

Abstract
Cytosolic Ca2+ concentration ([Ca2+]i) is reduced in cultured neurons undergoing neuronal death caused by inhibitors of the ubiquitin proteasome system. Activation of calcium entry via voltage-gated Ca2+ channels restores cytosolic Ca2+ levels and reduces this neuronal death (Snider et al. 2002). We now show that this reduction in [Ca2+]i is transient and occurs early in the cell death process, before activation of caspase-3. Agents that increase Ca2+ influx such as activation of voltage-gated Ca2+ channels or stimulation of Ca2+ entry via the plasma membrane Na-Ca exchanger attenuate neuronal death only if applied early in the cell death process. Cultures treated with proteasome inhibitors had reduced current density for voltage-gated Ca2+ channels and a less robust increase in [Ca2+]i after depolarization. Levels of endoplasmic reticulum (ER) Ca2+ were reduced and capacitative Ca2+ entry was impaired early in the cell death process. Mitochondrial Ca2+ was slightly increased. Preventing the transfer of Ca2+ from mitochondria to cytosol increased neuronal vulnerability to this death while blockade of mitochondrial Ca2+ uptake via the uniporter had no effect. Programmed cell death induced by proteasome inhibition may be caused in part by an early reduction in cytosolic and endoplasmic reticulum (ER) Ca2+, possibly mediated by dysfunction of voltage-gated Ca2+ channels. These findings may have implications for the treatment of disorders associated with protein misfolding in which proteasome impairment and programmed cell death may occur.
Keywords: endoplasmic reticulum, mitochondria, calcium imaging, caspase, capacitative calcium entry
Inhibition of the ubiquitin proteasome system (UPS) causes programmed cell death in many cell types, including neurons. We hypothesized that the neuronal death caused by proteasome inhibition might be associated with dysregulation of calcium (Ca2+) homeostasis, specifically with a reduction, rather than an increase, in neuronal Ca2+. Many studies have focused on increases in intracellular Ca2+ in disorders including stroke, epilepsy, trauma and neurodegenerative disorders (for recent reviews see Arundine and Tymianski 2004; Berliocchi et al. 2005). The role of reductions in intracellular Ca2+ in neuronal death has also been studied, primarily in developing neurons, and a “calcium set point” has been hypothesized to play a key role in neuronal survival (Johnson et al. 1992). We have previously reported that free cytosolic Ca2+ ([Ca2+]i) is reduced in cultured murine neocortical neurons after 1-4 hr treatment with proteasome inhibitors. We observed a similar reduction in [Ca2+]i in cultures treated with staurosporine, a protein kinase inhibitor that causes neuronal death associated with activation of programmed cell death pathways (Snider et al. 2002; Canzoniero et al. 2004). However, other investigators have reported that treatment with proteasome inhibitors increases [Ca2+]i (Landowski et al. 2005; Lee et al. 2005; Nawrocki et al. 2005b; Nawrocki et al. 2005a). These apparently conflicting results could represent differences in cell type, intensity of injury or timing of assessment of [Ca2+]i relative to injury. For example, there could be an early increase and subsequent decline in [Ca2+]i levels, or Ca2+ could be reduced in some cellular compartments and increased in others, as reported in cultured mesencephalic neurons treated with ceramide (Darios et al. 2005). Understanding the nature and timing of these derangements in Ca2+ homeostasis might have significant implications for designing rational treatment strategies for conditions where neuronal proteasome activity is impaired. We hypothesized that the neuronal death associated with impairment of the UPS was caused in part by transfer of Ca2+ between cytosol, ER and mitochondria and set out to clarify the timing of changes in [Ca2+]i relative to cell death and to identify mechanisms responsible for the alterations in [Ca2+]i and neuronal death under these conditions. A preliminary report of these studies has appeared in abstract form (Wu et al. 2006).
Materials and Methods
Preparation of murine neocortical cultures
Mixed neocortical cultures containing both neurons and astrocytes were prepared from mouse cortices as previously described (Rose et al. 1993; Lee et al. 2004). Briefly, dissociated neocortices obtained from fetal Swiss-Webster mice at 14-16 days gestation were plated onto a previously established glial monolayer. Unless otherwise indicated, experiments were performed on cultures after 13-14 days in vitro. Near-pure neuronal cultures were prepared by plating neocortices directly onto culture vessels that had been coated with poly-D-lysine (100 μg/ml) and laminin (4 μg/ml) prior to plating. These cultures contain less than 5% astrocytes; experiments on these cultures were performed 7-8 days after plating.
Calcium imaging
Measurements of [Ca2+]i were performed using fura-2 video microfluorimetry as previously reported (Snider et al. 2002; Canzoniero et al. 2004). All washes and imaging were carried out in HEPES buffered saline (144 mM NaCl, 10 mM glucose, 10 mM HEPES, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 μM glycine, pH = 7.2) in room air unless otherwise indicated. Cells were incubated for 45 min at room temperature with 5 μM fura-2 AM (Invitrogen, Carlsbad, CA), and incubated in buffer without fura-2 for another 30 min. Cultures treated with proteasome inhibitors and other compounds were processed in parallel with sham washed (treated with vehicle only) cultures. Cultures were imaged with a Nikon Eclipse TE300 inverted microscope (Nikon Instruments, Melville, NY) using UV illumination via a monochrometer (Till Photonics Polychrome II) and a 40X oil immersion objective, fitted with a 12 bit SensiCam CCD camera (Cooke Corp., Tonawanda, NY). Excitation wavelengths were alternated between 340 and 380 nm, and emissions were measured at ≥ 510 nm. Because neuronal cell bodies sit well above the underlying glial monolayer, it was possible to detect the 340/380 fluorescence originating in these cell bodies with minimal interference from underlying glia. This data is presented as excitation ratios in the figures. For comparison, in text we provide the [Ca2+]i estimated by translating fluorescence ratios (340/380 nm) to [Ca2+]i using the method of Grynkiewicz et al. (Grynkiewicz et al. 1985). Briefly, the calcium concentrations were determined by the formula:
Where Kd is the fura-2 apparent dissociation constant for calcium, Q is the ratio of Fmin to Fmax at 380 nm and R is the ratio of fluorescence intensity excited at 340 and 380 nm at the given time point during the experiment. The calibration values were determined in situ in the presence of 1.8 mM free Ca2+ (Rmax) or 2 mM EGTA (Rmin) with the co-application of 4 μM ionomycin.
[Ca2+]mito was measured similarly, except that cultures were washed, prior to imaging, with buffer lacking Ca2+ and containing ethylene glycol-bis(2-aminoethylether)-N,N,N,N’-tetraacetic acid (EGTA, 50 μM). Images were captured before and after application of the mitochondrial uncoupling agent protonophore carbonylcyanide-p-trifluoromethoxy-phenylhydrazone (FCCP, 10 μM for 10 min) as a measure of Ca2+ released from depolarized mitochondria, similar to the methods described by Brocard et al. (2001) and Thayer and Miller (1990).
[Ca2+]ER was measured as described by (Darios et al. 2005) using mag-fura-2 (furaptra). Mag-fura-2 has relatively low affinity for Ca2+ (Kd reported between 25-100 μM, Raju et al. 1989; Ravin et al. 1997) and tends to accumulate in intracellular compartments, making it useful for measurement of [Ca2+]ER (Solovyova et al. 2002). Cultures were loaded with mag-fura-2 (10 uM) and Pluronic F-127 (0.05%) for 1 hr at 37 °C in buffer containing 125 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1mM Na2HPO4, 5.5 mM glucose, 20 mM NaHCO3, 2 mM L-glutamine, and 20 mM HEPES, pH 7.2. The cells were washed and kept in dye-free media for 1 hr prior to imaging. Images were obtained as described above for fura-2. In our experiments, mag fura-2 Kd for Ca2+ as determined by in situ calibration was 184 μM, somewhat higher than reported values.
