

Summary
Aquaporins are a family of ubiquitous membrane proteins that form a pore for the permeation of water. Both electron and X-ray crystallography played major roles in determining the atomic structures of a number of aquaporins. This review focuses on electron crystallography, and its contribution to the field of aquaporin biology. We briefly discuss electron crystallography and the two-dimensional crystallization process. We describe features of aquaporins common to both electron and X-ray crystallographic structures; as well as some structural insights unique to electron crystallography, including aquaporin junction formation and lipid-protein interactions.
Keywords: aquaporin, electron crystallography, lipid-protein interactions, membrane junction, channel
Aquaporins (AQPs) are a family of ubiquitous membrane proteins that form a pore for the permeation of water across biological membranes (1). The aquaporin family is divided into two subcategories: the aquaporins—those channels that allow only water to permeate the pore; and the aquaglyceroporins—channels that also allow glycerol and other small solutes to permeate (2, 3). A number of research groups worked to determine the structure of these water channels since their discovery, and now the atomic structures of a number of AQPs have been characterized (4). These structures played pivotal roles in deciphering key aspects of AQP regulation, substrate selectivity, and the proton exclusion mechanism. While X-ray crystallography played a major role in the history of the structural biology of AQPs, this review focuses on the role that electron crystallography played in determining the atomic structures of a number of AQPs.
ELECTRON CRYSTALLOGRAPHY
Electron crystallography is an evolving field of structural biology. Recent advancements in methodology and technology have led to a number of success stories of membrane protein structures that have been determined to resolutions that rival those achieved by X-ray crystallography. These recent advancements were mainly in sample preparation, in particular the double carbon sandwich technique (5); improved charge couple device (CCD) cameras that boost signal and reduce noise; development of the field emission electron source; as well as the development of a highly sophisticated helium-cooled stage (6). In addition, the development of molecular replacement and structure refinement protocols for electron crystallography has accelerated the data analysis (7, 8). These advancements have been perhaps most evident in the structural studies of aquaporins, in particular the case of the structure of AQP0 that was determined to 1.9 Å resolution (9).
The most widely used method of producing two-dimensional (2D) crystals for electron crystallography is by dialysis (6, 10, 11). Typically, cell membranes enriched in the membrane protein of interest are solubilized with a mild detergent, and the solubilized protein is purified by a combination of affinity, ion exchange, and/or size exclusion chromatography. Lipids are then added to the protein/detergent solution at a specific lipid-to-protein ratio (LPR). The LPR is chosen such that the lipid amount is low enough to stabilize the protein, but at the same time high enough to facilitate membrane formation and 2D crystallization. The lipid/protein/detergent solution is then placed in a dialysis button, sealed with a dialysis membrane and placed in a beaker containing crystallization buffer lacking the detergent. The lipids selectively associate with the protein while the detergent is slowly dialyzed out of the button. During this process, membranes or vesicles begin to form and proteins are inserted within the lipid bilayer. A number of parameters can be changed to facilitate this 2D crystallization process, such as the LPR, lipid type, buffer pH, salt concentration, divalent cation concentration, temperature, and the presence of any number of precipitants. Under the right crystallization conditions, the protein will form lateral crystal contacts within the vesicles, resulting in 2D crystals that can be used for electron crystallography.
Once 2D lipid-protein crystals are formed, conditions for specimen preparation for cryomicroscopy and for data collection must be chosen and carefully optimized. For cryogenic specimen preparation, molybdenum electron microscopy grids are overlaid with a clean layer of carbon support immersed in a thin layer of a cryo protectant such as glucose or trehalose. A small amount of the 2D crystals are then pipetted onto the grid and in this way become embedded by the cryo protectant. Excess sample is blotted off with a clean sheet of filter paper and the preparation is then flash-frozen by plunging the grid into liquid nitrogen. The grid is then transferred into a specialized electron microscopy cryo holder, and inserted into an electron cryomicroscope. High-resolution images of 2D crystals can then be recorded on film, or by a CCD camera, under low-dose conditions to minimize electron beam damage to the specimens. Alternatively, high-resolution electron diffraction data can be recorded directly.
