggest that (±) SKF83959 acts at an extracellular site of the delayed rectifier K+ channel.
Voltage dependence of inhibition of the delayed rectifier K+ current by (±) SKF83959
The current-voltage (I/V) relationship of I K from a representative neuron in control and in the presence of (±) SKF83959 (100 µM) was plotted in Fig. 5A. (±) SKF83959 did not significantly change the threshold for activation of I K, but caused a remarkable downward shift of the I/V curve, and reduced its amplitude over the entire period of activation. The inhibition of I K by (±) SKF83959 seems to be greater at more depolarizing potentials. Thus, we plotted the relative amplitudes of I K as a function of the test potential. As shown in Fig. 5B, the inhibition of I K by (±) SKF83959 steeply increased between −20 mV and +20 mV. In the presence of (±) SKF83959, the relative currents in steps to −20 mV and to +20 mV, respectively, were 43.5±6.1% and 18.1±1.8% (n = 7, P = 0.0019, t = 3.96, df = 12). Between +20 mV and +80 mV the inhibition remained relatively constant. The results demonstrated that the inhibition of I K by (±) SKF83959 was voltage-dependent.
Figure 5. Effects of (±) SKF83959 on the current-voltage relationships of the delayed rectifier K+ current in rat hippocampal neurons.

(A) Current-voltage (I/V) curves of the delayed rectifier K+ current (I K) obtained in a representative neuron in control and during superfusion of (±) SKF83959 (100 µM). (B) Plot of the relative amplitudes of I K during superfusion of (±) SKF83959 (100 µM) as a function of the test potential. Each symbol represents the mean±S.E.M. (n = 7). The solid line is the linear fit, while the dashed line is the activation curve obtained in control condition. *P<0.01 (one-way ANOVA test) versus the value at −20 mV.
Effects of (±) SKF83959 on kinetic behaviors of the delayed rectifier K+ current
In addition to suppressing the amplitude of I K, (±) SKF83959 (100 µM) markedly accelerated the decay of the current (Fig. 6A&6B). The time course of the decay of the current trace was fitted with a single exponential function. The decay time constant (τ) for I K was 145.9±11.9 ms in control and 63.4±4.9 ms in the presence of 100 µM (±) SKF83959 (n = 7, P = 0.0002, t = 6.42, df = 12, vs. control) (Fig. 6C). Moreover, as depicted in Fig. 6D, (±) SKF83959 suppressed the amplitude of tail current, but did not cause a crossover of the tail current.
Figure 6. Effects of (±) SKF83959 on the decay of the delayed rectifier K+ current in rat hippocampal neurons.

(A) Superimposed traces of the delayed rectifier K+ current (I K) in a representative neuron prior to and during superfusion with (±) SKF83959 (100 µM). (B) The trace with (±) SKF83959 in (A) is scaled up. (C) Decay time constants of I K prior to and during superfusion with (±) SKF83959 (100 µM). n = 7, *P<0.01 versus the control. (D) Tail current traces evoked at −40 mV after a 500-ms depolarizing steps to +40 mV prior to and during superfusion with (±) SKF83959 (100 µM). Similar results were obtained in 3 neurons.
(±) SKF83959 (100 µM) elicited a marked hyperpolarizing shift (nearly 20 mV) of the voltage dependence of steady-state activation curve of I K (Fig. 7A). The value of V 1/2 for activation was changed from −1.7±2.6 mV to −22.2±3.1 mV (n = 8, P = 0.0002, t = 5.10, df = 14), whereas the value of k for activation was from 16.8±0.9 to 13.6±1.1, n = 8, P = 0.038, t = 2.29, df = 14). (±) SKF83959 (100 µM) had no significant effect on its steady-state inactivation (Fig. 7B). The value of V 1/2 for inactivation was nearly identical (−85.8±1.3 mV in control vs.−86.3±3.9 mV in the presence of (±) SKF83959, n = 8, P = 0.91, t = 0.11, df = 12). The value of k for inactivation has no significant change (−11.9±0.7 in control vs.−16.7±4.8 in the presence of (±) SKF83959, n = 8, P = 0.35, t = 1.00, df = 6). The same treatment accelerated the recovery of I K from inactivation (Fig. 7C). The time constant of recovery was found to reduce from 294.1±30.4 ms to 107.7±10.4 ms (n = 6, P = 0.0002, t = 5.81, df = 10).