In some experiments, [Ca2+]ER was also measured indirectly. Prior to imaging, cultures were washed with buffer lacking Ca2+ and containing EGTA (50 μM). Images were captured before and after application of the thapsigargin (5 μM) to block ER Ca2+ uptake. After 5 min, Ca2+ was added to the extracellular bathing media and images were captured for an additional 5 min.
Electrophysiology
Whole-cell recordings were performed using an Axopatch 1D amplifier (Molecular Devices, Sunnyvale, CA) and a Digidata 1322 acquisition board (Molecular Devices). pClamp software, version 9 (Molecular Devices) was used for data acquisition. Electrodes had resistances of 4-6 MΩ. In all instances, cells were excluded from analysis if a leak current >200 pA was observed.
For recording, the culture medium was exchanged for a saline solution containing (in mM): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, and 0.025 D-2-Amino-5-phosphonovalerate (D-APV), pH 7.25. For Ca2+ current recordings, 3 mM Ba2+ was used as the charge carrier to increase the current size and to improve the passive properties of the cell. Also, 500 nM tetrodotoxin (TTX), 1 μM 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), and 25 μM bicuculline were included to block sodium currents and spontaneous synaptic currents. All Ba2+ currents were digitally subtracted using a trace recorded in the presence of 50 μM Cd2+. The whole-cell pipette contained (in mM): 140 cesium methanesulfonate, 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, pH 7.25. Cells were stimulated with 50 ms pulses to 0 mV from the holding potential of -70 mV. Capacitance was estimated as described previously (Xu et al. 2000; Moulder et al. 2002).
Treatment with drugs and assessment of caspase activity and cell death
Cultures were treated with proteasome inhibitors and other drugs in Minimal Essential Media (MEM; with Earle’s salts, with 2 mM glutamine and 25 mM glucose) in a 5% CO2 incubator maintained at 37°C. Following the treatment period (typically 48 hr), cell death was analyzed using propidium iodide (PI) fluorescence or by analyzing efflux of lactate dehydrogenase (LDH) into the bathing media as previously described (Trost and Lemasters 1994; Sattler et al. 1997; Snider et al. 2002).
Caspase activity was analyzed by measuring degradation of a fluorogenic caspase-3 substrate, acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC) using a commercially available kit (Sigma Chemical Co., Saint Louis, MO). Cleavage of the substrate results in the release of the aminomethylcoumarin (AMC) fluorescent moiety. The assays were performed in a microplate format as recommended by the manufacturer. Background activity (activity not inhibited by addition of 2 μM Acetyl-Asp-Glu-Val-Asp-al, a caspase inhibitor) was subtracted.
Replication and Statistics
Unless otherwise noted, all data reported here represent at least n= 12 sister cultures (each sister culture is a culture well) from at least three independent replications. Data were analyzed for significance (P<0.05) by ANOVA and Bonferroni post-hoc test for multiple comparisons.
Results
[Ca2+]i is reduced during the early, reversible stages of proteasome inhibitor-induced neuronal death
We studied neuronal death occurring over 24-48 hr of moderate proteasome inhibition. We have previously shown that neuronal cultures treated with 3 μM MG-132 have about a 50% reduction in proteasome activity as compared to sham-washed cultures; 1 μM MG-132 reduces proteasome activity by 30-40% (Snider et al. 2002); this is similar to the level of proteasome impairment observed in brain regions affected by neurodegenerative disorders (Keller et al. 2000; McNaught and Jenner 2001; Keck et al. 2003). This process has many features of programmed cell death, including morphological changes such as cell body shrinkage and nuclear condensation and sensitivity to inhibition of protein and DNA synthesis and caspases (Qiu et al. 2000; Snider et al. 2002). As shown in Figure 1A (red lines), cultured neocortical neurons treated with MG- 132 (1 μM), a reversible proteasome inhibitor, began to die after about 24 hr treatment and were almost all killed by 44 hr treatment. Similar results were obtained with 3 μM MG-132 (data not shown). There was no significant death in sham-washed cultures over the 48 hr period. We next examined the time course of activation of caspase-3, a cysteine protease that plays a key role in many forms of programmed cell death (Jacobsen et al. 1996). Blue lines in Figure 1A show that, as expected, caspase-3 activity in MG-132 treated cultures increased after 12 hr treatment, before cell death was detectable, with a maximal increase at 24 hr, the last time point assessed.
Figure 1. Time course of neuronal death, caspase activity and [Ca2+]i following treatment with proteasome inhibitor (MG-132).

A. Cell death (red lines and symbols; Y-axis shown at right): Cultures were treated with proteasome inhibitors for 48 hr or sham-washed. Values shown are mean ± SEM cell death as assayed by propidium iodide fluorescence at the indicated time points. Values in sham-washed cultures were subtracted so death in sham-washed cultures =0%; there was no significant change in absolute fluorescence values for sham-washed cultures at any time point throughout the 48 hr treatment period. For all 3 measures shown in Panel A, * indicates death in MG-132-treated cultures differs significantly from that in sham-washed cultures; significance (P<0.05) was analyzed by ANOVA and Bonferroni post-hoc test. Caspase-3 activity (Blue line and symbols; Y-axis at far right): Cultures were treated as in the cell death assay. At the indicated time intervals after the start of the MG-132 treatment or sham wash, extracts were prepared and degradation of Ac-DEVD-AMC was determined. Values shown are mean ± SEM activity in pmol AMC cleaved/μg protein/min. Sham wash values did not differ significantly between any time points. Calcium imaging (Black line and symbols; Y-axis at left): cultures were sham-washed or treated with MG-132 and then returned to the incubator. At the indicated time after the start of drug treatment, cultures were loaded with fura-2 and images were collected. Values shown are mean ± SEM 340/380 ratios for n= 87-200 cells for each data point. Values shown are average of three consecutive measurements taken for each cell. MG-132 was included in the loading and washing buffers for treated cultures. B. Acute changes in [Ca2+]i. Cultures were loaded with fura-2 as in A. The solid line indicates when the cultures were sham washed. [Ca2+]i was measured every 30 seconds for an additional 30 min after the sham wash. Values shown are mean ± SEM 340/380 ratio for 15 neurons in two separate experiments. C. Similar to B, but MG-132 (2 μM) was added at the indicated time. Values shown are mean ± SEM 340/380 ratio for 17 neurons in two separate experiments.
We next measured free cytosolic Ca2+ ([Ca2+]i, black line in Figure 1A) during treatment with proteasome inhibitor. [Ca2+]i remained stable in sham-washed cultures immediately after washing (also see Figure 1B) and throughout the 24 hr observation period (data not shown) . The estimated [Ca2+]i in sham-washed cultures averaged 119 ± 9.5 nM, similar to previous studies. [Ca2+]i did not change immediately after application of MG-132 (Figure 1C), but was decreased after 4 hr (black lines in Figure 1A), to an estimated value of 101 ± 2.9 nM (85% of control, P<0.05). After 8 hr treatment with proteasome inhibitor, [Ca2+]i was reduced to 49 ± 4 nM (41% of control, P<0.001). This decrease in [Ca2+]i was transient as [Ca2+]i in MG-132-treated cultures increased to 146 ± 19 nM after 16 hr (not significantly different than sham-washed cultures). We did not measure [Ca2+]i or caspase activation at time points > 24 hr as some of the cells began to lose membrane integrity and loading and retention of dye was compromised. This reduction in [Ca2+]i thus began before activation of caspase-3 and before neuronal death was detectable by propidium iodide fluorescence.