For 2D protein crystals, the resolution of an image or of the electron diffraction data is not limited by the resolution of the microscope. Rather, the major limiting factor is damage to the specimen caused by the electron beam. Radiation damage is likely a two-step process involving the deposition of energy on the specimen by inelastic scattering leading to breakage of chemical bonds. Radicals are produced and ions or fragments of the molecule diffuse away resulting in a collapse of the initial structure. Radiation damage can be reduced significantly by lowering the temperature of the sample (6), and the development of electron microscopes equipped with a helium-cooled specimen stage has greatly improved the success in obtaining high-quality images and diffraction data. In the case of AQP0, electron diffraction data were obtained to a resolution of 3 Å under liquid nitrogen temperatures, but were improved to 1.9 Å resolution when the same 2D crystals were examined under helium temperatures at 4 K (7, 9). This significant improvement in resolution yielded a density map that was of high enough quality to resolve water molecules as well as a shell of lipids surrounding, and interacting with, the membrane protein (9).
AQUAPORIN STRUCTURE AND WATER PERMEABILITY
Aquaporin-1 was the first membrane protein identified as a water channel (12). AQP1 is a highly efficient water-selective aquaporin found in red blood cells, and since its atomic model was reported it has served as a structural archetype for studying the mechanism of water channel permeability and selectivity. The first atomic model of AQP1 was determined by the year 2000, to 3.8 Å resolution (13). At this resolution, amino acid side chains were visible and could be placed with reasonable accuracy. In fact, the 2.2 Å resolution X-ray structure of AQP1 that was determined a year later (14) was astonishingly similar to the 3.8 Å structure determined by electron crystallography (15).
Electron crystallographic studies of AQP1 2D crystals revealed that AQPs are square-shaped tetrameric assemblies (13) (Fig. 1A). All AQPs studied to date form similar tetrameric assemblies. Permeability studies using functional chimeras showed that the AQP1 tetramers contain four independent water channels (16). Each water channel is composed of a bundle of six membrane-traversing α-helices (H1-6) connected by five loops (loops A-E). The six transmembrane helices pack against one another as in a barrel, forming a hydrophilic water pore at the center of each monomer (Fig. 1B). Lining the water pore are two hydrophobic loops, loops B and E, which contain the conserved NPA motifs that are critical for maintaining proton gradients across the lipid bilayer (reviewed in ref. 4). These two loops insert into the channel from opposite sides of the membrane, placing their NPA motifs opposite each other at the center of the AQP channel. The loops then form a short helix and exit the channel on the same side of the membrane from which they entered (Fig. 1B). Both the amino and carboxy termini of the protein are located in the cytoplasm. While all AQPs share the same structural core architecture, there are some distinct structural variations in the loop regions as well as the cytoplasmic N- and C-termini suggesting functional and/or regulatory roles (4).
Figure 1.

Structure of aquaporin-1. A: Aquaporins are tetramers but each monomer forms a functional water pore (asterisk). B: Side view of an aquaporin-1 monomer. Six transmembrane helices pack against one another forming a barrel-like structure with the hydrophilic water pore at the center (green). The ar/R constriction site, or selectivity filter, is shown in red (Protein Data Bank accession No. 1FQY).
The key mechanism for substrate specificity in AQPs is a physical constriction in pore diameter (14). The water pathway in the AQP channels includes large extracellular and cytoplasmic vestibules connected by a narrow hydrophilic pore (Fig. 1B). AQP1 contains a constriction site, the ar/R site, also known as the selectivity filter. The ar/R site refers to conserved aromatic and arginine positions located just above the NPA motifs and correspond to the narrowest region of the water pore. In AQP1, the pore narrows to ∼3 Å in diameter—a diameter which is only marginally larger than the 2.8 Å diameter of a water molecule. In sharp contrast, the bacterial aquaglyceroporin GlpF is less restrictive, and residues at the constriction site of GlpF are substituted by larger and more hydrophobic residues, thus allowing glycerol to permeate the pore in addition to water (14, 17).