Figure 7. Effects of (±) SKF83959 on the activation and steady-state inactivation of the delayed rectifier K+ current in rat hippocampal neurons.

(A) Activation curves of the delayed rectifier K+ current (I K) prior to and during superfusion with 100 µM (±) SKF83959 (n = 8). (B) Steady-state inactivation curves of I K prior to and during superfusion with 100 µM (±) SKF83959 (n = 8). (C) Time courses of recovery of I K from inactivation prior to and during superfusion with 100 µM (±) SKF83959 (n = 6). For studying the activation, neurons were held at −50 mV, currents were elicited with a series of 400-ms step from −80 mV to +80 mV in 10 mV increments following a 600-ms hyperpolarizing prepulse to −110 mV and a 50-ms interval at −50 mV, delivered every 10 s. For studying the steady-state inactivation, neurons were held at 0 mV, currents were elicited with a series of 600-ms prepulses at different hyperpolarizing potentials followed by a 50-ms interval at −50 mV and a 400-ms step to +40 mV, then back to 0 mV, delivered every 10 s. For studying the time course of recovery from inactivation, neurons were held at 0 mV, currents were elicited on return from hyperpolarizing prepulses of varying durations at −110 mV to +40 mV, delivered every 10 s.
Discussion
(±) SKF83959 is a putative phosphatidylinositol (PI)-linked D1-like receptor agonist [3], [4]. The agent has been shown to possess potent anti-parkinsonian effects in a variety of animal models for Parkinson's disease with less severe dyskinesia and motor fluctuation [6]–[8], [15], [18]. In the present study, we demonstrated for the first time that (±) SKF83959 and other substituted phenylbenzazepines inhibited the delayed rectifier K+ current (I K) in rat hippocampal neurons with the highest inhibition produced by (±) SKF83959. (±) SKF83959 is nearly 25-fold more potent than tetraethylammonium (TEA), a classical blocker of I K, which had an IC 50 value of 1.05±0.21 mM in the same preparations [28].
To elucidate the mechanisms underlying (±) SKF83959-induced inhibition on I K, we found that the antagonists of D1, D2 or 5-HT1A receptors did not block the inhibition of I K by (±) SKF83959 (Fig. 3A), indicating that the inhibitory effect was a receptor-independent event. Furthermore, the fast nature in the onset of inhibition and recovery (Fig. 2B) implicates that the inhibition results from a direct interaction of (±) SKF83959 with the K+ channel. Moreover, we found that intracellular application of (±) SKF83959 had no effect on I K (Fig. 4B), suggesting that the agent is most likely to act at the outer mouth of the pore of K+ channel. In an effort to determine how (±) SKF83959 blocks the K+ channel. We demonstrated that the inhibition of I K by (±) SKF83959 was voltage-dependent (Fig. 5). Furthermore, the agent markedly accelerated the decay of I K in addition to suppressing its amplitude (Fig. 6A), although lack of the crossover of tail current (Fig. 6D). The results suggest that the agent preferentially binds to the open state of the K+ channel [30], [31]. It is conceivable that (±) SKF83959 acts as an open-channel blocker at the delayed rectifier K+ channel. (±) SKF83959 probably binds to the K+ channel with 2∶1 stoichometry (Hill coefficient = 1.81±0.13).
