

Abstract
Repopulation of cancer cells escaping lethal chemotherapy is a critical factor hindering treatment success. In this study, we tested the hypothesis that the antiprogestin mifepristone (MF) should be efficacious inducing cytostasis and preventing repopulation of ovarian cancer cells if given among rounds of cisplatin (CDDP) treatment. We established an in vitro approach wherein ovarian cancer cells with high or low sensitivity to CDDP were exposed to lethal rounds of CDDP for 1 h, 12 or 24 days apart. Every 4 or 8 days cell number, cell viability, cell cycle traverse, and colony-forming capacity of viable cells was analyzed. Although CDDP killed the majority of cells, there were remnant cells escaping CDDP lethality and repopulating the culture, as evidenced by increased cell number, improved clonogenic capacity of viable cells, and normalization of DNA synthesis. Conversely, when cells were exposed to CDDP for 1 h, and MF was present in the culture medium after CDDP removal, the number, clonogenic capacity, and DNA synthesis ability of the cells were reduced in a dose-dependent manner. The blockage by MF of post-CDDP repopulation was accompanied by a remarkable increase in the percentage of cells expressing the cell death marker cleaved poly (ADP-ribose) polymerase and the mitotic marker phospho-histone H3, suggesting that MF also potentiated CDDP lethality and that the cells likely die due to mitotic failure. In summary, this is the first study reporting that presence of MF after courses of lethal doses of CDDP prevents repopulation of remnant ovarian cancer cells surviving CDDP treatment.
Keywords: mifepristone, ovarian cancer, repopulation, cisplatin, cell death
Introduction
Ovarian cancer is the most deadly disease of the female reproductive tract. This is mainly due to the difficulty in achieving an early diagnosis and in identifying the ambiguous symptoms when the disease is confined to the ovaries (1,2). When the disease is diagnosed, in most cases abnormal growths have already progressed beyond the confines of the ovaries and into the nearby fallopian tubes, uterus, and various other sites within the peritoneal cavity (3). Consequently, the majority of patients diagnosed with ovarian cancer require debulking surgery followed by chemotherapy with platinum-based regimens (1,2,4,5). However, the efficacy of platinum-based chemotherapy is hindered by the elevated toxicity of the platinum derivatives (6), the development of mechanisms to evade drug toxicity (6,7), and the repopulation of cells between treatment intervals (8). Hence, finding therapeutic interventions that enhance the killing efficacy of platinum derivatives is of critical clinical relevance.
Repopulation of tumor cells refers to the continuing proliferation of surviving tumor cells with the capacity to regenerate the tumor that can occur during a course of chemotherapy or fractionated radiotherapy (8). Tumor cell repopulation between rounds of lethal chemotherapy takes place in the absence of any change in the intrinsic sensitivity of the tumor cells to the drugs, limits treatment effectiveness decreasing overall tumor cell reduction to a significant degree, and may be a critical cause of clinical drug resistance (8–10). It has been proposed that utilization of short-acting selective compounds in between courses of lethal therapy should be a proper strategy to inhibit repopulation of tumor cells (8). Following this rationale there is evidence that molecular targeted agents with relatively low toxicities can be excellent cytostatic anti-repopulation agents. For instance, the selective estrogen receptor modulators 4-hydroxy tamoxifen and arzoxifene inhibited repopulation of cultured MCF-7 and T47-D breast cancer cells in between courses of treatment with 5-fluorouracil or methotrexate (11). Arzoxifene also inhibited repopulation of MCF-7 breast cancer cells xenografted in nude mice when given between rounds of 5-fluorouracil or paclitaxel chemotherapy (12). In another example, the mammalian target of rapamycin inhibitor CCI-779 delayed the growth of PTEN-negative PC-3 prostate cancer cell xenografts in nude mice, when given between courses of mitoxantrone or docetaxel (13).
Mifepristone (MF), popularly known as “RU486” or “abortion pill,” is a prototype antiprogestin compound designed to be used as a blocker of progesterone action in the uterus (14). Yet, apart from this known contraceptive effect, MF has been reported to be effective as an anti-growth agent in endometriosis (15), leiomyomata (16), and breast (17,18), endometrial (19), prostate (20), gastric (21), and ovarian cancer cells (22). Our laboratory recently studied the molecular mechanisms of growth inhibition induced by MF in ovarian cancer cells and further defined its efficacy in an in vivo preclinical setting (23). We demonstrated that the cytostatic effect of MF in ovarian cancer is associated with inhibition of DNA synthesis, blockage of the cell cycle at the G1-to-S phase transition, upregulation of cyclin dependent kinase (Cdk) inhibitors p21cip1 and p27kip1, and inhibition of cyclin dependent kinase 2 (Cdk2) activity. In vivo in nude mice, we also showed that MF significantly delayed the growth of ovarian carcinoma xenografts in a dose-dependent manner. Based on these previous results, we hypothesized that MF should be efficacious in preventing ovarian cancer repopulation if applied in between rounds of cisplatin (CDDP) therapy. We established an in vitro model of ovarian cancer cell repopulation occurring during courses of lethal CDDP therapy. Using this in vitro model system, we studied whether intertwining cytostatic concentrations of the antiprogestin MF in between courses of CDDP treatment is an efficacious strategy to prevent repopulation of ovarian cancer cells leading to a better treatment outcome.
Materials and methods
Cell lines and in vitro exposure of cells to drugs
The human ovarian carcinoma cell line OV2008 was obtained from Dr. Stephen Howell (University of California, San Diego) and was maintained in RPMI 1640 (Mediatech, Herndon, VA) supplemented with 5% heat inactivated fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 10 mM HEPES (Mediatech), 4 mM L-glutamine (Mediatech), 1 mM sodium pyruvate (Mediatech), 1 X non-essential amino acids (Mediatech), 100 IU penicillin (Mediatech) and 100 µg/ml streptomycin (Mediatech). SK-OV-3 ovarian cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were routinely maintained in RPMI 1640 (Mediatech) supplemented with 5% fetal bovine serum (Atlanta Biologicals), 10 mM HEPES (Mediatech), 4 mM L-glutamine (Mediatech), 0.45% D (+) glucose (Sigma Chemical Company, St. Louis, MO), 1 mM sodium pyruvate (Mediatech), 1 X non-essential amino acids (Mediatech), 100 IU penicillin (Mediatech), 100 µg/ml streptomycin (Mediatech), and 0.01 mg/ml human insulin (Roche, Indianapolis, IN). Both cell lines were cultured at 37°C in a humidified atmosphere in the presence of 5% CO2.
Treatment of the cells with MF (Sigma) was done from a 116.5 mM stock solution in DMSO. The maximal concentration of DMSO in medium was 0.02 % (v/v). In the long-term experiments, MF-containing fresh medium was replaced every two days. CDDP (cis-diamminedichloroplatinum II) (Sigma) treatment was done from a 3 mM stock solution in 0.9% NaCl. Cells were exposed to CDDP for only 1 h. Thereafter, the medium was replaced with fresh CDDP-free medium either containing or not containing MF.
Cell proliferation and viability
Triplicate cultures were trypsinized, pelleted by centrifugation at 500 g for 5 min, and washed with PBS. The cells were resuspended in the ViaCount reagent (Guava Technologies, Hayward, CA) and studied using the Guava ViaCount application in the Guava EasyCyte Mini microcapillary cytometer (Guava Technologies). This assay provides an absolute cell count and viability data on a cell suspension, automating results like cell counts in a hemocytometer chamber with the trypan blue dye exclusion method for assessing cell viability. The cells are drawn into a capillary flow cell of known dimensions at a precisely controlled rate for measured periods of time. Absolute cell counts are obtained by knowing the exact sampling volumes. Viable and non-viable cells are assessed by the differential permeability of two DNA-binding dyes in the reagent. One dye is membrane permeable and stains all nucleated cells. The other dye only penetrates cells with compromised membrane integrity (i.e. non-viable cells). The data are acquired and analyzed using the CytoSoft 4.1 software (Guava Technologies).
4’, 6-diamidino-2-phenylindole dihydochloride (DAPI) staining and phase contrast microscopy
Cells cultured on 8-well chamber slides were exposed to vehicle (0.9 % NaCl), 20 (OV2008 cells) or 50 (SK-OV-3 cells) µM CDDP for 1 h, and maintained in CDDP-free media thereafter. After 96 h, detached cells were collected, centrifuged at 500 g for 5 min, fixed and resuspended in 100% methanol, adhered to a microscope slide, and stained for 10 min with DAPI (Molecular Probes, Eugene, OR). Nuclear morphology was observed and photographed using a Zeiss Axiovert M200 inverted fluorescence microscope (Carl Zeiss, Thornwood, NY). Cells that remained adherent to the original chamber slide were also fixed in 100% methanol, stained with DAPI and photographed. All cell preparations were simultaneously photographed using a phase contrast objective.