One of the most remarkable features of AQPs is their ability to allow water to permeate the pore at very high rates while preventing the passage of protons, thereby maintaining proton gradients across biological membranes. It is well accepted that protons can transfer efficiently along a continuous line of hydrogen bonded water molecules through the proton wire mechanism (18). A continuous line of water molecules is seen in the water pores of many AQP structures, yet protons are prohibited from passing. The mechanism of proton exclusion is now well established following extensive structural studies coupled with molecular dynamic simulations (reviewed in ref. 4). Water molecules within the pore of AQPs form an intricate network of hydrogen bonds with hydrophilic residues that line the pore. These residues orchestrate the permeation of water through the channel and orient the water molecules on either side of the two NPA motifs to create an opposing dipole moment. Central to this arrangement is a uniquely oriented water molecule that is stabilized by hydrogen bonds to the asparagine residues from the two NPA motifs. The orientation of water molecules induced by these interactions results in a net negative charge at either entrance to the pore, which effectively excludes positively charged protons from permeating the channel.
JUNCTION FORMING AQUAPORINS
All aquaporins form pores for water conductance, but AQP0 and AQP4 are unique in that they also form cell-cell adhesive junctions, and electron crystallography has provided structural insights into the junctional forms of these two channels. AQP0 was originally identified as the major intrinsic protein (MIP) of the eye lens (19, 20). Freeze-fracture experiments on lens membranes, as well as reconstitution experiments using purified AQP0 and atomic force microscopy on 2D crystals, all suggest that AQP0 is an adhesive protein (21-24). In fact, AQP0 predominantly localizes to the 11-13 nm “thin” junctions of lens fiber cells, where it forms densely packed square arrays in the lens core. Indeed, it was the adhesive role of AQP0 in lens fiber cells that confused researchers into believing that AQP0 was a lens gap junction protein. We have recently shown that AQP0 junction formation is triggered by C-terminal truncation (25), a post-translational modification that occurs in the lens in an age dependent manner (26). When lens AQP0 was purified from old fiber cells of the lens core, where a fraction of the AQP0s contain truncated C-termini, large double-layered 2D crystals formed. These double-layered crystals contained the same unit cell dimensions as the lens “thin” junctions and therefore likely recapitulate the AQP0-mediated “thin” membrane junctions that are abundant in the lens core.
AQP0 junction formation is primarily mediated by interactions between the extracellular loops A and C. Loop A is an important structural feature of AQP0 that facilitates junction formation, since it is not glycosylated as in other AQPs and is also much shorter. The result is that AQP0 tetramers have a relatively flat extracellular surface that allows two AQP0 tetramers from opposing membranes to interact with each other in a head-to-head fashion (Fig. 2A). One of the major junction-forming interactions is formed by proline 38. Eight symmetry-related Pro 38 residues pack against each other at the very center of the AQP0 junction, forming a rosette-like structure (Fig. 2B). This proline residue is conserved in AQP0 from different species but is substituted in other AQPs (7). When viewed from the cytosolic side, the water channels from adjoining tetramers are perfectly aligned and could potentially form a continuous cell-to-cell water pore. However, the electron crystallographically determined structure of AQP0 revealed that the water channels are closed in the junctional form of AQP0. While open AQP structures contain a continuous line of six to nine water molecules in the water channel (9), the junctional structure of AQP0 only contained three water molecules in the pore. These water molecules were too far apart to participate in hydrogen bond interactions. Pore closure is primarily the result of Met 176, which swings into the water pathway, obstructing the entrance to the pore. In the X-ray crystallographically determined structure of nonjunctional AQP0, the channel is in an open conformation and Met 176 points away from the channel pore (9).
Figure 2.