Accumulating evidence shows that loss of intracellular K+ ions through enhanced delayed rectifier K+ channel (mainly Kv2.1 channel) mediates apoptosis of cortical neurons induced by a variety of treatments, such as serum deprivation, exposure to staurosporine or β–amyloid peptide fragment, etc. [23], [25]. In 6-hydroxydopamine -induced neurotoxicity, voltage-dependent potassium channels were found to play a vital role [32], since blockage of I K by TEA was shown to effectively against the neurotoxicity and improve the neuronal viability. In other hand, the neuroprotective effects of classical I K blocker TEA were also demonstrated in animal models of transient focal ischemia [26], [27]. Thus, the drug discovery targeted to specific K+ channel has been proposed as a potential therapeutic approach in treatment of neurodegenerative diseases [33]–[35]. We recently demonstrated that (±) SKF83959 protected rat cortical neurons against H2O2-induced injury, which was partially mediated though a putative PI-linked D1-like receptor-dependent mechanism via inhibition of GSK3β pathway [19]. The finding of (±) SKF83959 and its analogs as a potent blocker for the delayed rectifier K+ channel in the present study not only reveals a role of those compounds in the modulation of K+ channel, but also suggests a potential new mechanism for the neuroprotection of phenylbenzazepine derivatives. In the case of (±) SKF83959, how the neuroprotective effects via blockage of the delayed rectifier K+ channel contributes to the observed attenuation in the development of dyskinesia and reduction in the occurrence of motor fluctuation and wearing-off dyskinesia in chronic (±) SKF83959-treated PD animals [7], [18] are worth of further study.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by 973 and 863-plan of the Ministry of Science and Technology of China (2007AA02z163, 2009CB522201), and grants from Natural Science Foundation of China (30770662, 30825042, 20872153) and the National Institute of Health Grant DA-14251 (JLN). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Arnt J, Hyttel J, Sanchez C. Partial and full dopamine D1 receptor agonists in mice and rats: relation between behavioural effects and stimulation of adenylate cyclase activity in vitro. Eur J Pharmacol. 1992;213:259–267. doi: 10.1016/0014-2999(92)90690-6. [DOI] [PubMed] [Google Scholar]
- 2.Cools AR, Lubbers L, van Oosten RV, Andringa G. SKF 83959 is an antagonist of dopamine D1-like receptors in the prefrontal cortex and nucleus accumbens: a key to its antiparkinsonian effect in animals? Neuropharmacol. 2002;42:237–245. doi: 10.1016/s0028-3908(01)00169-1. [DOI] [PubMed] [Google Scholar]
- 3.Jin L-Q, Goswami S, Cai G, Zhen XC, Friedman E. SKF83959 selectively regulates phosphatidylinositol-linked D1 dopamine receptors in rat brain. J Neuroche. 2003;85:378–386. doi: 10.1046/j.1471-4159.2003.01698.x. [DOI] [PubMed] [Google Scholar]
- 4.Panchalingam S, Undie AS. SKF83959 exhibits biochemical agonism by stimulating [35S]GTP[gamma]S binding and phosphoinositide hydrolysis in rat and monkey brain. Neuropharmacol. 2001;40:826–837. doi: 10.1016/s0028-3908(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 5.Neumeyer JL, Kula NS, Bergman J, Baldessarini RJ. Receptor affinities of dopamine D1 receptor-selective novel phenylbenzazepines. Eur J Pharmacol. 2003;474:137–140. doi: 10.1016/s0014-2999(03)02008-9. [DOI] [PubMed] [Google Scholar]
- 6.Deveney AM, Waddington JL. Pharmacological characterization of behavioural responses to SK&F83959 in relation to ‘D1-like’ dopamine receptors not linked to adenylyl cyclase. Brit J Pharmacol. 1995;116:2120–2126. doi: 10.1111/j.1476-5381.1995.tb16420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gnanalingham KK, Erol DD, Hunter AJ, Smith LA, Jenner P, et al. Differential anti-parkinsonian effects of benzazepine D1 dopamine agonists with varying efficacies in the MPTP-treated common marmoset. Psychopharmacol (Berl) 1995;117:275–286. doi: 10.1007/BF02246102. [DOI] [PubMed] [Google Scholar]