Calcium influx via voltage gated Ca2+ channels can attenuate proteasome inhibitor-induced injury even if delayed up to 16 hr
We have previously reported that co-application of the calcium channel agonist (-) Bay K8644(BayK) throughout a 48 hr treatment attenuates proteasome inhibitor-induced neuronal death, while blockade of NMDA-subtype glutamate receptor mediated Ca2+ influx with NMDA receptor antagonists (MK-801, D-APV) increases this death (Snider et al. 2002). We now show that co-application of the glutamate receptor agonist kainate (KA) increased [Ca2+]i in sham-washed cultures and reversed the MG-132 induced decrease in [Ca2+]i (Figure 2). The increase in [Ca2+]i observed with KA is similar to that we have reported for BayK, and both compounds reversed the reduction in [Ca2+]i caused by proteasome inhibitors (compare Figure 2A here with Figure 7A in Snider et al., 2002). We reasoned that increasing [Ca2+]i using BayK or KA would only attenuate neuronal death if the Ca2+-raising agent was added during the critical time period when Ca2+ was reduced. We tested this hypothesis by delaying the timing of addition of KA or BayK. Cultures were treated with proteasome inhibitor for 48 hr as in Figure 1. (-) Bay K8644 (10 μM) or kainate (10 μM) were present throughout the treatment period in the (0 hr) condition, or added at the indicated time 4-20 hr after the start of the treatment period. Cell death was analyzed at the end of the 48 hr exposure. BayK attenuated proteasome inhibitor-induced neuronal death even when added 20 hr after the start of the 48 hr treatment period; later application (≥ 24 hr) did not reduce neuronal death. Addition of kainate (KA, 10 μM) also attenuated cell death when added up to 16 hr after the start of treatment (Figure 2B). We did not test addition at time points later than 24 hr since significant neuronal death was already detectable at that time (Figure 1). Treatment with BayK or kainate alone for 48 hr did not result in detectable neuronal death (data not shown).
Figure 2. Activating Ca2+ influx via voltage gated Ca2+ channels prevents proteasome inhibitor-induced [Ca2+]i depletion and attenuates neuronal death.

A. Murine neocortical cultures were sham-washed (vehicle only) or treated with MG-132 (2 μM) in the presence or absence of kainate (10 μM) or with kainate alone. [Ca2+]i was imaged after 4 hr. Values shown are mean ± SEM 340/380 ratios for n= 40 cells for each data point. * indicates significant difference (P<0.05) from SHAM condition. B. Murine neocortical cultures were treated with MG-132 (1 μM) for 48 hr. (-) Bay K8644 (BayK, 10 μM) or kainate (KA, 10 μM) was added at the beginning of the treatment period at the indicated time after the start of the treatment period. Compounds added at time “0” were co-applied with the MG-132 and were present throughout the treatment period. Values shown are mean ± SEM cell death analyzed at the end of the 48 hr treatment period. * indicates values for KA or BayK treated cultures differed significantly (P<0.05) from those of sister cultures treated with MG-132 alone.
Figure 7. Blockade of mitochondrial Ca2+ efflux exacerbates the proteasome inhibitor induced reduction in [Ca2+]i and neuronal death.

A. Murine neocortical cultures were sham-washed (Sham) or treated with 0.3 μM MG-132, 10 μM CGP-37157 (CGP), or both. [Ca2+]i was measured after 4 hr as in Figure 1. Values shown are mean ± SEM 340/380 ratio for 22-37 cells per condition. * indicates significant difference (P<0.001) from sham condition. B. Cultures were treated with MG-132 (0.3 μM) or NMDA (12.5 μM) alone or with the indicated concentration of CGP-37157; values shown are mean ± SEM cell death as measured by propidium iodide efflux at the end of the treatment period (48 hr for MG-132, 24 hr for NMDA). * indicates that neuronal death in cultures treated with MG-132 + the indicated concentration of CGP-37157 differed significantly from that in cultures treated with MG-132 without CGP-37157 (P<0.05 by ANOVA with Bonferroni post hoc). None of the NMDA+CGP-37157 conditions differed significantly from NMDA alone. Similar results were obtained using lactate dehydrogenase efflux as a measure of neuronal death. C. Cultures were treated with the indicated concentration of clastolactacystin for 48 hr alone or with 10 μM CGP-37157. Values shown are mean ± SEM cell death as measured by propidium iodide efflux; * indicates values in +CGP condition differ significantly from those in clastolactacystin only condition. D. Neuronally-enriched neocortical cultures (>95% neurons) were treated with 0.1 μM MG-132 for 48 hr alone, or in the presence of 10 μM BayK, kainate (KA) or CGP-37157 (CGP). Values shown are mean ± SEM cell death measured by propidium iodide fluorescence at the end of the treatment period. * indicates significant difference (P<0.05 by ANOVA with Bonferroni post hoc) from MG only condition.
MG-132 is relatively non-specific reversible proteasome inhibitor, so we analyzed [Ca2+]i in neuronal cultures after 4 hr treatment with clastolactacystin β-lactone, a highly specific irreversible proteasome inhibitor (Dick et al. 1997; Fenteany and Schreiber 1998). We observed a reduction in [Ca2+]i similar to that seen with MG-132. 340/380 nm ratio for sham-washed cultures was 0.972 ± 0.024, compared to 0.825 ± 0.035 for cultures treated with 3 μM clastolactacystin (differs significantly by ANOVA, P<0.001). Estimated [Ca2+]i was 120 ± 6.4 nM in sham-washed cultures; this was reduced to 82.0 ± 10.1 nM in clastolactacystin-treated cultures (data not shown). Consistent with our previous observations (Snider et al. 2002), co-application of 10 μM BayK modestly reduced neuronal death caused by 48 hr treatment with 3 μM clastolactacystin. Kainate did not significantly reduce this death, but kainate did block the ability of NMDA receptor antagonists to potentiate clastolactacystin-induced neuronal death (Supplemental Figure 1). NMDA antagonists alone were not toxic under these conditions, although toxicity has been reported in a similar culture system (Hwang et al. 1999).
We have previously reported that reductions in [Ca2+]i, either by treatment with Ca2+-free media or with the Ca2+-chelator BAPTA, cause a caspase-dependent death in cultured cortical neurons (Canzoniero et al. 2004). The period during which application of Ca2+-raising agents is neuroprotective overlaps with that during which [Ca2+]i is reduced (compare Figure 2B to Figure 1A) and parallels the timing of caspase activation, consistent with the idea that caspase activation represents the “commitment point” beyond which cell death is inevitable. We confirmed that co-application of BayK blocked caspase activation by measuring caspase activity 24 hr after the start of MG-132 treatment in the presence or absence of BayK. In this series of experiments, caspase activity was 4321 ± 486 pmol/μg/min after 24 hr treatment with 1 μM MG-132; this increase was nearly completely blocked by the co-application of 10 μM BayK (caspase activity after 24 hr in BayK+ MG cultures was 161 ± 16 pmol/μg/min, differs from MG-132 alone P<0.001; for comparison, caspase activity in sham-washed cultures after 24 hr was 72 ± 1 pmol/μg/min, data not shown).
Increasing calcium influx via the Na-Ca exchanger attenuates proteasome inhibitor-induced neuronal death
These findings suggest that the reduction in [Ca2+]i may be an important, potentially reversible event that ultimately contributes to proteasome inhibitor-induced neuronal death. Both BayK and kainate elevate [Ca2+]i by increasing Ca2+ entry through L-type voltage-gated Ca2+ channels. Another major route of Ca2+ flux in neuronal cells is the plasma membrane Na-Ca exchanger (NCX). The exchanger is bidirectional; it can reduce [Ca2+]i in “forward mode” by transferring Ca2+ out of the cell in exchange for Na+, or increase cytoplasmic Ca2+ in “reverse mode.” The role of the exchanger in maintaining Ca2+ homeostasis under resting conditions remains unclear, but a control experiment demonstrated that cultures treated with the NCX inhibitor benzamil (10 μM for 4 hr) had an increase in [Ca2+]i, suggesting that the exchanger functions predominantly in “forward mode” (transferring Ca2+ out of the cell) under resting conditions (Figure 3, Sham condition at left). In cultures treated with the proteasome inhibitor MG-132, the addition of benzamil normalized [Ca2+]i so that [Ca2+]i after 4 hr treatment was not significantly different from that in sham-washed cultures. Such co-application of benzamil also reduced the neuronal death caused by 48 hr treatment with MG-132 (Figure 3, right panel) or with clastolactacystin (Supplemental Figure 1).