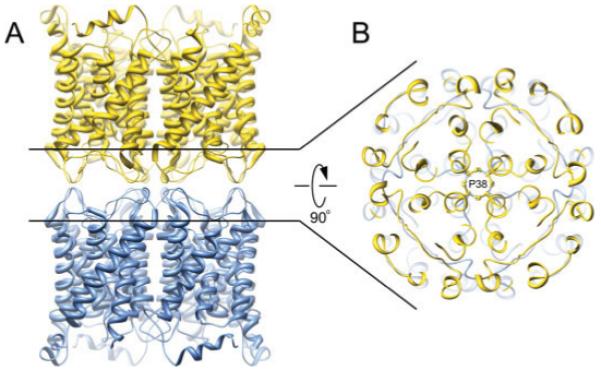
Structure of the aquaporin-0 mediated membrane junction. A: Side view of the aquaporin-0 junction. Two opposing tetramers interact in a head-to-head fashion. B: A major adhesive contact is mediated by proline 38, forming a rosette-like structure at the very center of the stacked tetramers (Protein Data Bank accession No. 2B6O).
AQP4 is expressed in glial cells, serving as the predominant water channel in the brain (27, 28). As with AQP0, reconstitution of AQP4 resulted in double-layered 2D crystals (8); however, the arrangement of opposing tetramers was significantly different than that of AQP0 (Fig. 3A). The orthogonal arrays of AQP4 tetramers stack in a head-to-head fashion, but are offset from one another, and a tongue into groove packing interaction involving the extracellular loop C facilitates AQP4 junction formation. The juxtaposed arrangement of the AQP4 crystal lattices places each AQP4 tetramer in contact with four opposing tetramers (Fig. 3B). This orientation blocks the extracellular vestibule of all AQP4 water channels, preventing the formation of continuous cell-to-cell water pores. An adhesive role for AQP4 had not been previously described, and aberrations in the crystallographic junctions suggested the interaction was weak. The weak interactions between double-layered arrays posed a technical challenge to overcome in obtaining the atomic model (8). Therefore, physiological evidence for AQP4 junction formation was pursued to determine if the electron crystallographic structure was biologically relevant, and indeed immunolabeling studies located AQP4 at junctional regions of glial lamellae. Furthermore, AQP4 expressed in fibroblast cell lines deficient in junction-forming proteins were shown to cluster, while AQP1 expression caused no such clustering effect. Although a definitive physiological role of AQP4 junction formation will require further characterization, the electron crystallographic structure of this protein represents a fantastic example of protein structure identifying an unforeseen function.
Figure 3.
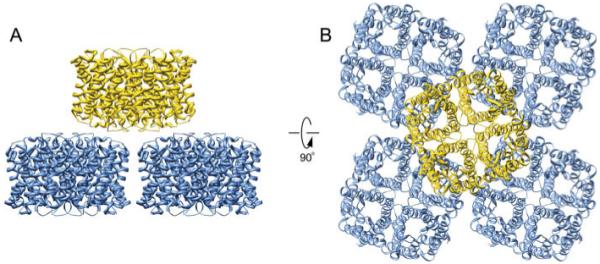
Double layered aquaporin-4 2D crystals. A and B: Side and top views of the double-layered 2D crystals of aquaporin-4, respectively. Each aquaporin-4 tetramer contacts four tetramers from the opposing layer (Protein Data Bank accession No. 2D57).
LIPID-PROTEIN INTERACTIONS
In 2D crystallography, the membrane protein of interest is crystallized within a lipid bilayer, where proteins and lipid form an intricate system fit for its function: the lipids maintain a hydrophobic and electrical seal around the protein, and the protein performs its function, which in the case of AQPs is to form a channel for water permeation. Electron crystallography therefore enables researchers to study the structure of a membrane protein while it is embedded in the near-native environment of the lipid bilayer, and in the case of AQPs, the channels are fully functional in 2D crystals (29).