- 8.Gnanalingham K K, Hunter AJ, Jenner P, Marsden CD. The differential behavioural effects of benzazepine D1 dopamine agonists with varying efficacies, co-administered with quinpirole in primate and rodent models of Parkinson's disease. Psychopharmacol (Berl) 1995;117:287–297. doi: 10.1007/BF02246103. [DOI] [PubMed] [Google Scholar]
- 9.Nutt JG, Carter JH, Lea ES, Woodward WR. Motor fluctuations during continuous levodopa infusions in patients with Parkinson's disease. Move Disord. 1997;12:285–292. doi: 10.1002/mds.870120304. [DOI] [PubMed] [Google Scholar]
- 10.Waddington J, O'Tuathaigh C, O'Sullivan G, Tomiyama K, Koshikawa N, et al. Phenotypic studies on dopamine receptor subtype and associated signal transduction mutants: insights and challenges from 10 years at the psychopharmacology–molecular biology interface. Psychopharmacol (Berl) 2005;181:611–638. doi: 10.1007/s00213-005-0058-8. [DOI] [PubMed] [Google Scholar]
- 11.Wirtshafter D, Osborn CV. The atypical dopamine D1 receptor agonist SKF 83959 induces striatal Fos expression in rats. Eur J Pharmacol. 2005;528:88–94. doi: 10.1016/j.ejphar.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Desai RI, Neumeyer JL, Paronis CA, Nguyen P, Berman J. Behavioral effects of R (+) and S (−) enantiomers of the dopamine D1-like partial receptor agonist. Eur J Pharmacol. 2007;558:98–106. doi: 10.1016/j.ejphar.2006.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai RI, Neumeyer JL, Bergman J, Paronis CA. Pharmacological characterization of the effects of dopamine D1 agonists on eye blinking in rats. Behav Pharmacol. 2007;18:745–754. doi: 10.1097/FBP.0b013e3282f14ee6. [DOI] [PubMed] [Google Scholar]
- 14.Mannoury la Cour C, Vidal S, Pasteau V, Cussac D, Millan MJ. Dopamine D1 receptor coupling to Gs/olf and Gq in rat striatum and cortex: A scintillation proximity assay (SPA)/antibody-capture characterization of benzazepine agonists. Neuropharmacol. 2007;52:1003–1014. doi: 10.1016/j.neuropharm.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 15.Andringa G, Stoof JC, Cools AR. Sub-chronic administration of the dopamine D1 antagonist SKF 83959 in bilaterally MPTP-treated rhesus monkeys: stable therapeutic effects and wearing-off dyskinesia. Psychopharmacol. 1999;146:328–334. doi: 10.1007/s002130051124. [DOI] [PubMed] [Google Scholar]
- 16.Clifford JJ, Tighe O, Croke DT, Kinsella A, Sibley DR, et al. Conservation of behavioural topography to dopamine D1-like receptor agonists in mutant mice lacking the D1A receptor implicates a D1-like receptor not coupled to adenylyl cyclase. Neurosci. 1999;93:1483–1489. doi: 10.1016/s0306-4522(99)00297-3. [DOI] [PubMed] [Google Scholar]
- 17.Zhen X, Goswami S, Friedman E. The role of the phosphatidyinositol-linked D1 dopamine receptor in the pharmacology of SKF83959. Pharmacol Biochem Behav. 2005;80:597–601. doi: 10.1016/j.pbb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 18.Zhang H, Ma L, Wang F, Chen J, Zhen X. Chronic SKF83959 induced less severe dyskinesia and attenuated L-DOPA-induced dyskinesia in 6-OHDA-lesioned rat model of Parkinson's disease. Neuropharmacol. 2007;53:125–133. doi: 10.1016/j.neuropharm.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Yu Y, Wang JR, Sun PH, Guo Y, Zhang ZJ, et al. Neuroprotective effects of atypical D1 receptor agonist SKF83959 are mediated via D1 receptor-dependent inhibition of glycogen synthase kinase-3B; and a receptor-independent anti-oxidative action. J Neruochem. 2008;104:946–956. doi: 10.1111/j.1471-4159.2007.05062.x. [DOI] [PubMed] [Google Scholar]