Figure 3. Blockade of the Na-Ca exchanger (NCX) increases [Ca2+]i and reduces vulnerability to MG-132-induced neuronal death.

Left panel: [Ca2+]i measurements. Murine neocortical cultures were sham-washed (SHAM) or treated with benzamil (BENZ, 10 μM) for 4 hr in the absence (left two SHAM bars) or presence of MG-132 (MG, 1 μM) and then [Ca2+]i was measured as in Figure 1A. * indicates values were significantly different (P<0.05 by ANOVA with Bonferroni post hoc) from those in sham-washed condition without benzamil (left most bar). # indicates that cultures with BENZ+MG-132 differed from those treated with MG-132 alone. Values for MG-132 cultures treated with BENZ did not differ significantly from those in sham-washed condition without BENZ. Right panel: Cultures were treated with MG-132 (MG, 0.3 or 1.0 μM) for 48 hr in the presence or absence of benzamil (BENZ, 10 μM). Values shown are mean ± SEM cell death as assayed by propidium iodide fluorescence at the end of the 48 hr treatment period. * indicates values in +BENZ condition differ significantly from treated with MG alone.
Proteasome inhibitors cause a reduction in Ca2+ influx via voltage-gated Ca2+ channels
The observation that proteasome inhibitors reduce [Ca2+]i and that increasing Ca2+ influx can attenuate proteasome inhibitor induced death suggests that proteasome inhibitor treated cultures have reduced Ca2+ intake or increased Ca2+ efflux. We examined basal Ca2+ efflux and saw no significant differences in proteasome-inhibitor treated cells, although there was a trend toward reduced basal Ca2+ efflux in proteasome inhibitor-treated neurons (Supplemental Figure 2). We next investigated whether proteasome inhibitors altered Ca2+ influx via voltage-gated Ca2+ channels. As shown in Figure 4A, proteasome inhibitor-treated neurons had a reduction in amplitude of currents through voltage-gated Ca2+ channels. There was no effect seen when only L-type channels were examined, suggesting this effect is mediated by other types of voltage-gated Ca2+ channels (e.g., N, P,Q, R or T type channels). We also observed a smaller increase in [Ca2+]i during depolarization in proteasome inhibitor-treated than in sham-washed neurons, a result consistent with reduced Ca2+ influx via voltage gated Ca2+ channels (Figure 4C).
Figure 4. Effect of proteasome inhibitor treatment on voltage-gated Ca2+ channel currents and Ca2+ influx.
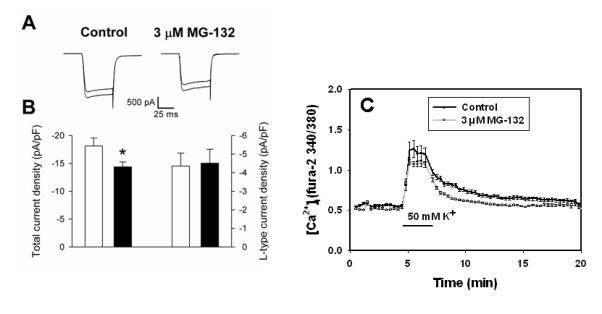
A. Representative Ca2+ currents elicited by voltage steps from -70 to 0 mV in a neuron treated with control medium and a neuron treated with 3 μM MG-132 for 4 hr. Currents were recorded in each neuron in the absence (larger trace) and presence (smaller trace) of 5 μM nifedipine. The capacitance of the control neuron was 24.7 pF and that of the MG-132-treated neuron was 25.6 pF. B. Summary of the total Ca2+ current density (two left-most bars) and the Ca2+ current density carried by L-type channels (two right-most bars) in control (open bars) and MG-132-treated (filled bars) neurons. n = 11-12; *P < 0.04. No statistically significant difference was observed in the L-type current density. C. Cultured neurons were sham-washed (Control) or treated with 3 μM MG-132 for 4 hr. Cultures were loaded with fura-2 and [Ca2+]i was imaged as in Figure 1. After 5 min, cultures were changed into media supplemented with 50 mM K+ for 2 min, then returned to control medium. Values shown are mean ± SEM 340/380 ratio for n= 96-100 cells per condition.
ER Ca2+ is also reduced following treatment with proteasome inhibitor
The reduction in [Ca2+]i caused by proteasome inhibitor may in part reflect a reduction in Ca2+ influx, but redistribution of Ca2+ from cytosol into other cellular compartments could also contribute. For example, a toxic redistribution of Ca2+ from cytosol to ER and ultimately to mitochondria has been reported in cultured neurons treated with ceramide (Darios et al. 2003; Darios et al. 2005). We directly measured ER Ca2+ ([Ca2+]ER) using the low affinity dye mag-fura-2. We confirmed that mag-fura-2 ratios reflected ER Ca2+ levels by showing that thapsigargin (2.5 μM for 30 min) reduced the putative [Ca2+]ER signal by about 50% (see Supplemental Figure 3A), similar to previous reports using this method in cultured neurons (Darios et al. 2005). In addition, permeabilization of the plasma membrane with digitonin (10 μM) reduced fura-2 fluorescence to background levels, while the mag-fura-2 signal was only slightly decreased, consistent with localization of most of the mag-fura-2 signal within organelles such as the ER (Supplemental Figure 3B). As shown in Figure 5A, [Ca2+]ER in cultures treated with proteasome inhibitor for 4 hr was reduced and reached a nadir at 8 hr, then returned to baseline levels by 16 hr, a pattern identical to that observed for [Ca2+]i.
Figure 5. Effect of proteasome inhibitors on endoplasmic reticulum (ER) Ca2+ ([Ca2+]ER).

A. Measurement of [Ca2+]ER with mag-fura-2. Cultures were treated with MG-132 (2 μM) and then loaded with mag-fura-2 (see Methods) and [Ca2+]ER was analyzed at the indicated time points after the start of the treatment period; images and data analysis were similar to that for fura-2 in Figures 1 and 3. n= 87-143 cells per time point. * indicates significant difference (P<0.05 by ANOVA) from sham-washed (time = 0) condition. B. Indirect measurement of [Ca2+]ER with fura-2 and thapsigargin. Cultures were treated with MG-132 (2 μM) for 4 hr or 16 hr (darker and lighter gray lines respectively) or sham-washed for 4 hr (black line). Cultures were loaded with fura-2; before imaging, the media was changed to calcium free buffer. Baseline [Ca2+]i was measured in Ca2+-free media and the cells were treated with thapsigargin (5 μM). After 5 min, thapsigargin was washed out, 1.8 mM calcium was added to the bathing media and [Ca2+]i was measured for another 5 minutes. A representative trace is shown for each condition. Similar results were obtained for n= 8-10 cells each condition. MG-132 was present in treated cultures throughout the experiment.
We also examined ER Ca2+ levels indirectly by analyzing changes in [Ca2+]i after blockade of ER Ca2+ uptake with thapsigargin (5 μM). Thapsigargin was added to cultures in nominally Ca2+-free media to eliminate increases in Ca2+ from extracellular sources. Using this method, we observed a robust thapsigargin-stimulated increase in [Ca2+]i in sham-washed cultures, but not in cultures treated with proteasome inhibitor for 4 or 16 hr (Figure 5B). After [Ca2+]i reached steady state, Ca2+ was added to the extracellular media and [Ca2+]i increased, consistent with influx via the store-activated channels. The increase in [Ca2+]i after addition of extracellular Ca2+ was blunted in cultures treated with proteasome inhibitor for 4 hr, but had recovered by 16 hr. Proteasome-inhibitor treated cultures thus have reduced levels of Ca2+ in the ER and, at least early in the injury, likely have reduced capacitative Ca2+ entry to refill these depleted stores.