An important feature of the electron crystallographically determined structure of AQP0 was that a lipid bilayer was visible in the density map, providing structural information on lipid-protein interactions (9). Some lipid molecules have been previously identified and characterized in several non-AQP X-ray crystallographic studies (reviewed in ref. 30). In all these cases, the lipids were co-purified and co-crystallized with the protein of interest. These lipids therefore represent highly stable and specific lipid-protein interactions, and indeed in some cases were required for the function of the protein (30). AQP0 has no such functional requirement, and solubilization and purification of the protein from native sources completely delipidated the protein (9). The 2D crystals used for AQP0 electron crystallographic studies were grown using the synthetic lipid dimyristoyl phosphatidyl choline (DMPC); thus providing unique structural information on how lipids interact nonspecifically with membrane proteins.
Nine unique DMPC lipid molecules were identified within the asymmetric unit of junctional AQP0, and formed a nearly complete lipid bilayer (Fig. 4A). Seven of these lipids (PC1-7) make direct protein interactions and represent the annular lipid shell that completely shields each tetramer from direct protein-protein interactions within the 2D crystalline array (Figs. 4A and 4B). PC8 and PC9 represent bulk-lipids: they have no direct contact with the protein and were found occupying the occluded space located at the fourfold axis between AQP0 tetramers. The acyl chains of the DMPC molecules forming the annular lipid shell adopt structurally unique conformations, presumably induced by their restricted environments. The conformations adopted by these lipids exquisitely match the outer contours of the protein transmembrane domain and thus create a hydrophobic seal around the protein. The negatively charged head groups of lipids observed in specific protein interactions are typically positioned close to positively charged amino acid sidechains such as arginine (9). The structure of AQP0 has been determined by both electron and X-ray crystallography (9, 31). Comparing the structure of AQP0 in lipid with that of AQP0 in detergent, it is observed that residues interacting with lipids in the electron crystallographic structure interact with detergent molecules in the X-ray structure. However, temperature B factors were much lower for these residues when they interacted with lipids rather than with detergent (reviewed in ref. 32) suggesting that lipids may have an important stabilizing role on membrane proteins.
Figure 4.
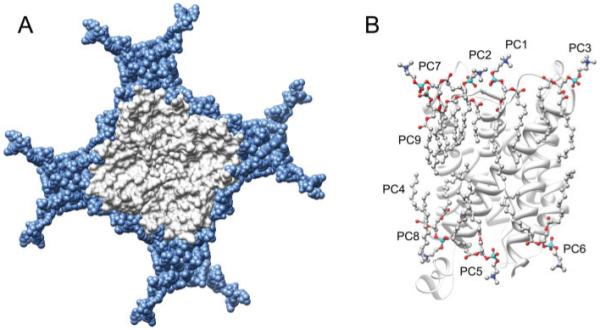
Lipid-protein interactions in aquaporin-0 double-layered 2D crystals. A: Lipids (blue) pack tightly around the aquaporin-0 tetramer (grey). Lipid acyl chains fit snuggly into irregularities on the protein surface. B: The nine unique DMPC lipid molecules (PC1-9) identified per aquaporin-0 monomer. PC1-7 are annular lipids while PC8 and 9 represent bulk lipids (Protein Data Bank accession No. 2B6O).
PERSPECTIVES
While both electron and X-ray crystallography, coupled with molecular dynamic simulations, have revealed structural and functional details of water channels, many questions remain unresolved. First, pore regulation is poorly understood. Only two aquaporin structures have been determined in a closed-pore conformation, AQP0 (7) and the plant SoPIP2;1 (33). However, channel permeability is dynamically modulated in a number of AQPs, although the structural details are unknown (reviewed in ref. 4). Second, while a number of aquaporin structures are known, the bacterial GlpF is currently the only aquaglyceroporin structure determined to date. None of the structures of the mammalian glycerol facilitators (AQP3, AQP7, AQP9, AQP10) are known in spite of their medical relevance (2). Finally, the biological function and structure of noncanonical AQPs is unknown: AQPs 11 and 12 are two mammalian AQPs that do not have the NPA motifs yet are still impermeable to protons; and the structure of AQP6 is needed to understand how this channel can conduct ions.