Clonogenic survival assay
Two-hundred fifty or 500 viable cells were placed in 6-well plates and cultured for 7 days until colonies were large enough to be clearly discerned. At this point, the medium was aspirated, the dishes washed once with PBS, fixed with 100 % methanol for 30 min, and stained with a filtered solution of 0.5 % (w/v) crystal violet (Sigma) for 10 min. The wells were then washed with tap water and dried at room temperature. The colonies, defined as groups of ≥ 30 cells, were scored manually with the aid of a Nikon Diaphot inverted microscope (Nikon, Garden City, NY). Clonogenic survival was expressed as the number of colonies formed during the different treatment paradigms.
Cell cycle analysis
After treatment, cells were trypsinized, pelleted by centrifugation at 500 g for 5 min, washed with PBS, and fixed with 4 % paraformaldehyde. Cells were once again washed with PBS and pelleted by centrifugation at 500 g for 5 min. Then, approximately 100,000–200,000 cells were resuspended in 200 µl of cell cycle buffer [3.8 mM sodium citrate (Sigma), 7 U/ml RNase A (Sigma), 0.1 % (v/v) Triton X-100 (Sigma), and 0.05 mg/ml propidium iodide (Sigma)] at a concentration of 500–1000 cells/µl. Cells were analyzed for the capacity of their DNA to bind propidium iodide utilizing the Guava EasyCyte Mini microcapillary cytometer and the cell cycle application of the CytoSoft 4.1 software (Guava Technologies).
Bromodeoxyuridine (BrdUrd) labeling
Cells were cultured on sterile four-well chamber slides at a concentration of 15,000 cells per well and subjected to the various treatments. Six hours before the end of the experiment, cells were exposed to 10 µM BrdUrd (Molecular Probes). At the end of the incubation, cells were fixed with 4 % paraformaldehyde, and BrdUrd incorporated into the DNA was visualized by immunocytochemistry.
Immunocytochemistry
The slide-containing cells were incubated with 2N HCl for 10 min at room temperature. The acid was aspirated and neutralized by washing thee times with borate buffer (pH = 8.6). The slides were then washed with PBS and the endogenous peroxidase activity was neutralized with 3 % H2O2 in PBS for 20 min at room temperature. The slides were once again washed with PBS and blocked with 2.5 % normal horse serum in 0.2 % Tween 20 and 0.1 % Triton X-100 for 20 min at room temperature. The slides were incubated overnight at 4°C with an anti-BrdUrd monoclonal antibody (2 µg/ml, A-21300; Molecular Probes), anti-phospho-histone H3 (Ser10) (1 µg/ml; #06–570; Upstate Cell Signaling Solutions, Lake Placid, NY), or anti-PARP p85 fragment (1:100; #7341; Promega, Madison, WI). The slides were then washed in PBS, once again treated with 3 % H2O2, re-washed, incubated with an anti-mouse or anti-rabbit Ig ImmPress reagent (Vector Laboratories, Burlingame, CA) for 30 min at room temperature, and stained with Vector NovaRed Substrate (Vector Laboratories) for developing the peroxidase activity. After washing with tap water, the sections were dehydrated in serial alcohols to xylene and mounted. In the negative control slides the primary antibody was replaced with blocking solution. Random fields were viewed with a Nikon Diaphot inverted microscope, and the number of labeled and unlabeled cells was determined. At least 500 cells or all of the cells remaining on the slide were counted for each sample. Pictures were taken using a Leica CM1850 microscope (Leica Microsystems, Plymouth, MN).
SDS-PAGE and western blotting
Cells were scraped, pelleted, washed twice with PBS, and lysed by the addition of two volumes of radioimmunoprecipitation assay buffer (RIPA) containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 % NP-40 (Sigma), 0.25 % sodium deoxycholate (Sigma), 1 mM EDTA, 1 mM PMSF (Sigma), 1 µg/ml pepstatin (Sigma), 1 mM orthovanadate (Sigma) and 1 mM sodium fluoride (Sigma). Cells were disrupted by passing them though a 21 gauge needle, and gently rocked on ice for 30 min. Lysates were centrifuged at 16,000 g for 15 min at 4°C, and the supernatant was considered the whole cell extract, which was assayed for protein content by using the bicinchoninic acid method (BCA; Pierce, Rockford, IL). The whole cell extracts were appropriately diluted in 6 X concentrated electrophoresis sample buffer, boiled for 10 min, and stored at −80°C until electrophoresed. Equivalent amounts of protein (50 µg) per point were loaded in 12 % (w/v) acrylamide gels, subjected to SDS-PAGE and transferred to PVDF membranes. The blots were blocked in 5 % (v/v) nonfat milk in TBS containing 0.1 % (v/v) Tween 20 (T). Blots were then probed overnight with primary antibodies against poly (ADP-ribose) polymerase (PARP) (#9542; 1:1000; Cell Signaling Technology, Danvers, MA) or caspase-3 (#;9662; 1:1000; Cell Signaling Technology). The membranes were washed 3 × 5 min in TBS-T and incubated with 1: 10,000 dilution of peroxidase-conjugate secondary antibody (#111-035-003; Jackson ImmunoResearch Laboratories, West Grove, PA) for 30 min at room temperature. The blots were again washed, developed by chemiluminescence, and exposed to radiographic film. Blots were stripped and reprobed with an antibody directed against β-Actin (clone AC-15; 1:20,000; Sigma) to control for protein loading.
DNA fragmentation
Floating and adherent cells were pelleted and digested overnight at 50°C in a buffer composed of 100 mM NaCl, 10 mM Tris HCl (pH 8.0), 25 mM EDTA (pH 8.0), 0.5 % SDS and 0.1 mg/ml proteinase K (Life Technologies, Rockville, MD). The genomic DNA was extracted from the digested cells with phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v), precipitated, and digested for 60 min at 37°C with 1 µg/ml ribonuclease (deoxyribonuclease-free; Roche). After extraction and precipitation, an equal amount of DNA for each sample (2 µg) was separated by electrophoresis on a 2.5 % agarose gel, impregnated with SYBR Gold nucleic acid gel stain (Molecular Probes), examined using an ultraviolet transilluminator, and photographed with the Amersham Typhoon Fluorescence imaging system. A 100 bp DNA ladder (Promega) was utilized for determining the size of the fragments of DNA.
Statistical analysis
All data are reported as means ± SEM, and statistical significance was defined as P < 0.05. To compare cell growth, clonogenic survival, cell cycle distribution, and BrdUrd, cleaved PARP and phospho-histone H3 labeled nuclei among the experimental groups, one-way ANOVA followed by the Newman-Keuls’ multiple comparison test or two-way ANOVA followed by the Bonferroni’s multiple comparison test were used as appropriate. Whenever the experiment involved the comparison of only two groups, the Student t-test was used.
Results
Exposure of cells to CDDP for 1 h induces substantial death, yet culture repopulation ensues with time
The first step towards studying repopulation of ovarian cancer cells was to establish an in vitro approach in which cells are exposed to successive rounds of a lethal dose of CDDP and repopulation of remnant clones that escape the drug takes place in between those rounds. This repopulation, however, should not be the result of the development of chemoresistance, which frequently occurs in vitro when cancer cells become exposed to platinum derivatives for extended periods of time. OV2008 cells that are highly sensitive to platinum were exposed to thee rounds of CDDP for 1 h within a period of 36 days. This allotted time is not sufficient for the cells to develop platinum resistance, which has been reported to occur after several months of drug exposure (24). In addition, in the in vitro approach of repopulation we should discriminate tumor cells that escape CDDP toxicity and can repopulate a tumor (i.e. clonogenic cells likely to represent tumor initiating cells), from cells that are alive yet lethally damaged and not able to proliferate. Such distinction was achieved by evaluating the clonogenic capacity of the cells along with the treatment protocol. The concentration of CDDP utilized for 1 h to achieve lethality in OV2008 cells was 20 µM. This concentration more than quadruples the reported 50% growth inhibition concentration (IC50) measured in this cell line [3.5 µM for a 1-h exposure to CDDP as assayed by clonogenic survival (25)], and has been shown to trigger cell death via a caspase-dependent apoptotic process (26). We confirmed this lethality in our laboratory. Fig. 1A shows that when OV2008 cells were exposed for 1 h to 20 µM CDDP, fragmented and sub-diploid genomic DNA accumulated 72 h later, which is indicative of the presence of apoptotic cells based on a deficit in their DNA content. The lethality of CDDP was further visualized by the morphological features of apoptosis displayed by the non-adherent cells 96 h after exposure to the drug (Fig. 1B). The involvement of the executor caspase-3 in the cell death process induced by CDDP was confirmed by the cleavage of inactive procaspase-3 to a presumably active 17 kDa cleaved form, which is compatible with the cleavage of the caspase-3 target, poly (ADP-ribose) polymerase (PARP) beginning 48 h after CDDP removal (Fig. 1C). The damage induced by CDDP was further visualized by phase contrast microscopy (Fig. 1D). Four days after the 1-h CDDP exposure most of the cells were non-adherent and dead, and of the few remaining adherent cells the majority was enlarged and vacuolated, likely undergoing degeneration. Only scarce normal-looking cells could be observed in the culture at this time. By day 8 after CDDP exposure, although some large vacuolated cells still remained adherent, cells of similar sizes to those of untreated cultures were evident. Twelve days after the exposure to CDDP, the population of normal-looking cells massively covered the culture plate, whereas the large vacuolated cells detached and were removed with the change in culture media. Collectively, results in Fig. 1 clearly show that exposure of OV2008 cells to 20 µM CDDP for 1 h is lethal for the vast majority of cells, yet there are remnant adherent cells that are capable of repopulating the culture.