Proteasome inhibitor-treated neurons have modestly increased levels of mitochondrial Ca2+
We next looked at the role of mitochondrial Ca2+ because mitochondria provide an important high capacity, low affinity cellular Ca2+ store. We measured mitochondrial Ca2+ ([Ca2+]mito) indirectly by monitoring fura-2 fluorescence before and after the application of the protonophore carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, 10 μM), a maneuver that dissipates the mitochondrial proton gradient and releases mitochondrial Ca2+ (Thayer and Miller 1990; Brocard et al. 2001). Neuronal cultures were treated with MG-132 and then changed to Ca2+-free buffer prior to imaging. Sham-washed cultures had little change in [Ca2+]i after exposure to FCCP (gray line in Figure 6A; basal [Ca2+]i in control cultures was 100 ± 6 nM, increasing to a maximum of 124 ± 8 nM after FCCP), consistent with previous reports using similar methods in rat forebrain cultures (Brocard et al. 2001). In contrast, cultures treated with proteasome inhibitor (MG-132, 2 μM for 4 hr) had a lower resting [Ca2+]i than control cultures (similar to the decrease shown in Figure 1A and our previous studies) but elevated [Ca2+]i after treatment with FCCP, consistent with increased Ca2+ release from mitochondrial stores (maximum [Ca2+]i estimated at 230 nM, black line Figure 6A). For comparison, we analyzed [Ca2+]mito in neurons treated with 300 μM NMDA for 10 min; this treatment causes a rapidly evolving neuronal death triggered in part by mitochondrial Ca2+ overload (Nicholls 2004). As expected, basal [Ca2+]i was increased in NMDA-treated cells (to an estimated level of about 1.05 μM) and [Ca2+]i increased massively after application of FCCP (Figure 6B). The NMDA-induced increase in [Ca2+]mito was much greater than that observed in cultures treated with MG-132 (same data for MG-132 shown in 6A and 6B, but scaling is different). These values likely underestimate the increase in NMDA-treated cells because fura-2, a relatively high affinity dye, may have reached saturation. Thus, although there is an increase in mitochondrial Ca2+ in proteasome inhibitor-treated cultures, this increase is of a much lower magnitude than that seen in cultures treated with NMDA, a situation in which mitochondrial Ca2+ overload likely plays an important role in cell death.
Figure 6. Effects of proteasome inhibitor on mitochondrial Ca2+ levels and role of mitochondrial Ca2+ in proteasome inhibitor and NMDA-induced neuronal death.

A. Murine neocortical cultures were sham-washed (gray line) or treated with MG-132 (1 μM, black line) as in Figure 1. After 3 hr drug treatment, cultures were loaded with fura-2; MG-132 was included in the loading and washing buffers for treated cultures. Cultures were washed into Ca2+-free buffer and basal ratios were obtained. Cultures were treated with FCCP (10 uM) for 10 min, then FCCP was washed out and image acquisition continued. A representative tracing is shown for each condition. Similar results were obtained for at least 40 individual cells per condition in three independent experiments (averaged data is shown in Panel C). B. Cultures were treated with NMDA (300 μM for 10 min, gray line) prior to imaging. Results for NMDA were obtained from n =10 cells in two separate experiments. Data shown here for MG-132 (black line) is same as shown in Panel A; it is reproduced here to demonstrate difference in scale. C. Pooled results for experiments shown in A-B and effect of blockade of the mitochondrial uniporter with ruthenium red. Cultures were treated with NMDA and MG-132 as in A and B. The indicated cultures were pretreated with ruthenium red (RuR, 10 μM) for 1 hr prior to the start of the MG-132 treatment; ruthenium red was present in the indicated cultures throughout the experiment. For NMDA treatment regimen: RuR was added 45 min before NMDA treatment; 300 μM NMDA was added to the cultures for 10 min before the buffers were changed to Ca2+ free media; all images were acquired in the Ca2+ free media. Values shown are average ± SEM basal 340/380 excitation ratio (time point just prior to addition of FCCP) and peak 340/380 excitation ratio at the end of the 10 min FCCP treatment for at least 40 cells per condition. * indicates “+FCCP” condition differs significantly (P<0.05) from “DRUG ALONE.” Differences between basal and peak levels after FCCP were analyzed for significance using Student’s t-test. There was no difference between + and − FCCP conditions in cultures treated with ruthenium red. D. Effect of ruthenium red on neuronal death caused by proteasome inhibitors or NMDA. Murine neocortical neurons were treated with 1 μM MG-132 or 10 μM clastolactacystin for 48 hr or with 15 μM NMDA for 24 hr. Ruthenium red was present during the treatment period in the indicated cultures (+RuR, 10 μM). Values shown are mean ± SEM cell death measured using propidium iodide staining at the end of the treatment period. Similar results were obtained using lactate dehydrogenase efflux as a measure of cell death. * indicates “+ RuR” condition differs significantly from “DRUG ALONE” (P<0.05 by ANOVA with Bonferroni post hoc).
Role of mitochondrial Ca2+ in neuronal death caused by proteasome inhibitor
As noted above, the increases in [Ca2+]mito observed in proteasome inhibitor-treated neurons were small and might not be physiologically significant. We addressed this issue by testing the effect of ruthenium red (RuR, 10 μM) on mitochondrial Ca2+ levels and neuronal death in proteasome inhibitor-treated cultures. RuR is a widely used, although potentially nonspecific and poorly cell permeable, antagonist of mitochondrial Ca2+ uptake via the uniporter (Lehninger et al. 1967). RuR has been reported to block the uniporter in cultured neurons (Wang and Thayer 2002). Cultured neurons were sham-washed or treated with RuR for 40 min; baseline cytoplasmic Ca2+ was measured with fura-2 and then mitochondrial Ca2+ was released with FCCP and cytoplasmic Ca2+ was measured again. As shown in Figure 6C, 40 min treatment with RuR (10 μM) increased basal [Ca2+]i levels and abolished the ability of FCCP to further increase [Ca2+]i under all conditions tested, consistent with blockade of mitochondrial Ca2+ uptake. The increase in [Ca2+]i (in the absence of FCCP) in RuR-treated cultures likely reflects inhibition of basal mitochondrial Ca2+ uptake.
We next tested the effects of RuR treatment (pretreatment and co-application) on neuronal vulnerability to proteasome inhibitor and NMDA-induced neuronal death. Blockade of mitochondrial Ca2+ uptake would be expected to block mitochondrial Ca2+ overload. Co-application of RuR attenuated NMDA-induced neuronal death, but did not attenuate MG-132- or clastolactacystin-induced death (Figure 6D). Co-application of RuR also failed to attenuate less severe insults, such as 3 μM clastolactacystin treatment for 24 hr (43.9 ± 6.2% neuronal death alone; 41.7 ± 5.9% neuronal death with co-application of 10 μM RuR). RuR does not inhibit NMDA receptors at the concentrations used here, as evidenced by lack of effect on [Ca2+]i when applied concurrently with NMDA (data not shown).
Mitochondria take up Ca2+ via the uniporter and then release it under physiological conditions via the mitochondrial Na-Ca exchanger. We tested the effect of inhibiting mitochondrial Ca2+ efflux on [Ca2+]i and neuronal vulnerability to proteasome inhibitor-induced injury using CGP-37157, a highly specific inhibitor of the mitochondrial Na-Ca exchanger (Cox et al. 1993; White and Reynolds 1997). Treatment with 10 μM CGP-37157 did not significantly reduce [Ca2+]i, nor did treatment with a sublethal concentration of MG-132 (0.3 μM) (Figure 7A). Co-application of CGP-37157 and 0.3 μM MG-132 caused a significant reduction in [Ca2+]i similar to that seen following treatment with higher, toxic concentrations of MG-132 (compare Figure 7A to Figures 1 and 2) and also potentiated MG-132 and clasto-lactacystin-induced neuronal death (Figure 7B-C). In contrast, co-application of CGP-37157 did not alter vulnerability to NMDA-induced death (7B, light gray line). Application of CGP-37157 alone caused no injury at concentrations up to 25 μM (data not shown).