ACKNOWLEDGEMENTS
Work on aquaporins in the Gonen laboratory is supported by NIH grant R01 GM079233. SA is supported by NIH molecular biophysics training grant 5 T32 GM083268-19.
REFERENCES
- 1.Agre P, Sasaki S, Chrispeels MJ. Aquaporins: a family of water channel proteins. Am. J. Physiol. 1993;265:F461. doi: 10.1152/ajprenal.1993.265.3.F461. [DOI] [PubMed] [Google Scholar]
- 2.Agre P, Kozono D. Aquaporin water channels: molecular mechanisms for human diseases. FEBS Lett. 2003;555:72–78. doi: 10.1016/s0014-5793(03)01083-4. [DOI] [PubMed] [Google Scholar]
- 3.Yang B, Verkman AS. Water and glycerol permeabilities of aquaporins 1-5 and MIP determined quantitatively by expression of epitope-tagged constructs in Xenopus oocytes. J. Biol. Chem. 1997;272:16140–16146. doi: 10.1074/jbc.272.26.16140. [DOI] [PubMed] [Google Scholar]
- 4.Gonen T, Walz T. The structure of aquaporins. Q. Rev. Biophys. 2006;39:361–396. doi: 10.1017/S0033583506004458. [DOI] [PubMed] [Google Scholar]
- 5.Gyobu N, Tani K, Hiroaki Y, Kamegawa A, Mitsuoka K, Fujiyoshi Y. Improved specimen preparation for cryo-electron microscopy using a symmetric carbon sandwich technique. J. Struct. Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv. Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- 7.Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004;429:193–197. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- 8.Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on array formation and cell adhesion. J. Mol. Biol. 2005;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 9.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional crystals of aquaporin-0. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasler L, Heymann JB, Engel A, Kistler J, Walz T. 2D crystallization of membrane proteins: rationales and examples. J. Struct. Biol. 1998;121:162–171. doi: 10.1006/jsbi.1998.3960. [DOI] [PubMed] [Google Scholar]
- 11.Stahlberg H, Fotiadis D, Scheuring S, Rémigy H, Braun T, Mitsuoka K, Fujiyoshi Y, Engel A. Two-dimensional crystals: a powerful approach to assess structure, function and dynamics of membrane proteins. FEBS Lett. 2001;504:166–172. doi: 10.1016/s0014-5793(01)02746-6. [DOI] [PubMed] [Google Scholar]
- 12.Preston GM, Carroll TP, Guggino WB, Agre P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science. 1992;256:385–387. doi: 10.1126/science.256.5055.385. [DOI] [PubMed] [Google Scholar]
- 13.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 14.Sui H, Han BG, Lee JK, Walian P, Jap BK. Structural basis of water-specific transport through the AQP1 water channel. Nature. 2001;414:872–878. doi: 10.1038/414872a. [DOI] [PubMed] [Google Scholar]
- 15.Fujiyoshi Y, Mitsuoka K, de Groot B, Philippsen A, Grubmüller H, Agre P, Engel A. Structure and function of water channels. Curr. Opin. Struct. Biol. 2002;12:509–515. doi: 10.1016/s0959-440x(02)00355-x. [DOI] [PubMed] [Google Scholar]
- 16.Shi LB, Skach WR, Verkman AS. Functional independence of monomeric CHIP28 water channels revealed by expression of wild-type mutant heterodimers. J. Biol. Chem. 1994;269:10417–10422. [PubMed] [Google Scholar]
- 17.Tajkhorshid E, Nollert P, Jensen MØ, Miercke LJ, O’Connell J, Stroud RM, Schulten K. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 2002;296:525–530. doi: 10.1126/science.1067778. [DOI] [PubMed] [Google Scholar]