Figure 1.

Lethality of 1-h exposure of OV2008 cells to 20 µM CDDP and the fate of residual cells that survive treatment. OV2008 cells were seeded and allowed to attach to the plate surface for 48 h. Cells were exposed to either vehicle (0.9 % NaCl) or 20 µM CDDP for 1 h and cultured in CDDP-free media thereafter. (a) 72 h following CDDP exposure, the cells were pelleted, and either permeabilized, labeled with propidium iodide, and the sub-G1 DNA content assessed by microcapillary cytometry, or the genomic DNA was isolated, separated by agarose electrophoresis, stained, and photographed. Bar, mean ± SEM. *, P < 0.05 relative to values at 0 h. (b) 96 h after CDDP treatment, the non-adherent cells were re-adhered to a microscope slide, stained with DAPI, photographed using a fluorescence microscope and compared against adherent cells of vehicle-treated cultures. Arrows, apoptotic nuclei. Arrowhead, cell undergoing mitosis. Scale bar = 20 µm. (c) 48 h after CDDP exposure, whole protein extracts where obtained and separated by electrophoresis, and immunoblots were probed with the indicated antibodies. (d) Cells that remained adherent following exposure to CDDP are shown as photographed by phase contrast microscopy. Arrows, large vacuolated cells likely undergoing degeneration. Arrowheads, cells undergoing repopulation. Scale bar = 20 µm.
We then exposed OV2008 cells to 3 rounds of 20 µM CDDP for 1 h, 12 days apart, and evaluated cell count and clonogenic capacity of surviving cells every 4 days. The first dose of CDDP reduced cell number by more than half when measured 4 days after CDDP exposure, yet by day 12, the number of cells was 6-fold higher than that measured on day 4 (Fig. 2A). The second dose of CDDP given on day 12 led to a reduced cell number 4 days later (day 16), but cell number was increased on days 20 and 24 to values even higher than those observed on day 12. The third CDDP treatment administered on day 24 radically reduced the number of cells by day 28, yet once again growth resumed thereafter. Fig. 2B shows, along with the treatment paradigm, that the percentages of cellular particles allocated to the sub-G1 and S regions of the cell cycle histogram displayed a clear inverse relationship to each other. After each time CDDP impacted the cells, a large increase in the percentage of cellular particles with hypodiploid DNA content was observed, in association with an abrupt decline in the percentage of cells with DNA content consistent with S phase transit. Conversely, when the cells began repopulating the culture, a decline in the percentage of hypodiploid DNA-containing cellular particles was observed in coincidence with an increased percentage of cells with S phase-like DNA content. The lethality induced by CDDP and the culture repopulation that followed is further supported by the decline in cell viability measured after each CDDP exposure, which was followed by an increase in culture viability just before the subsequent CDDP treatment (Fig. 2C). When the viable cells shown in Fig. 2C were subjected to a clonogenic survival assay to assess their long-term proliferation capacity, it became apparent that the number of colonies derived from presumably repopulating cells that escaped CDDP lethality paralleled changes in the number of cells depicted in Fig. 2A. Large declines in clonogenic survival were observed any time CDDP was given, followed by progressive increases in clonogenicity in between the CDDP treatment intervals (Fig. 2D). To determine the growth capacity of cells present on the plates after the cultures had been exposed to 3 rounds of CDDP, cells on day 42 after being subjected to the intermittent 1-h CDDP treatment paradigm were trypsinized, and their proliferation capacity was compared against that of cells never exposed to CDDP. Results shown in Table 1 indicate that growth rates of untreated or CDDP-pretreated cells after 48 or 72 h of culture were indistinguishable from one another, suggesting that the repopulating cells escaping CDDP lethality did not accumulate any apparent damage that could affect their subsequent capacity to reproduce. Collectively, results in Fig. 2 and Table 1 suggest that although CDDP kills a large proportion of OV2008 cells, there are remnant cells that escape CDDP lethality and repopulate the culture at a pace indistinguishable from that of cells never treated with CDDP.
Figure 2.

Effect of thee time-spaced courses of 20 µM CDDP for 1 h on number, cell cycle kinetics, cell viability, and clonogenic survival of adherent OV2008 cells. Cells were seeded and allowed to attach to the plate for 48 h. Plates were then treated with vehicle (0.9 % NaCl) or 20 µM CDDP for 1 h (Day 0). CDDP was then removed and fresh medium was replaced. The treatment with CDDP was repeated on days 12 and 24. Every second day the floating cells were removed and fresh medium was added. Every 4 days, one aliquot of cells was exposed to exclusion fluorochomes to determine number of cells (a) and their percent viability (c) using a microcapillary cytometer. (b) A second cell aliquot was subjected to permeabilization, labeling with propidium iodide, and the cell cycle distribution was assessed. Represented are the kinetic distribution of cells with sub-G1 DNA content and cells with DNA content pertinent to S phase. (d) Five hundred viable cells were placed in 6-well plates and cultured for 7 days to assess for colony formation (clonogenic survival). Arrows, intermittent 1-h CDDP treatment. Values represent the means of a representative experiment performed in triplicates ± SEM. This experiment was repeated five times with similar outcomes. *, P < 0.05 relative to values on day 16; #, P < 0.05 relative to values on day 4; †, P < 0.05 relative to values on day 28; §, P < 0.05 relative to values on days 16 and 20.
Table 1.
Cells that repopulated after 3 rounds of CDDP grow at a similar pace as cells never treated with CDDP.
| Number of Cells (× 105) | ||
|---|---|---|
| Hours | OV2008 (Untreated) | OV2008 (CDDP pretreated) |
| 0 | 2.28 ± 0.16 | 1.83 ± 0.17 |
| 48 | 3.98 ± 0.37 | 3.88 ± 0.48 |
| 72 | 8.08 ± 0.57 | 7.58 ± 0.32 |
Cells were treated for 1 h with 20 µM CDDP every 12 days for a total of 3 rounds on days 0, 12, and 24. Cells that were adherent on day 42 were trypsinized and replated in CDDP-free media. Once seeding was completed, the number of cells was assessed after 0, 48 or 72 h of culture. The growth of CDDP-pretreated cells was compared against the growth of an equal number of cells that never received CDDP and that were cultured in parallel under similar conditions. At the end of the incubation, cells were trypsinized and counted in a microcytometer. Data are expressed as the mean ± SEM of triplicate cultures.