Although astrocytes are less vulnerable to proteasome inhibitor-induced death than neurons (Snider et al. 2002), some of the effects of Ca2+ altering strategies on neuronal vulnerability could be mediated indirectly by astrocytes in these mixed neuronal-astrocyte cultures. “Pure” neuronal cultures containing >95% neurons were more sensitive to proteasome inhibitors (MG-132 LD50 ∼ 0.1 μM) than cultures containing both neurons and astrocytes MG-132 LD50 1-3 μM) but, similar to mixed astrocyte-neuronal cultures, proteasome inhibitor-induced death in pure neuronal cultures was attenuated by co-application of (-) Bay K8644 (10 μM) or benzamil (10 μM) and exacerbated by co-application of CGP-37157 (10 μM). Co-application of kainate did not reduce neuronal death, but this could in part be due to the increased vulnerability of these cultures to excitotoxic neuronal injury.
Discussion
We report here that cultured neurons treated with proteasome inhibitors had reduced cytosolic and ER Ca2+ levels 4-8 hr after the start of the injury, before or concurrent with activation of caspase-3 and before cell death was detectable by propidium iodide staining. We show a reduction in voltage-gated Ca2+ channel current density and in the depolarization-stimulated increase in [Ca2+]i in proteasome inhibitor-treated neurons, and show that maneuvers that increase Ca2+ influx via the voltage-gated Ca2+ channel or the Na-Ca2+ exchanger blocked the decrease in Ca2+ levels and reduced cell death. The “therapeutic window” for application of compounds that increased Ca2+ entry via the voltage-gated Ca2+ channel corresponds roughly to the time period of reduced cellular Ca2+ and ended as caspases were activated. These findings suggested that proteasome inhibitor-induced neuronal death is mediated by a reduction in Ca2+ influx that may be in part due to dysfunction of voltage-gated Ca2+ channels and an overall cellular Ca2+ deficiency. Interestingly, proteasome inhibitor treatment did not alter current density of L-type voltage-gated Ca2+ channels, so the inhibitory effects of proteasome inhibition may be on N, P, Q, R or T type channels. The protective effects of (-) Bay K8644, an L-type channel agonist, were consistent with the idea that L-type channels remain functional in proteasome-inhibitor treated cells.
Both a reversible (MG-132) and a more specific irreversible (clastolactacystin β-lactone) proteasome inhibitor reduced neuronal [Ca2+]i. Neuronal death induced by these inhibitors was similarly attenuated by co-application of BayK8644 and benzamil and exacerbated by NMDA antagonists. There was a difference in the effects of co-application of kainate; kainate modestly attenuated neuronal death caused by MG-132 (Figure 2B) but the injury-reducing effects of kainate against clastolactacystin-induced death did not achieve statistical significance (compare Figure 2B and Supplemental Figure 1).
To our knowledge, this is the first study to assess [Ca2+]ER and [Ca2+]mito in cells undergoing insults associated with a reduction in [Ca2+]i. We initially hypothesized that the reduction in cytosolic [Ca2+]i would be accompanied by increased sequestration of Ca2+ in ER and mitochondria; this was clearly not the case for ER Ca2+ as it was reduced to a similar extent as [Ca2+]i. Store-operated Ca2+ uptake was impaired after 4 hr treatment with proteasome inhibitor and was restored after 16 hr, raising the possibility that a defect in coupling of ER and cytosolic Ca2+ levels to capacitative Ca2+ entry underlies the derangement in Ca2+ homeostasis in proteasome inhibitor-treated neurons.
An increase, rather than a decrease, in [Ca2+]i has been reported during the first hr of treatment with proteasome inhibitors in undifferentiated PC12 cells or myeloma cells (Landowski et al. 2005; Lee et al. 2005). We did not observe any increase in [Ca2+]i in proteasome inhibitor treated neurons (Figure 1B-C). Interestingly, the acute increase in [Ca2+]i observed in myeloma cells was followed by an apparent reduction in [Ca2+]i over the next 30 min (see Landowski et al. 2005). It is thus possible that proteasome inhibitor-mediated toxicity in other cell types may also be mediated by a reduction in [Ca2+]i.
We considered the possibility that mitochondrial Ca2+ overload might play a role in proteasome inhibitor-induced death, perhaps through transfer of Ca2+ from cytosol and ER to mitochondria as suggested by Darios et al. for ceramide-induced neuronal death (2003; 2005). Although we observed a modest increase in [Ca2+]mito in neurons undergoing proteasome inhibitor-induced neuronal death, the magnitude of increase in [Ca2+]mito was much lower than that seen following a toxic NMDA treatment and blockade of mitochondrial Ca2+ uptake via the uniporter with ruthenium red did not reduce proteasome inhibitor-induced death, in contrast to the effects of ruthenium red in NMDA-treated neurons (Figures 6B and 6D). Conversely, blockade of mitochondrial Ca2+ efflux with CGP-37157 increased proteasome inhibitor-induced neuronal death but did not alter vulnerability to NMDA (Figure 7B), suggesting that mitochondrial Ca2+ uptake might contribute to proteasome inhibitor induced death by sequestering Ca2+ from other compartments rather than by a toxic effect on mitochondria.
The injury-reducing effects of increased Ca2+ entry were not restricted to entry via voltage-gated Ca2+ channels, as MG-132 and clastolactacystin-induced neuronal death was also reduced by co-application of benzamil, a relatively nonspecific blocker of the plasma-membrane Na-Ca exchanger. Although nonspecific effects of this compound cannot be ruled out, these studies are consistent with the idea that reductions in [Ca2+]i play a role in cell death.
There are some potential confounds with the measures of Ca2+ used here, particularly the measures of ER and mitochondrial Ca2+. We imaged [Ca2+] in cell bodies; alterations in cytosolic, ER and mitochondrial Ca2+ in neuronal processes are likely to play an important role in neuronal survival and were not assessed in these studies. Mag-fura-2 also binds Mg2+ (KD ∼ 1.5 mM for Mg2+ as compared to ∼ 50-190 μM for Ca2+) so some of the signal detected could possibly reflect Mg2+ rather than Ca2+ (Raju et al. 1989; Hyrc et al. 2000) . However, studies in astrocytes and myocytes have shown that this dye is relatively selective for Ca2+ at concentrations > 50 μM (Golovina and Blaustein 1997). There can be averaging of signal between compartments with higher (e.g., ER) and those with lower Ca2+ (e.g., cytosol) so this technique may underestimate [Ca2+]ER (Hyrc et al. 2007), but such averaging is unlikely to account for the observed decrease in [Ca2+]ER in proteasome inhibitor-treated cultures.
The use of FCCP-mediated depolarization to assess mitochondrial Ca2+ also has potential drawbacks; for example, FCCP could cause acidification (Wang et al. 1995; White and Reynolds 1997) or plasma membrane depolarization (Buckler and Vaughan-Jones 1998). Despite the potential confounds, the increase in [Ca2+]i seen acutely after application of FCCP has been widely used as an indirect measurement of [Ca2+]mito in cultured cortical neurons. The concentration of FCCP used here (10 μM) is relatively high, but within the range of concentrations (0.75 – 10 μM) used in other studies of cultured neurons (Duchen et al. 1990; Thayer and Miller 1990; Kiedrowski and Costa 1995; Reynolds and Hastings 1995; Brocard et al. 2001). We observed similar results using 1 μM FCCP. We used fura-2 here rather than a lower affinity dye in order to measure the relatively modest increases in [Ca2+]mito seen following proteasome inhibitor treatment. The disadvantage of this approach is that we likely underestimated the NMDA-induced increase in [Ca2+]mito.