- 20.Agnati LF, Leo G, Vergoni AV, Martinez E, Hockemeyer J, et al. Neuroprotective effect of L-DOPA co-administered with the adenosine A2A receptor agonist CGS 21680 in an animal model of Parkinson's disease. Brain Res Bull. 2004;64:155–164. doi: 10.1016/j.brainresbull.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Obeso JA, Rodriguez-Oroz M, Marin C, Alonso F, Zamarbide I, et al. The origin of motor fluctuations in Parkinson's disease: importance of dopaminergic innervation and basal ganglia circuits. Neurology. 2004;62:S17–30. doi: 10.1212/wnl.62.1_suppl_1.s17. [DOI] [PubMed] [Google Scholar]
- 22.Yu SP, Farhangrazi ZS, Ying HS, Yeh CH, Choi DW. Enhancement of outward potassium current may participate in β-amyloid peptide-induced cortical neuronal death. Neurobiol Dis. 1998;5:81–88. doi: 10.1006/nbdi.1998.0186. [DOI] [PubMed] [Google Scholar]
- 23.Yu S-P, Yeh C-H, Sensi SL, Gwag BJ, Canzoniero LM, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 24.Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franciosi S, Ryu JK, Choi HB, Radov L, Kim SU, et al. Broad-spectrum effects of 4-aminopyridine to modulate amyloid β1-42-induced cell signaling and functional responses in human microglia. J Neurosci. 2006;26:11652–11664. doi: 10.1523/JNEUROSCI.2490-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Gao TM, Gong L-W, Zhuang Z-Y, Li X. Potassium channel blocker TEA prevents CA1 hippocampal injury following transient forebrain ischemia in adult rats. Neurosci Lett. 2001;305:83–86. doi: 10.1016/s0304-3940(01)01821-3. [DOI] [PubMed] [Google Scholar]
- 27.Wei L, Yu SP, Gottron F, Snider BJ, Zipfel, et al. Potassium channel blockers attenuate hypoxia- and ishemia-induced neuronal death in vitro and in vivo. Stroke. 2003;34:1281–1286. doi: 10.1161/01.STR.0000065828.18661.FE. [DOI] [PubMed] [Google Scholar]
- 28.Song MK, Liu H, Jiang HL, Yue JM, Hu GY. Electrophysiological characterization of 14-benzoyltalatisamine, a selective blocker of the delayed rectifier K+ channel found in virtual screening. Eur J Pharmacol. 2006;531:47–53. doi: 10.1016/j.ejphar.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 29.Hu GY, Biro Z, Hill RH, Grillner S. Intracellular QX-314 causes depression of membrane potential oscillations in lamprey spinal neurons during fictive locomotion. J Neurophysiol. 2002;87:2676–2683. doi: 10.1152/jn.2002.87.6.2676. [DOI] [PubMed] [Google Scholar]
- 30.Delpon E, Valenzuela C, Gay P, Franqueza L, Snyders D J, et al. Block of human cardiac Kv1.5 channels by loratadine: voltage-, time- and use-dependent block at concentrations above therapeutic levels. Cardiovascul Res. 1997;35:341–350. doi: 10.1016/s0008-6363(97)00121-1. [DOI] [PubMed] [Google Scholar]
- 31.Ahn H, Kim S, Jang H-J, Kim M-J, Rhie D-J, et al. Open channel block of Kv1.3 by rosiglitazone and troglitazone: Kv1.3 as the pharmacological target for rosiglitazone. Naunyn-Schmiedeberg's Arch Pharmacol. 2007;374:305–309. doi: 10.1007/s00210-006-0118-6. [DOI] [PubMed] [Google Scholar]
- 32.Redman PT, Jefferson BS, Ziegler CB, Mortensen OV, Torres GE, et al. A vital role for voltage-dependent potassium channels in dopamine transporter-mediated 6-hydroxydopamine neurotoxicity. Neurosci. 2006;143:1–6. doi: 10.1016/j.neuroscience.2006.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shieh C-C, Coghlan M, Sullivan JP, Gopalakrishnan M. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol Rev. 2000;52:557–594. [PubMed] [Google Scholar]
- 34.Wickenden AD. K+ channels as therapeutic drug targets. Pharmacol & Therapeut. 2002;94:157–182. doi: 10.1016/s0163-7258(02)00201-2. [DOI] [PubMed] [Google Scholar]
- 35.Zaks-Makhina E, Kim Y, Aizenman E, Levitan ES. Novel Neuroprotective K+ Channel Inhibitor Identified by High-Throughput Screening in Yeast. Mol Pharmacol. 2004;65:214–219. doi: 10.1124/mol.65.1.214. [DOI] [PubMed] [Google Scholar]