Intertwining cytostatic doses of MF in between CDDP-free treatment intervals prevents repopulation of cells escaping the lethality of CDDP
OV2008 cells were treated for 1 h with 20 µM CDDP. The treatment was removed and the cells were incubated in media containing 0, 5, 10 or 20 µM MF for 12 days. We have previously shown that these concentrations of MF induce cell cycle arrest without cell killing (23). Fig. 3A shows that the number of adherent cells in cultures not receiving MF declined robustly on days 4 and 8 following the removal of CDDP, yet on day 12 repopulation is apparent as shown by the increase in cell number. When MF was present in the culture media, the reduction in number of adherent cells on days 4 and 8 following CDDP removal was significantly enhanced in a dose-dependent manner. Remarkably on day 12, MF was able to completely abrogate the repopulation that occurred in the MF untreated cultures. The effect of MF was further confirmed in a clonogenic survival assay (Fig. 3B). Viable cells from previous experiment and obtained on day 4 after CDDP removal show very low clonogenic capacity, which however increased on day 8, followed by an even higher increase on day 12. Conversely, the clonogenic capacity of cells chronically exposed to MF after the removal of CDDP was negligible on day 4, and remained very low on days 8 and 12 compared to cells only exposed to CDDP. Because the number of OV2008 cells that remained in the cultures receiving MF after the first round of CDDP was almost negligible, the following rounds with CDDP treatment were not conducted. In order to confirm the long-term effects of CDDP treatment plus MF, we modeled repopulation following lethal CDDP in a less sensitive cell line, SK-OV-3 (27,28), aiming to perform successive rounds of CDDP and, at the same time work with a manageable number of remaining cells when MF was also present. Because the growth rate of SK-OV-3 cells is slower than OV2008, we modeled repopulation in between 24-day intervals. Preliminary experiments with SK-OV-3 cells allowed us to select 1-h exposure to 50 µM CDDP as a lethal treatment paradigm. Fig. 4A shows that when SK-OV-3 cells were exposed to 50 µM CDDP for 1 h, cellular fragments with hypodiploid DNA content accumulated 72 h later. The lethality of CDDP at this concentration in SK-OV-3 cells was further confirmed by the fragmentation of the genomic DNA and the apoptotic morphology of the non-adherent cells. Upon removal of the first dose of CDDP, the lethality induced led to more than 7-fold reduction in number of adherent cells 16 days later, yet repopulation began occurring on day 24 (Fig. 4B), which was further confirmed by the clonogenic survival capacity of the cells (Fig. 4D). The second dose of CDDP on day 24 did not cause any apparent reduction in the number of adherent cells by day 28 (Fig. 4D), although the long-term lethality of CDDP was manifested by the reduced clonogenicity of viable cells taken from day 28 cultures (Fig. 4D). The repopulation of SK-OV-3 cells following the second round of CDDP was manifested more clearly on days 40 and 48, when the clonogenic capacity of adherent cells increased to levels similar to those of day 24, demonstrating a long-term inefficacy for the second round of CDDP in these cells. Seven days after the first 1-h CDDP exposure, many of the cells were non-adherent and dead. Of the remaining adherent cells the majority was enlarged (Fig. 4C, upper left corner). On day 15 following the first CDDP exposure, an emerging population of cells was observed (Fig. 4C, upper right corner) with similar appearance to those of untreated cultures (Fig. 4C, inset). When 20 µM MF was added to the culture media following the removal of the first CDDP treatment, the repopulation of SK-OV-3 cells, clearly quantified on day 24 in CDDP-only treated cultures, was markedly abrogated (Figs. 4B and D). Furthermore, repopulation was also inhibited in the MF treated cultures following the second round of CDDP (Figs. 4B and D). Moreover, MF modified notably the morphology of adherent cells. When 20 µM MF was present in the culture media for 7 or 15 days following CDDP treatment, most of the remaining adherent cells presented with spindle-shaped morphology (Fig. 4C, lower panels). Altogether, results in Fig. 3 and Fig. 4 clearly demonstrate that the presence of cytostatic concentrations of MF following removal of a lethal dose of CDDP, prevents the repopulation that otherwise occurs from remnant cells that escape the lethality of the platinum agent.
Figure 3.

Effect of chronic cytostatic concentrations of MF on the fate of OV2008 cells after exposure to one round of CDDP. Cells were seeded and allowed to attach to the plate surface for 48 h and thereafter treated with 20 µM CDDP for 1 h. The drug-containing medium was then removed and fresh medium with 0, 5, 10, or 20 µM MF was replaced every 2 days. Every 4 days number of adherent cells was measured by microcapillary cytometry (a), whereas the clonogenic capacity of 250 viable cells was studied in a clonogenic survival assay (b). Values represent the means of a representative experiment performed in triplicates ± SEM. This experiment was repeated 3 times with similar outcomes. *, P < 0.05 relative to MF-free control values on day 4; †, P < 0.05 relative to time-matched MF-free controls.
Figure 4.

Effect of chronic cytostatic concentrations of MF on the fate of SK-OV-3 cells exposed to 2 rounds of CDDP. SK-OV-3 were seeded, allowed to attach to the plate surface for 48 h, exposed to vehicle (0.9 % saline) or 50 µM CDDP for 1 h, and subsequently cultured in CDDP-free medium. (a) 72 h or 96 h after CDDP treatment, cells were respectively either pelleted, permeabilized, labeled with propidium iodide, and the sub-G1 DNA content was assessed by microcapillary cytometric analysis, or instead the genomic DNA was isolated, separated by agarose electrophoresis, stained, and photographed (upper panels). *, P < 0.05 relative to values at 0 h. Bar = mean ± SEM. 96 h after CDDP treatment, non-adherent cells were re-adhered to a microscope slide, stained with DAPI, photographed using a fluorescence microscope and compared against adherent cells of vehicle-treated cultures (lower panels). Arrows, apoptotic nuclei. Scale bar = 20 µm. In a second experiment, cells were treated with 50 µM CDDP for 1 h on days 0 and 24. After each treatment, CDDP was removed and the cells were cultured without or with MF every 2 days. Every 4 days number of adherent cells was measured by microcapillary cytometry (b), whereas the clonogenic capacity of viable cells was studied in a clonogenic survival assay (d). Values in panels (b) and (d) represent the means of a representative experiment performed in triplicates ± SEM. This experiment was repeated 4 times with similar outcomes. *, P < 0.05 relative to MF-free control values on day 4; #, P < 0.05 relative to MF-free control values on day 28; †, P < 0.05 relative to time-matched MF-free controls. (c) Morphology of adherent cells observed 7 or 15 days following exposure to CDDP alone (Control) or to the combination of CDDP and 20 µM MF. The inset in the upper right panel represents normally growing, untreated SK-OV-3 cells. Scale bar = 20 µm.
MF inhibits cell cycle progression inducing S + G2/M phase arrest in ovarian cancer cells following CDDP exposure
To examine the effect of MF on the kinetics of the cell cycle following CDDP treatment, the proportion of cells cycling was assessed by analyzing DNA synthesis using BrdUrd labeling in OV2008 cell cultures. Cells were exposed to 20 µM CDDP for 1 h and then incubated in the presence of 0, 5, 10 or 20 µM MF for 10 days. Six hours before the end of the experiment, the cells were pulsed with 10 µM BrdUrd and the incorporation of BrdUrd into newly synthesized DNA was assessed by immunocytochemistry. Figure 5 shows that the percentage of BrdUrd-labeled nuclei significantly increased in the cultures that had received CDDP compared to untreated OV2008 cells growing exponentially. MF significantly reduced, in a dose-dependent manner, the percentage of BrdUrd-labeled nuclei when compared to cultures receiving only CDDP. Presence of 20 µM MF for 10 days in cells not receiving CDDP also led to a significant inhibition of DNA synthesis, similar to what we observed when OV2008 cells were exposed to MF for only 24 h (23). To further evaluate the effect of MF on cell cycle traverse in cells pre-treated with CDDP, OV2008 cells were exposed to CDDP for 1 h and then to CDDP-free medium containing 20 µM MF for 4 or 8 days. The cell cycle distribution of OV2008 cells untreated or exposed to only MF was also included in the study. At the end of the experiment the cells were stained with propidium iodide and the allocation of cells within the cell cycle was analyzed by microcytometry. Results in Table 2 indicate that MF-only treatment for 4 or 8 days induced an accumulation of cells in G1 confirming our previous observation after 3-day exposure (23). Four days after the 1-h CDDP exposure there was a significant increase in sub-G1 DNA content, a large decline in the fraction of cells transiting G1, and an increase in the proportion of cells with hyperploid DNA content. Eight days after CDDP treatment, although the proportion of cells with hypodiploid and hyperploid DNA contents were still elevated when compared to untreated cultures, there was a significant proportion of cells transiting G1 compared to the values observed on day 4. When MF followed CDDP, there was a higher proportion of cells transiting G1 compared to cultures receiving only CDDP 4 days after cisplatin removal. On day 8, however, the proportion of cells transiting G1 in the CDDP plus MF group was lower compared to the CDDP-only counterpart, and most notably, the combination treatment was characterized by a robust increase in the number of cells allocated to the S + G2/M phases of the cell cycle. Together, DNA synthesis studies and cytometry data demonstrate that abrogation by MF of the repopulation following CDDP-induced lethality associates with inhibition of DNA synthesis and accumulation of the ovarian cancer cells at the S + G2/M transit.
Figure 5.