Taken together, these observations support a model in which neurons undergoing proteasome inhibitor-induced neuronal death have an overall Ca2+ deficiency so that increasing Ca2+ entry from extracellular sources provides neuroprotection and blocking Ca2+ entry with glutamate antagonists or blocking mitochondrial Ca2+ release with CGP-37157 exacerbates injury. The contribution of Ca2+ deficiency to neuronal death occurs during the early steps of the cell death cascade before the activation of caspases and commitment to die. We hypothesize that proteasome inhibition impairs uptake via voltage-gated Ca2+ channels, perhaps via multiple N, P,Q, R or T type channels, but not through L-type channels. Proteasome inhibition may perturb normal homeostatic mechanisms that regulate capacitative Ca2+ entry. These findings raise the possibility that Ca2+ deficiency, rather than Ca2+ overload, may contribute to neuronal death in disorders where programmed cell death pathways are activated or proteasomal protein degradation pathways are impaired. Such a situation may occur in peripheral nerve or CNS in patients treated with proteasome inhibitors (e.g., for multiple myeloma), or perhaps in neurodegenerative disorders, although the role of proteasome impairment in these disorders has not yet been established and these disorders evolve over years rather than hours. Further studies of proteasome inhibition and intracellular Ca2+ deficiency in neurological disorders are important as some currently contemplated therapeutic strategies, such as glutamate receptor antagonists, could increase neuronal injury during the early Ca2+-deficient period, while treatments that increase Ca2+ influx could prove unexpectedly effective in delaying neuronal death.
Supplementary Material
Supplemental Figure 1. Effect of Ca2+ influx modulators on clastolactacystin β-lactone induced neuronal death A. Murine neocortical cultures were treated with clastolactacystin-β-lactone (CL, 3 μM) for 48 hr. (-) Bay K8644 (BayK, 10 μM), kainate (KA, 1 μM) or benzamil (BENZ, 10 μM) were added to the indicated cultures at the beginning of the treatment period. Values shown are mean ± SEM cell death analyzed by PI fluorescence at the end of the 48 hr treatment period. * indicates values for BayK and benzamil treated cultures differed significantly from CL alone (P=0.007 for BayK; P=0.019 for benzamil). The injury reduction in kainate-treated cultures did not reach significance. B. Similar to A. except that cultures were treated with 1 μM clastolactacystin alone (CL) or with the addition of MK-801 (10 μM), D-APV (1 mM) or MK-801 + kainate (5 μM) during the 48 hr treatment with proteasome inhibitor. * indicates significant difference from cultures treated with CL alone; # indicates CL+MK+KA condition differs significantly from CL+MK condition.
Supplemental Figure 2. Proteasome inhibition does not alter basal neuronal Ca2+ efflux. Neuronal cultures were loaded with 45Ca2+ for 48 hr, then cultures were washed and returned to standard media, either with no additions (Sham) or 1 uM or 3 uM MG-132. 45Ca2+ efflux into the media was measured at 1 hr intervals. Values shown are mean ± SEM % cpm released (cpm in media/cpm in cells at start of efflux period).
Supplemental Figure 3. Validation of mag-fura 2 fluorescence as a measure of [Ca2+]ER A. Murine neocortical cultures were loaded with Mag-fura-2, washed into Ca2+-free buffer and then treated with thapsigargin (2.5 μM for 30 min, Thapsigargin) or sham-washed (Sham). Values shown are mean ± SEM 340/380 ratio for at least 40 cells for each condition. * indicates value in thapsigargin condition differs significantly with that in control condition (P< 0.01). B. Similar to A, except cultures were loaded with Mag-fura-2 (black line) or fura-2 (gray line). Images were obtained for 5 min, then digitonin (10 μM) was added and imaging continued for an additional 30-35 min. Values shown are fluorescence intensity (ex360 (isosbestic point)/em510) from a representative tracing for each condition. n= 8 neurons imaged with Mag-fura-2 and 5 for Fura-2.
Acknowledgements
This work was supported by the NIH/NINDS R01 NS44923 (BJS), NIH Neuroscience Blueprint Core Grant P30 NS057105 to Washington University and by the Charles and Joanne Knight Alzheimer Research Initiative of Washington University’s Alzheimer’s Disease Research Center. We thank Dr. Eugene M. Johnson, Jr., for his helpful comments on the manuscript.
Abbreviations used
- (AMC)
aminomethylcoumarin
- (BayK)
(-)Bay-K8644
- (Ca2+)
calcium
- (FCCP)
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- (ER)
endoplasmic reticulum
- ([Ca2+]i)
free cytosolic Ca2+ concentration
- ([Ca2+]ER)
free ER Ca2+ concentration
- ([Ca2+]mito)
free mitochondrial Ca2+ concentration
- (UPS)
ubiquitin proteasome system
REFERENCES
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell Mol Life Sci. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliocchi L, Bano D, Nicotera P. Ca2+ signals and death programmes in neurons. Philos.Trans. R. Soc. Lond. B Biol. Sci. 2005;360:2255–2258. doi: 10.1098/rstb.2005.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocard JB, Tassetto M, Reynolds IJ. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J. Physiol. 2001;531:793–805. doi: 10.1111/j.1469-7793.2001.0793h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of mitochondrial uncouplers on intracellular calcium, pH and membrane potential in rat carotid body type I cells. J. Physiol. 1998;513(Pt 3):819–833. doi: 10.1111/j.1469-7793.1998.819ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzoniero LM, Babcock DJ, Gottron FJ, et al. Raising intracellular calcium attenuates neuronal apoptosis triggered by staurosporine or oxygen-glucose deprivation in the presence of glutamate receptor blockade. Neurobiol. Dis. 2004;15:520–528. doi: 10.1016/j.nbd.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Cox DA, Conforti L, Sperelakis N, et al. Selectivity of inhibition of Na(+)-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- Darios F, Lambeng N, Troadec JD, et al. Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J. Neurochem. 2003;84:643–654. doi: 10.1046/j.1471-4159.2003.01590.x. [DOI] [PubMed] [Google Scholar]
- Darios F, Muriel M-P, Khondiker ME, et al. Neurotoxic Calcium Transfer from Endoplasmic Reticulum to Mitochondria Is Regulated by Cyclin-Dependent Kinase 5-Dependent Phosphorylation of Tau. J. Neurosci. 2005;25:4159–4168. doi: 10.1523/JNEUROSCI.0060-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick LR, Cruikshank AA, Destree AT, et al. Mechanistic studies on the inactivation of the proteasome by lactacystin in cultured cells. J. Biol. Chem. 1997;272:182–188. doi: 10.1074/jbc.272.1.182. [DOI] [PubMed] [Google Scholar]
- Duchen MR, Valdeolmillos M, O’Neill SC, et al. Effects of metabolic blockade on the regulation of intracellular calcium in dissociated mouse sensory neurones. J. Physiol. 1990;424:411–426. doi: 10.1113/jphysiol.1990.sp018074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Schreiber SL. Lactacystin, proteasome function, and cell fate. J. Biol. Chem. 1998;273:8545–8548. doi: 10.1074/jbc.273.15.8545. [DOI] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hwang JY, Kim YH, Ahn YH, et al. N-Methyl-D-aspartate receptor blockade induces neuronal apoptosis in cortical culture. Exp. Neurol. 1999;159:124–130. doi: 10.1006/exnr.1999.7126. [DOI] [PubMed] [Google Scholar]
- Hyrc KL, Bownik JM, Goldberg MP. Ionic selectivity of low-affinity ratiometric calcium indicators: mag-Fura-2, Fura-2FF and BTC. Cell Calcium. 2000;27:75–86. doi: 10.1054/ceca.1999.0092. [DOI] [PubMed] [Google Scholar]