- 18.Pomes R, Roux B. Structure and dynamics of a proton wire: a theoretical study of H1 translocation along the single-file water chain in the gramicidin A channel. Biophys. J. 1996;71:19–39. doi: 10.1016/S0006-3495(96)79211-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloemendal H, Zweers A, Vermorken F, Dunia I, Benedetti EL. The plasma membranes of eye lens fibres. Biochemical and structural characterization. Cell Differ. 1972;1:91–106. doi: 10.1016/0045-6039(72)90032-2. [DOI] [PubMed] [Google Scholar]
- 20.Alcala J, Lieska N, Maisel H. Protein composition of bovine lens cortical fiber cell membranes. Exp. Eye Res. 1975;21:581–595. doi: 10.1016/0014-4835(75)90040-8. [DOI] [PubMed] [Google Scholar]
- 21.Bok D, Dockstader J, Horwitz J. Immunocytochemical localization of the lens main intrinsic polypeptide (MIP26) in communicating junctions. J. Cell. Biol. 1982;92:213–220. doi: 10.1083/jcb.92.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zampighi G, Simon SA, Robertson JD, McIntosh TJ, Costello MJ. On the structural organization of isolated bovine lens fiber junctions. J. Cell Biol. 1982;93:175–189. doi: 10.1083/jcb.93.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasler L, Walz T, Tittmann P, Gross H, Kistler J, Engel A. Purified lens major intrinsic protein (MIP) forms highly ordered tetragonal two-dimensional arrays by reconstitution. J. Mol. Biol. 1998;279:855–864. doi: 10.1006/jmbi.1998.1796. [DOI] [PubMed] [Google Scholar]
- 24.Fotiadis D, Hasler L, Müller DJ, Stahlberg H, Kistler J, Engel A. Surface tongue-and-groove contours on lens MIP facilitate cell-to-cell adherence. J. Mol. Biol. 2000;300:779–789. doi: 10.1006/jmbi.2000.3920. [DOI] [PubMed] [Google Scholar]
- 25.Gonen T, Cheng Y, Kistler J, Walz T. Aquaporin-0 membrane junctions form upon proteolytic cleavage. J. Mol. Biol. 2004;342:1337–1345. doi: 10.1016/j.jmb.2004.07.076. [DOI] [PubMed] [Google Scholar]
- 26.Ball LE, Garland DL, Crouch RK, Schey KL. Post-translational modifications of aquaporin 0 (AQP0) in the normalhuman lens: spatial and temporal occurrence. Biochemistry. 2004;43:9856–9865. doi: 10.1021/bi0496034. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J. Biol. Chem. 1994;269:5497–5500. [PubMed] [Google Scholar]
- 28.Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA. 1994;91:13052–13056. doi: 10.1073/pnas.91.26.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walz T, Smith BL, Zeidel ML, Engel A, Agre P. Biologically active two-dimensional crystals of aquaporin CHIP. J. Biol. Chem. 1994;269:1583–1586. [PubMed] [Google Scholar]
- 30.Wiener M. A census of ordered lipids and detergents in X-ray crystal structures of integral membrane proteins. In: Tamm LK, editor. Protein-Lipid Interactions: From Membrane Domains to Cellular Networks. Wiley-VCH Verlag GmbH and Co.; Weinheim: 2005. pp. 29–49. [Google Scholar]
- 31.Harries WE, Akhavan D, Miercke LJ, Khademi S, Stroud RM. The channel architecture of aquaporin 0 at a 2.2-A resolution. Proc. Natl. Acad. Sci. USA. 2004;101:14045–14050. doi: 10.1073/pnas.0405274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hite RK, Gonen T, Harrison SC, Walz T. Interactions of lipids with aquaporin-0 and other membrane proteins. Pflugers Arch. doi: 10.1007/s00424-007-0353-9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Törnroth-Horsefield S, Wang Y, Hedfalk K, Johanson U, Karlsson M, Tajkhorshid E, Neutze R, Kjellbom P. Structural mechanism of plant aquaporin gating. Nature. 2005 doi: 10.1038/nature04316. [DOI] [PubMed] [Google Scholar]