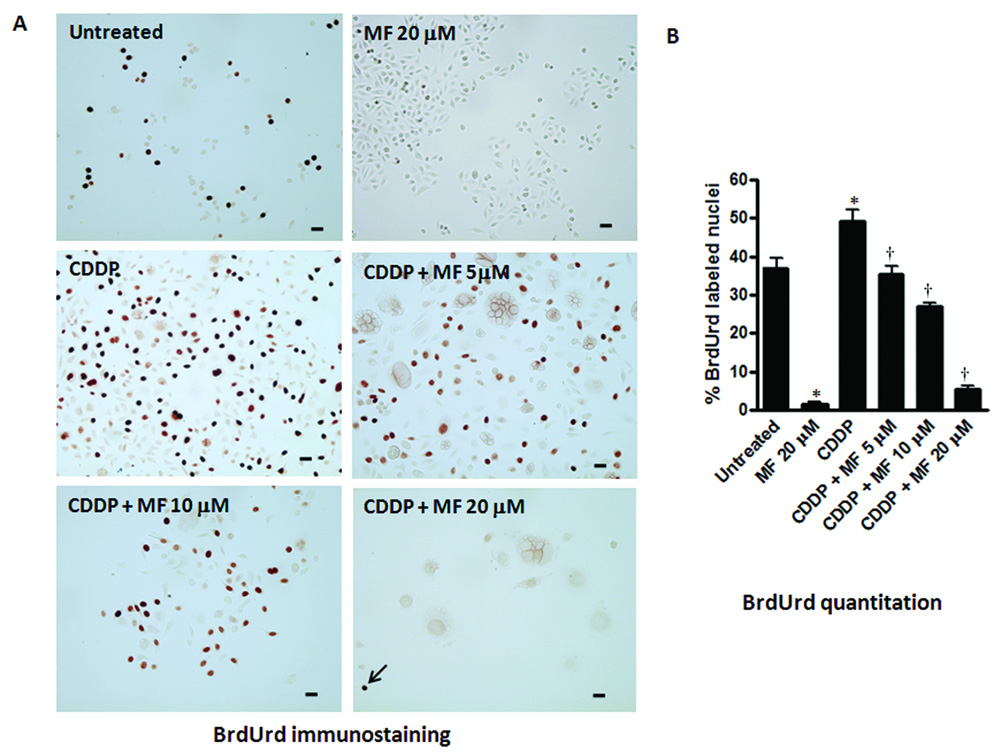
Effect of cytostatic concentrations of MF on DNA synthesis in OV2008 cells studied 10 days after being exposed to a lethal concentration of CDDP for 1 h. Cells were seeded and allowed to attach to the plate surface for 48 h, and exposed to vehicle (0.9 % NaCl) or 20 µM CDDP for 1 h. The treatment was then removed and fresh media without or with 5, 10, or 20 µM MF was replaced every 2 days. Ten days after CDDP exposure, the cells were pulsed with 10 µM BrdUrd for 6 h, washed with PBS, fixed with 4% paraformaldehyde, and subjected to immunocytochemistry using an anti-BrdUrd antibody (a). BrdUrd positive nuclei were quantified using a microscope with 400 X magnification and at least 500 cells were counted for each sample (b). The arrow indicates a scattered BrdUrd positive cell in the culture receiving CDDP plus 20 µM MF. Untreated cells in their logarithmic phase of growth, and cells that received only 20 µM MF for 10 days were also included in the study. The experiment was done in triplicates and the results are expressed as percentage of BrdUrd positive cells. Scale bar = 20 µm. Bars, mean ± SEM. *, P < 0.05 compared to untreated; †, P < 0.05 compared to CDDP.
Table 2.
Effect of MF on cell cycle kinetics in OV2008 cells repopulating after CDDP exposure.
| Untreated | MF | CDDP | CDDP + MF | MF | CDDP | CDDP + MF | |
|---|---|---|---|---|---|---|---|
| Day 4 |
Day 8 |
||||||
| % Sub-G1 | 2.03 ± 0.14 | 2.23 ± 0.18 | 52.3 ± 1.67* | 20.6 ± 1.45*† | 15.7 ± 4.93* | 48.7 ± 1.58* | 19.8 ± 1.56*† |
| % G1 | 56.1 ± 1.72 | 66.1 ± 0.92* | 3.22 ± 0.37* | 48.8 ± 1.32*† | 65.1 ± 2.46* | 17.1 ± 1.89*# | 7.98 ± 1.60*† |
| % S + G2/M | 33.5 ± 1.21 | 25.3 ± 0.90* | 21.6 ± 1.67* | 21.8 ± 1.61* | 14.8 ± 0.79* | 19.5 ± 1.31* | 53.3 ± 1.73*† |
| % > 4N | 8.13 ± 0.61 | 6.40 ± 1.04 | 22.9 ± 1.44* | 8.95 ± 0.86 | 4.43 ± 2.15 | 14.7 ± 0.81* | 21.6 ± 2.05*† |
Cells were treated with vehicle (0.9 % NaCl) or 20 µM CDDP for 1 h. The treatment was removed and fresh media without (untreated) or with 20 20 µM MF was replaced every 2 days. Four or 8 days after CDDP exposure, the cells were analyzed for the capacity of their DNA to bind propidium iodide by microcytometric analysis. In the groups receiving CDDP + MF we were unable to discriminate with certainty the cells that uptake propidium iodide with intensity consistent with DNA content pertaining the S or the G2/M phases of the cell cycle. Consequently, the two cell populations were measured as a whole in all experimental groups to allow comparisons. Data are mean ± SEM (n = 6, except for the group receiving MF alone with n = 3).
P < 0.05 compared to untreated
P < 0.05 compared to the time-matched CDDP.
P < 0.05 compared to CDDP on day 4. The experiment was repeated twice with similar outcomes.
Chronic exposure MF enhances the lethality of CDDP in association with accumulation of cells transiting mitosis
Results in Fig. 3 show that OV2008 cultures exposed to CDDP for 1 h followed by chronic exposure to 20 µM MF did not have sufficient remaining viable cells to allow performing a second treatment with CDDP, suggesting an added lethality to the cytostatic effect of MF. To study whether the abrogation by MF of cell repopulation that followed CDDP-induced lethality is only limited to a cytostatic effect of the antiprogestin that prevents the cells from reproducing, or there is an added lethality of MF when it follows CDDP, OV2008 cells were either cultured in the presence of vehicle for 8 days, treated with only 20 µM MF for 8 days, treated with CDDP for 1 h and then in CDDP-free media for 8 days, or exposed to CDDP for 1 h followed by 20 µM MF for 8 days. At the end of the experiment the cells were subjected to immunocytochemical analysis for cleaved poly (ADP-ribose) polymerase (PARP), a widely accepted marker for cell death (29). Results displayed in Fig. 6A and B show that untreated cultures do not have cells expressing cleaved PARP and that cultures receiving MF-only treatment for 8 days show rare cells labeled for the marker. Instead, in CDDP-treated cultures, ~ 10% of the cells labeled for cleaved PARP. This labeling appears to be limited to the large vacuolated cells previously described by phase contrast and shown in Fig. 1D. Remarkably, cultures exposed to MF following CDDP show a highly significant ~ 4-fold increase in the percentage of cleaved PARP positive cells, which was not limited to the large vacuolated cells, but also observed in cells of intermediate and apparently normal sizes. There is evidence in the literature for a targeted compound which when acting individually induces G1 cell cycle arrest, but when given after another drug that causes genotoxic stress – such as CDDP – it programs the cell to bypass the G2 DNA damage checkpoint, triggering an unscheduled mitosis (30). Thus, we evaluated whether the observed increase in the proportion of cells with DNA content consistent with S + G2/M involved an increased percentage of cells within the mitotic phase. Normally growing OV2008 cells, cells receiving 20 µM MF for 10 days, cells receiving CDDP for 1 h and then CDDP-free media for 10 days, or cells treated with the combination of CDDP for 1 h followed by 20 µM MF for 10 days were studied for their immunocytochemical expression of Ser-10 phosphorylated histone H3, a hallmark for mitotic transit (31,32). Results depicted in Fig. 6C and D show that, as expected, the percentage of cells transiting mitosis in an untreated unsynchonized culture is ~ 2%. This value does not change with MF and slightly increases with CDDP exposure alone, suggesting that the cells may be undergoing accelerated repopulation (8). Most interestingly, when MF treatment followed CDDP exposure, the percentage of cells expressing phosphorylated histone H3 increased ~ 30-fold compared to untreated controls, suggesting that the vast majority of cells that still remain adherent to the culture plate are transiting mitosis. The remarkable accumulation of cleaved PARP positive cells and the increase in percentage of cells expressing phosphorylated histone H3 (Fig. 6), together with the reduced clonogenic capacity of the surviving cells (Fig. 3B), suggest that when MF treatment follows CDDP exposure, it potentiates CDDP-induced lethality and the cells die most likely as a consequence of an abnormal mitosis.
Figure 6.