- Hyrc KL, Rzeszotnik Z, Kennedy BR, et al. Determining calcium concentration in heterogeneous model systems using multiple indicators. Cell Calcium. 2007 doi: 10.1016/j.ceca.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen MD, Weil M, Raff MC. Role of Ced-3/ICE-family proteases in staurosporine-induced programmed cell death. J. Cell Biol. 1996;133:1041–1051. doi: 10.1083/jcb.133.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EM, Jr., Koike T, Franklin J. A “calcium set-point hypothesis” of neuronal dependence on neurotrophic factor. Exp. Neurol. 1992;115:163–166. doi: 10.1016/0014-4886(92)90242-i. [DOI] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, et al. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Costa E. Glutamate-induced destabilization of intracellular calcium concentration homeostasis in cultured cerebellar granule cells: role of mitochondria in calcium buffering. Mol Pharmacol. 1995;47:140–147. [PubMed] [Google Scholar]
- Landowski TH, Megli CJ, Nullmeyer KD, et al. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005;65:3828–3836. doi: 10.1158/0008-5472.CAN-04-3684. [DOI] [PubMed] [Google Scholar]
- Lee CS, Han ES, Han YS, et al. Differential effect of calmodulin antagonists on MG132-induced mitochondrial dysfunction and cell death in PC12 cells. Brain Res. Bull. 2005;67:225–234. doi: 10.1016/j.brainresbull.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Lee CS, Tee LY, Warmke T, et al. A proteasomal stress response: pretreatment with proteasome inhibitors increases proteasome activity and reduces neuronal vulnerability to oxidative injury. J. Neurochem. 2004;91:996–1006. doi: 10.1111/j.1471-4159.2004.02813.x. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Carafoli E, Rossi CS. Energy-linked ion movements in mitochondrial systems. Adv. Enzymol. Relat. Areas Mol. Biol. 1967;29:259–320. doi: 10.1002/9780470122747.ch6. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Fu T, Melbostad H, et al. Ethanol-induced death of postnatal hippocampal neurons. Neurobiol. Dis. 2002;10:396–409. doi: 10.1006/nbdi.2002.0523. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Pino MS, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005a;65:11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Dunner K, Jr., et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005b;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr. Mol. Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- Qiu JH, Asai A, Chi S, et al. Proteasome inhibitors induce cytochrome c-caspase-3-like protease- mediated apoptosis in cultured cortical neurons. J. Neurosci. 2000;20:259–265. doi: 10.1523/JNEUROSCI.20-01-00259.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju B, Murphy E, Levy LA, et al. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. 1989;256:C540–548. doi: 10.1152/ajpcell.1989.256.3.C540. [DOI] [PubMed] [Google Scholar]
- Ravin R, Spira ME, Parnas H, et al. Simultaneous measurement of intracellular Ca2+ and asynchronous transmitter release from the same crayfish bouton. J. Physiol. 1997;501(Pt 2):251–262. doi: 10.1111/j.1469-7793.1997.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose K, Choi DW, Goldberg MP. Cytotoxicity in murine neocortical cell culture. In: Tyson C, Frazier J, editors. In Vitro Biological Methods. Vol. 1A. Academic Press; San Diego, CA: 1993. pp. 46–60. [Google Scholar]
- Sattler R, Charlton MP, Hafner M, et al. Determination of the time course and extent of neurotoxicity at defined temperatures in cultured neurons using a modified multiwell plate fluorescence scanner. J. Cereb. Blood Flow Metab. 1997;17:455–463. doi: 10.1097/00004647-199704000-00011. [DOI] [PubMed] [Google Scholar]
- Snider BJ, Tee LY, Canzoniero LM, et al. NMDA antagonists exacerbate neuronal death caused by proteasome inhibition in cultured cortical and striatal neurons. Eur. J. Neurosci. 2002;15:419–428. doi: 10.1046/j.0953-816x.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- Solovyova N, Veselovsky N, Toescu EC, et al. Ca(2+) dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca(2+)-induced Ca(2+) release triggered by physiological Ca(2+) entry. EMBO J. 2002;21:622–630. doi: 10.1093/emboj/21.4.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J. Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost LC, Lemasters JJ. A cytotoxicity assay for tumor necrosis factor employing a multiwell fluorescence scanner. Anal. Biochem. 1994;220:149–153. doi: 10.1006/abio.1994.1311. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA. NMDA-induced calcium loads recycle across the mitochondrial inner membrane of hippocampal neurons in culture. J. Neurophysiol. 2002;87:740–749. doi: 10.1152/jn.00345.2001. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Richardson SR, Thayer SA. Intracellular acidification is not a prerequisite for glutamate-triggered death of cultured hippocampal neurons. Neurosci. Lett. 1995;186:139–144. doi: 10.1016/0304-3940(95)11305-g. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J. Physiol. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Hyrc K, Lin Y, et al. Proteasome inhibitor-induced neuronal death is associated with low intracellular calcium: role of endoplasmic reticulum and mitochondrial calcium. Neuroscience Meeting Planner; Atlanta, GA. Society for Neuroscience, 2006. Online; 2006. Program No. 824.826. [Google Scholar]
- Xu W, Cormier R, Fu T, et al. Slow death of postnatal hippocampal neurons by GABA(A) receptor overactivation. J. Neurosci. 2000;20:3147–3156. doi: 10.1523/JNEUROSCI.20-09-03147.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Effect of Ca2+ influx modulators on clastolactacystin β-lactone induced neuronal death A. Murine neocortical cultures were treated with clastolactacystin-β-lactone (CL, 3 μM) for 48 hr. (-) Bay K8644 (BayK, 10 μM), kainate (KA, 1 μM) or benzamil (BENZ, 10 μM) were added to the indicated cultures at the beginning of the treatment period. Values shown are mean ± SEM cell death analyzed by PI fluorescence at the end of the 48 hr treatment period. * indicates values for BayK and benzamil treated cultures differed significantly from CL alone (P=0.007 for BayK; P=0.019 for benzamil). The injury reduction in kainate-treated cultures did not reach significance. B. Similar to A. except that cultures were treated with 1 μM clastolactacystin alone (CL) or with the addition of MK-801 (10 μM), D-APV (1 mM) or MK-801 + kainate (5 μM) during the 48 hr treatment with proteasome inhibitor. * indicates significant difference from cultures treated with CL alone; # indicates CL+MK+KA condition differs significantly from CL+MK condition.
Supplemental Figure 2. Proteasome inhibition does not alter basal neuronal Ca2+ efflux. Neuronal cultures were loaded with 45Ca2+ for 48 hr, then cultures were washed and returned to standard media, either with no additions (Sham) or 1 uM or 3 uM MG-132. 45Ca2+ efflux into the media was measured at 1 hr intervals. Values shown are mean ± SEM % cpm released (cpm in media/cpm in cells at start of efflux period).
Supplemental Figure 3. Validation of mag-fura 2 fluorescence as a measure of [Ca2+]ER A. Murine neocortical cultures were loaded with Mag-fura-2, washed into Ca2+-free buffer and then treated with thapsigargin (2.5 μM for 30 min, Thapsigargin) or sham-washed (Sham). Values shown are mean ± SEM 340/380 ratio for at least 40 cells for each condition. * indicates value in thapsigargin condition differs significantly with that in control condition (P< 0.01). B. Similar to A, except cultures were loaded with Mag-fura-2 (black line) or fura-2 (gray line). Images were obtained for 5 min, then digitonin (10 μM) was added and imaging continued for an additional 30-35 min. Values shown are fluorescence intensity (ex360 (isosbestic point)/em510) from a representative tracing for each condition. n= 8 neurons imaged with Mag-fura-2 and 5 for Fura-2.