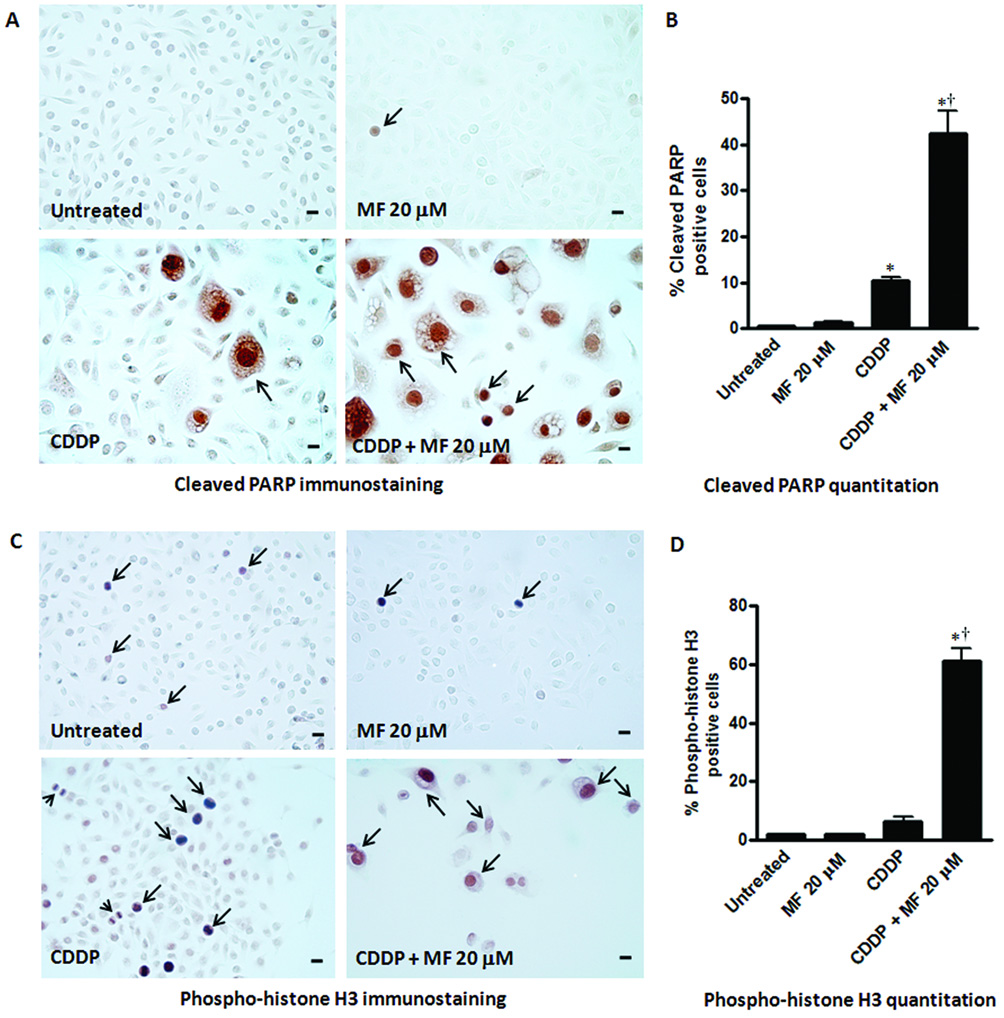
Effect of a cytostatic concentration of MF on the expression of cleaved poly (ADP-ribose) polymerase (PARP) and phospho-histone H3 in OV2008 cells studied respectively 8 or 10 days after being exposed to a lethal concentration of CDDP for 1 h. Cells were seeded for 48 h, and then treated with vehicle (0.9 % NaCl) or 20 µM CDDP for 1 h. CDDP was then removed and fresh media without or with 20 µM MF was replaced every 2 days. As control groups, untreated cells in their logarithmic phase of growth, and cells that received only 20 µM MF for 8 or 10 days were included in the study. At the end of the experiment, adherent cells were washed, fixed with 4% paraformaldehyde, and subjected to immunocytochemistry using an antibody that recognizes only the cleaved form of PARP (a), or phospho-histone H3 (c). Quantitation of cleaved PARP (b) and phospho-histone H3 (d) positive nuclei were done using a microscope with 400 X magnification and at least 500 cells were counted for each sample. The experiment was done in triplicates and the results are expressed as percentage of cleaved PARP or phospho-histone H3 positive cells. Arrows indicate examples of positive labeling. In panel (a) notice that in cultures receiving CDDP + MF 20 µM, cleaved PARP positive staining is observed not only in large degenerated cells, but also in smaller cells. In panel (c), arrowheads represent figures of mitosis. Bars, mean ± SEM. Scale bar = 20 µm. *, P < 0.05 compared to untreated; †, P < 0.05 compared to CDDP.
Discussion
We have established an in vitro approach whereby repopulation of ovarian cancer cells following multiple rounds of CDDP therapy ensues. We also confirmed our hypothesis by demonstrating that MF was efficacious in preventing the repopulation of ovarian cancer cells occurring between platinum treatment intervals. In addition, we found that chronic exposure to MF after platinum treatment seems to enhance platinum killing efficacy.
The majority of in vitro studies reported in the literature involving treatment of ovarian cancer cells with CDDP were performed analyzing the response to one-time exposure to the drug (26,33–36). An array of doses and exposure times can be found in those studies. Whereas some investigators exposed ovarian cancer cells to CDDP for 24, 48 or 72 h (34–36), others intended to mimic the in vivo therapeutic approach in humans by exposing ovarian cancer cells to CDDP for only 1 h (26,33). Even though this second approach appears closer to in vivo therapy, it does not allow determining if tumor repopulation occurs in between successive treatments with CDDP. Thus, our objective was first to determine if tumor cell repopulation occurred between CDDP rounds. To model repopulation of ovarian cancer cells in vitro we selected the OV2008 cell line, which has been reported to be highly sensitive to CDDP (25,33,37), and the SK-OV-3, a cell line less sensitive to CDDP (27). We utilized the 1-h exposure paradigm and a previously established lethal dose, which was, as expected, higher for SK-OV-3 cells than for OV2008 cells. The results show that 1-h exposure to CDDP was sufficient to induce lethal damage as demonstrated by morphological and biochemical features of apoptotic cell death displayed by OV2008 and SK-OV-3 cultures within 96 h following CDDP removal. Although the majority of the cells in the culture succumbed to the lethality of CDDP, there were isolated cells that survived the treatment. These cells, because of their scarcity on the culture plate, may be easily missed in routine cell cultures if long-term follow-up is not conducted. When such a population of remnant cells is exposed to normal culture conditions, they reproduce and repopulate the culture. We were able to document repopulation of highly sensitive OV2008 cells after 3 episodes of CDDP treatment and of less sensitive to CDDP, SK-OV-3 cells after 2 episodes of drug exposure. This is the first study to document a model system of ovarian cancer cell repopulation in vitro following CDDP treatment. Very recently repopulation of SK-OV-3 ovarian cancer cells in response to 3 intermittent doses of paclitaxel was reported (38). An increase in cell proliferation and clonogenic survival was observed in the intermittent group with each treatment gap, indicating a gradual acceleration in repopulation rate. In our study, OV2008 cells repopulating after CDDP exposure show an apparent increased ability to incorporate BrdUrd into their DNA when compared with exponentially growing, untreated control cells. These data suggest that in this model system there is either a synchonization of the cells in culture after the CDDP exposure, which could explain the large number of cells synthesizing DNA at the same time, or instead, the increased number of cells synthesizing DNA is a product of accelerated cell repopulation. The nature of the cells that escape CDDP lethality remains to be determined. When repopulation is studied in vivo, the regrowth phenomenon is usually attributed to cells that are more distant from the tumor blood supply, consequently being exposed to lower concentration of the drug compared to cells closer to blood vessels (8). In a cultured cell line, however, because cells usually grow in a monolayer, it is difficult to utilize such an explanation to account for cellular repopulation considering that we utilized a concentration of CDDP several fold higher than the IC50 documented for this cell line (25). Another possibility is that cells escaping platinum toxicity are indeed resistant to the drug. The short experimental time (a total of 36 days for OV2008 cells and of 48 days for SK-OV-3 cells), however, makes the repopulation as a consequence of emerging clones that resist CDDP very unlikely. It has been amply documented that development of CDDP resistance in vitro occurs when cells are exposed intermittently to CDDP over several months (24). Another explanation for the repopulation of ovarian cancer cells post-CDDP that we observed in vitro is based on the fact that the culture was not synchonized and that cells are more prone to CDDP killing at particular times during the cell cycle. For instance, tumor testicular germ cells in vitro are more sensitive to the lethality induced by CDDP during the G2/M phase of the cell cycle than in other phases (39). Finally, there is a possibility that indeed CDDP is killing the population of differentiated cancer cells representing the bulk of the culture, but not the scarce tumor initiating cells with the capacity to regenerate the culture, and that appear to be resistant to most common DNA damaging agents (40). The presence of tumor initiating cells in ovarian cancer cell lines, however, has yet to be determined. It is interesting to note that the cells repopulating the culture after being exposed to 3 rounds of CDDP treatment in our model system grow at the same pace as cells never exposed to CDDP, indicating that the cells do not seem to be genetically damaged by previous CDDP treatment.
MF has been known for displaying anti-growth properties in different human cell types (15–17,19–22). We recently began to elucidate the molecular mechanisms involved in this effect on ovarian cancer cell growth (23). We have shown that exposure of OV2008 cells to 20 µM MF for 24 h reduced synthesis of DNA more than 10-fold (23). A similar concentration of MF utilized chronically in between CDDP treatment intervals was sufficient to abrogate DNA synthesis occurring in repopulating cells that had been exposed to CDDP. The efficacy of MF preventing repopulation following CDDP was also manifested by reduced cell number and reduced clonogenic survival in both, the more sensitive to CDDP OV2008 cells, and the less sensitive to CDDP, SK-OV-3 cells. When ovarian cancer cells are exposed to only MF, the cell cycle is arrested in the G1 phase (22,23). In this study we confirm this assumption, yet it is noticeable that when MF was added to cells previously treated with CDDP, they tended to accumulate at the S + G2/M phases rather than in G1. This is interesting because the result may provide the rationale for a drug interaction between CDDP and MF, leading to a potentiation of platinum therapy by the antiprogestin. It is known that CDDP treatment leads to a transitory G2 cell cycle arrest, which is utilized by the cells as an opportunity to repair any damaged DNA. However, if the DNA damage is significant and the DNA repair mechanism cannot operate, the cells usually trigger their own demise (41). Thus, MF may be a disruptor of the DNA damage and repair pathways operating after CDDP exposure. Consequently the cells would enter an unscheduled mitosis with damaged DNA, which usually would trigger a cell death mechanism due to mitotic failure (42). Partial support for this hypothesis is that cells receiving the combination treatment of CDDP followed by MF show an elevated percentage of cells allocated to the M phase of the cell cycle suggested by 6-fold overexpression of the mitotic marker, phospho-histone H3, when compared to CDDP-only treated cultures. This result, together with the data showing that the cultures receiving CDDP followed by MF express 4-fold more cleaved PARP compared with cultures receiving only CDDP, suggest that cells chronically exposed to MF after receiving lethal platinum therapy not only are unable to repopulate, but also are likely to die by transiting into an unscheduled mitosis that could trigger death (i.e. mitotic death) by either apoptosis or necrosis. These two modalities of cell death have been recently reported to occur in ovarian carcinoma cell lines in response to DNA damaging agents (43). Thus, it is apparent that the cell death pathway triggered by the combination of CDDP plus MF needs further investigation.
MF may be interfering with early steps in the DNA damage response pathway that lead to a failure of cells to arrest in the G2 phase when challenged with CDDP. This rationale is supported by data generated utilizing the kinase inhibitor UCN-01 (7-hydroxystaurosporine), which as a single agent is able to induce G1 growth arrest in non-small-cell lung carcinoma similar to the effect observed in ovarian cancer cells treated with MF (23). When UCN-01 was used after CDDP, however, the combination was synergistic in terms of growth inhibition likely by reducing the time cells spend in the S or G2 phases to operate the DNA damage check point in order to allow for repair of the DNA damage induced by platinum (30).
We demonstrated that the cytostatic effect of MF in ovarian cancer cells associates with an abrupt reduction in the activity of Cdk2 (23). One of the most well-studied and accepted effects of Cdk2 is its critical involvement in promoting the transition of the cells in the cell cycle from G1 to the S phase (44). However, Cdk2 has also been implicated in DNA repair. For instance, the DNA repair machinery is dysfunctional in Cdk2 deficient cells and cells lacking the DNA repair component of BRCA1 are prone to cell death in response to Cdk2 inhibition (45). This role played by Cdk2 in the DNA repair process provides the rationale for a synergistic interaction of Cdk2 inhibition and DNA damaging agents in the killing of cancer cells and could also explain the potentiation by MF of CDDP-induced lethality of ovarian cancer cells. Another hypothesis as to how MF could facilitate the lethal effects of CDDP is based upon its capacity to abrogate the expression of the E2F1 transcription factor as recently reported by our laboratory (23). E2F1 is needed to regulate the expression of genes involved in the nucleotide excision DNA repair pathway (NER) (46), which is one major mechanism involved in the repair of CDDP-DNA adducts (6,7,47,48). Because in ovarian cancer increased expression of the endonuclease ERCC1 (excision repair cross-complementing-1) involved in NER has been correlated with CDDP resistance (49) and antisense RNA against ERCC1 sensitizes ovarian cancer cells to the lethality of CDDP (50), it is possible that MF potentiates CDDP lethality by interfering with the operation of the NER pathway.
The remarkable accumulation of cleaved PARP positive cells and the increase in percentage of phospho-histone H3 positive cells when MF follows CDDP, together with the reduced clonogenic capacity of surviving cells, suggests that MF potentiates CDDP lethality and that the combination of CDDP followed by MF triggers cell death as consequence of a failed mitosis. If our hypothesis holds true in vivo, the strategy of adding MF to the CDDP chemotherapeutic schedule in ovarian cancer should allow reducing either the number of CDDP cycles or the dose of CDDP without losing efficacy in terms of inhibition of tumor growth. Consequently, the scheduling of MF between courses of platinum-based therapy for human ovarian cancer has reasonable potential for improving treatment success.
Acknowledgments
This research was supported by Grant Number K22CA121991 from the National Cancer Institute, Grant Number 2 P20 RR016479 from the INBRE Program of the National Center for Research Resources, and funds from the University of South Dakota. We thank Ms. Micaela Telleria for editing the manuscript and Dr. Barbara Goodman for the critical reading of the manuscript.
References
- 1.DiSaia PJ, Bloss JD. Treatment of ovarian cancer: New strategies. Gynecol Oncol. 2003;90:S24–S32. doi: 10.1016/s0090-8258(03)00341-x. [DOI] [PubMed] [Google Scholar]
- 2.Martin VR. Ovarian cancer: An overview of treatment options. Clin J Oncol Nurs. 2007;11:201–207. doi: 10.1188/07.CJON.201-207. [DOI] [PubMed] [Google Scholar]
- 3.Naora H, Montell DJ. Ovarian cancer metastasis: Integrating insights from disparate model organisms. Nat Rev Cancer. 2005;5:355–366. doi: 10.1038/nrc1611. [DOI] [PubMed] [Google Scholar]
- 4.Ozols RF. Challenges for chemotherapy in ovarian cancer. Ann Oncol. 2006. pp. v181–v187. [DOI] [PubMed]
- 5.Bukowski RM, Ozols RF, Markman M. The management of recurrent ovarian cancer. Semin Oncol. 2007;34:S1–S15. doi: 10.1053/j.seminoncol.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Perez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anti-cancer agents in medicinal chemistry. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 7.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 8.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: An important cause of treatment failure. Nat Rev Cancer. 2005;5:516–525. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 9.Davis AJ, Tannock JF. Repopulation of tumour cells between cycles of chemotherapy: A neglected factor. Lancet Oncol. 2000;1:86–93. doi: 10.1016/s1470-2045(00)00019-x. [DOI] [PubMed] [Google Scholar]
- 10.Brade AM, Tannock IF. Scheduling of radiation and chemotherapy for limited-stage small-cell lung cancer: Repopulation as a cause of treatment failure? J Clin Oncol. 2006;24:1020–1022. doi: 10.1200/JCO.2005.04.9676. [DOI] [PubMed] [Google Scholar]
- 11.Licun W, Tannock IF. Selective estrogen receptor modulators as inhibitors of repopulation of human breast cancer cell lines after chemotherapy. Clin Cancer Res. 2003;9:4614–4618. [PubMed] [Google Scholar]
- 12.Wu L, Tannock IF. Effect of the selective estrogen receptor modulator arzoxifene on repopulation of hormone-responsive breast cancer xenografts between courses of chemotherapy. Clin Cancer Res. 2005;11:8195–8200. doi: 10.1158/1078-0432.CCR-05-1258. [DOI] [PubMed] [Google Scholar]
- 13.Wu L, Birle DC, Tannock IF. Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005;65:2825–2831. doi: 10.1158/0008-5472.CAN-04-3137. [DOI] [PubMed] [Google Scholar]
- 14.Philibert D, Moguilewsky M, Mary I, Lecaque D, Tournemine C, Secchi J, Deraedt R. Pharmacological profile of ru486 in animals. In: Baulieu EE, Segal SJ, editors. The antiprogesterone steroid ru486 and human fertility control. NY: Plenum Press; 1985. pp. [Google Scholar]
- 15.Fedele L, Berlanda N. Emerging drugs for endometriosis. Expert Opin Emerg Drugs. 2004;9:167–177. doi: 10.1517/eoed.9.1.167.32945. [DOI] [PubMed] [Google Scholar]
- 16.Steinauer J, Pritts EA, Jackson R, Jacoby AF. Systematic review of mifepristone for the treatment of uterine leiomyomata. Obstet Gynecol. 2004;103:1331–1336. doi: 10.1097/01.AOG.0000127622.63269.8b. [DOI] [PubMed] [Google Scholar]
- 17.Liang Y, Hou M, Kallab AM, Barrett JT, El Etreby F, Schoenlein PV. Induction of antiproliferation and apoptosis in estrogen receptor negative mda-231 human breast cancer cells by mifepristone and 4-hydroxytamoxifen combination therapy: A role for tgfbeta1. Int J Oncol. 2003;23:369–380. [PubMed] [Google Scholar]
- 18.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science. 2006;314:1467–1470. doi: 10.1126/science.1130471. [DOI] [PubMed] [Google Scholar]
- 19.Schneider CC, Gibb RK, Taylor DD, Wan T, Gercel-Taylor C. Inhibition of endometrial cancer cell lines by mifepristone (ru 486) J Soc Gynecol Investig. 1998;5:334–338. doi: 10.1016/s1071-5576(98)00037-9. [DOI] [PubMed] [Google Scholar]
- 20.El Etreby MF, Liang Y, Johnson MH, Lewis RW. Antitumor activity of mifepristone in the human LNCaP, LNCaP-C4, and LNCaP-C4-2 prostate cancer models in nude mice. Prostate. 2000;42:99–106. doi: 10.1002/(sici)1097-0045(20000201)42:2<99::aid-pros3>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 21.Li DQ, Wang ZB, Bai J, Zhao J, Wang Y, Hu K, Du YH. Effects of mifepristone on proliferation of human gastric adenocarcinoma cell line SGC-7901 in vitro. World J Gastroenterol. 2004;10:2628–2631. doi: 10.3748/wjg.v10.i18.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose FV, Barnea ER. Response of human ovarian carcinoma cell lines to antiprogestin mifepristone. Oncogene. 1996;12:999–1003. [PubMed] [Google Scholar]
- 23.Goyeneche AA, Caron RW, Telleria CM. Mifepristone inhibits ovarian cancer cell growth in vitro and in vivo. Clin Cancer Res. 2007;13:3370–3379. doi: 10.1158/1078-0432.CCR-07-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coley HM. Development of drug-resistant models. Methods Mol Med. 2004;88:267–273. doi: 10.1385/1-59259-406-9:267. [DOI] [PubMed] [Google Scholar]
- 25.Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M, Kuo YM, Rochdi M, Howell SB. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–6565. [PubMed] [Google Scholar]
- 26.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem. 2003;278:19245–19256. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 27.Ormerod MG, O'Neill C, Robertson D, Kelland LR, Harrap KR. Cis-diamminedichloroplatinum(ii)-induced cell death through apoptosis in sensitive and resistant human ovarian carcinoma cell lines. Cancer Chemother Pharmacol. 1996;37:463–471. doi: 10.1007/s002800050413. [DOI] [PubMed] [Google Scholar]
- 28.Melichar B, Hu W, Patenia R, Melicharova K, Gallardo ST, Freedman R. rIFN-gamma-mediated growth suppression of platinum-sensitive and -resistant ovarian tumor cell lines not dependent upon arginase inhibition. J Transl Med. 2003;1:5. doi: 10.1186/1479-5876-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scovassi AI, Poirier GG. Poly(ADP-ribosylation) and apoptosis. Mol Cell Biochem. 1999;199:125–137. doi: 10.1023/a:1006962716377. [DOI] [PubMed] [Google Scholar]
- 30.Mack PC, Gandara DR, Lau AH, Lara PN, Jr, Edelman MJ, Gumerlock PH. Cell cycle-dependent potentiation of cisplatin by ucn-01 in non-small-cell lung carcinoma. Cancer Chemother Pharmacol. 2003;51:337–348. doi: 10.1007/s00280-003-0571-6. [DOI] [PubMed] [Google Scholar]
- 31.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone h3 initiates primarily within pericentromeric heterochromatin during g2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 32.Taylor WR. Facs-based detection of phosphorylated histone h3 for the quantitation of mitotic cells. Methods Mol Biol. 2004;281:293–299. doi: 10.1385/1-59259-811-0:293. [DOI] [PubMed] [Google Scholar]
- 33.Mansouri A, Zhang Q, Ridgway LD, Tian L, Claret FX. Cisplatin resistance in an ovarian carcinoma is associated with a defect in programmed cell death control through xiap regulation. Oncol Res. 2003;13:399–404. doi: 10.3727/096504003108748410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Feng Q, Kim JM, Schneiderman D, Liston P, Li M, Vanderhyden B, Faught W, Fung MF, Senterman M, Korneluk RG, Tsang BK. Human ovarian cancer and cisplatin resistance: Possible role of inhibitor of apoptosis proteins. Endocrinology. 2001;142:370–380. doi: 10.1210/endo.142.1.7897. [DOI] [PubMed] [Google Scholar]
- 35.Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, Sakata M, Motoyama T, Kurachi H, Tasaka K, Murata Y. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279:23477–23485. doi: 10.1074/jbc.M313709200. [DOI] [PubMed] [Google Scholar]
- 36.Rasola A, Anguissola S, Ferrero N, Gramaglia D, Maffe A, Maggiora P, Comoglio PM, Di Renzo MF. Hepatocyte growth factor sensitizes human ovarian carcinoma cell lines to paclitaxel and cisplatin. Cancer Res. 2004;64:1744–1750. doi: 10.1158/0008-5472.can-03-2383. [DOI] [PubMed] [Google Scholar]
- 37.Fraser M, Leung B, Jahani-Asl A, Yan X, Thompson WE, Tsang BK. Chemoresistance in human ovarian cancer: The role of apoptotic regulators. Reprod Biol Endocrinol. 2003;1:66. doi: 10.1186/1477-7827-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vassileva V, Allen CJ, Piquette-Miller M. Effects of sustained and intermittent paclitaxel therapy on tumor repopulation in ovarian cancer. Mol Cancer Ther. 2008;7:630–637. doi: 10.1158/1535-7163.MCT-07-2117. [DOI] [PubMed] [Google Scholar]
- 39.Mueller S, Schittenhelm M, Honecker F, Malenke E, Lauber K, Wesselborg S, Hartmann JT, Bokemeyer C, Mayer F. Cell-cycle progression and response of germ cell tumors to cisplatin in vitro. Int J Oncol. 2006;29:471–479. [PubMed] [Google Scholar]
- 40.Kvinlaug BT, Huntly BJ. Targeting cancer stem cells. Expert opinion on therapeutic targets. 2007;11:915–927. doi: 10.1517/14728222.11.7.915. [DOI] [PubMed] [Google Scholar]
- 41.Sorenson CM, Barry MA, Eastman A. Analysis of events associated with cell cycle arrest at G2 phase and cell death induced by cisplatin. J Natl Cancer Inst. 1990;82:749–755. doi: 10.1093/jnci/82.9.749. [DOI] [PubMed] [Google Scholar]
- 42.Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: Mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 43.Vakifahmetoglu H, Olsson M, Tamm C, Heidari N, Orrenius S, Zhivotovsky B. DNA damage induces two distinct modes of cell death in ovarian carcinomas. Cell Death Differ. 2008;15:555–566. doi: 10.1038/sj.cdd.4402286. [DOI] [PubMed] [Google Scholar]
- 44.Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–149. doi: 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deans AJ, Khanna KK, McNees CJ, Mercurio C, Heierhorst J, McArthur GA. Cyclin-dependent kinase 2 functions in normal DNA repair and is a therapeutic target in BRCA1-deficient cancers. Cancer Res. 2006;66:8219–8226. doi: 10.1158/0008-5472.CAN-05-3945. [DOI] [PubMed] [Google Scholar]
- 46.Berton TR, Mitchell DL, Guo R, Johnson DG. Regulation of epidermal apoptosis and DNA repair by E2F1 in response to ultraviolet b radiation. Oncogene. 2005;24:2449–2460. doi: 10.1038/sj.onc.1208462. [DOI] [PubMed] [Google Scholar]
- 47.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 48.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, Yu JJ, Mu C, Yunmbam MK, Slavsky D, Cross CL, Bostick-Bruton F, Reed E. Association between the level of ERCC-1 expression and the repair of cisplatin-induced DNA damage in human ovarian cancer cells. Anticancer Res. 2000;20:645–652. [PubMed] [Google Scholar]
- 50.Selvakumaran M, Pisarcik DA, Bao R, Yeung AT, Hamilton TC. Enhanced cisplatin cytotoxicity by disturbing the nucleotide excision repair pathway in ovarian cancer cell lines. Cancer Res. 2003;63:1311–1316. [PubMed] [Google Scholar]