

Abstract
Patients with poorly differentiated (PDTC), anaplastic (ATC) and radioactive iodine-refractory (RAIR) differentiated thyroid cancers have a high mortality, particularly if positive on 18F-fluorodeoxyglucose-positron emission tomography (FDG-PET). To obtain comprehensive genetic information on advanced thyroid cancers, we designed an assay panel for mass spectrometry genotyping encompassing the most significant oncogenes in this disease: 111 mutations in RET, BRAF, NRAS, HRAS, KRAS, PIK3CA, AKT1 and other related genes were surveyed in 31 cell lines, 52 primary tumors (34 PDTCs and 18 ATCs) and 55 RAIR, FDG-PET positive recurrences and metastases (nodal and distant) from 42 patients. RAS mutations were more prevalent than BRAF (44 vs 12%; p=0.002) in primary PDTC, whereas BRAF was more common than RAS (39 vs 13%, p=0.04) in PET-positive metastatic PDTC. BRAF mutations were highly prevalent in ATC (44%) and in metastatic tumors from RAIR PTC patients (95%). Among patients with multiple metastases, 9/10 showed between-sample concordance for BRAF or RAS mutations. By contrast, 5/6 patients were discordant for mutations of PIK3CA or AKT1. AKT1_G49A was found in 10 specimens, exclusively in metastases. This is the first documentation of AKT1 mutation in thyroid cancer. Thus, RAIR FDG-PET-positive metastases are enriched for BRAF mutations. If BRAF is mutated in the primary, it is likely that the metastases will harbor the defect. By contrast, absence of PIK3CA/AKT1 mutations in one specimen may not reflect the status at other sites since these mutations arise during progression, an important consideration for therapies directed at PI3K effectors.
Keywords: Mass spectrometry genotyping, Thyroid Cancer, PIK3CA, AKT1, BRAF
Introduction
Despite the favorable prognosis of well-differentiated thyroid cancer, ∼ 5% of them progress to radioactive iodine-refractory (RAIR), 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET)-positive disease, which commonly leads to death within 5 years. The histology of RAIR FDG-PET positive thyroid carcinomas at metastatic and primary sites has been characterized; 50% are poorly differentiated thyroid cancer (PDTC), 23% well-differentiated papillary thyroid cancers (WD-PTC) and 20% tall cell variant (TCV) PTC (1). Anaplastic carcinomas are invariably RAIR and have a median survival of 0.5 years. Conventional treatment is of marginal benefit for advanced thyroid cancers, emphasizing the importance of developing novel effective therapies.
Progress in our understanding of the genetic alterations underlying the development of thyroid cancer has opened the way for patient-specific therapy by targeting the mutated genes that are causally implicated in this disease. In PTC, non-overlapping mutations of RET, NTRK, RAS and BRAF, genes encoding effectors which activate MAPK, are found in about 70% of cases (2-4). RAS mutations are also found in 50% of follicular thyroid cancer (FTC), which together with PAX8/PPAR rearrangements are seen in 85% of these cancers (5). PDTC and ATC can arise from preexisting well-differentiated thyroid cancers (WDTC), particularly from PTC. Accordingly, mutations of genes considered to be early events in development of WDTC are also found in PDTC and ATC. Indeed, RET rearrangements and mutations of RAS and BRAF are found in PDTC (13%, 46−55% and 12−17%, respectively), whereas the latter two have been detected in ATC (6−52% and 25−29%) (6-9). Alterations of effectors of the phosphoinositide-3 kinase (PI3K) signaling pathway, namely PIK3CA and PTEN, have also been found in thyroid cancer. In contrast to MAPK, these genetic alterations are commonly associated with the later stages of thyroid malignant progression, being more frequent in ATC (16% and 14%, respectively) than in WD-PTC (2%, 2%) or FTC (8%, 7%) (10). Recently, a transforming mutation (E17K) in the pleckstrin homology domain of the AKT1 gene has been detected in breast, colon, ovarian and lung cancer (11-13). Alterations of PIK3CA, PTEN and AKT1 are mutually exclusive in breast cancer (11, 12). To our knowledge, there are no studies examining the prevalence of PIK3CA and AKT1 mutations in PDTC. A single study investigating for AKT1 mutations in FTC and ATC yielded negative findings (14).
Compounds that target oncoproteins in the MAPK and PI3K pathways are currently in preclinical and clinical development. It is likely that mapping of genetic alterations in cancer specimens will help determine how patients should be treated. This is particularly important in RAIR cancers, and even more so in those that are also FDG-PET positive, as these are in greatest need of new effective therapies. We designed a thyroid cancer-dedicated platform for matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (MS) genotyping (Sequenom®) of a comprehensive set of oncogenic mutations. This method for high-throughput genotyping is very sensitive for detection of sequence alterations (15, 16). This may be important for detection of mutations in advanced thyroid cancers, which commonly have intermingled stromal tissue, including massive infiltration of tumor-associated macrophages (17). Here we focused on genotyping “druggable” oncogenes encoding signaling effectors in MAPK and PI3K pathways in advanced thyroid cancers.
Material and Methods
Histopathologic analysis
Tumors were classified according to WHO 2004 criteria with the exception of TCV-PTC and PDTCs. PDTCs were defined by proliferative grading features: i.e. ≥5 mitoses/10 high-power fields (HPF) and/or tumor necrosis regardless of architectural pattern (18). This definition differs from the most recent Turin proposal that requires the presence of a solid/trabecular/insular growth pattern in addition to proliferative grading (19). Tumors were classified as TCV if they contained ≥50% tall cells. The predominant type of tumor cells present in the PDTC was classified as papillary-like, follicular-like, tall cell or oncocytic.
Thyroid cancer tissues and cell lines studied
We analyzed 52 primary tumors (34 PDTC and 18 ATC) and 55 recurrent and nodal and/or distant metastatic samples from 42 patients with RAIR FDG-PET positive thyroid carcinomas, diagnosed between 1983 and 2007 at MSKCC. We previously characterized the histopathology of 70 patients with recurrent/metastatic RAIR FDG-PET positive thyroid carcinomas (1). A patient was deemed RAIR if they had an elevated serum thyroglobulin with structural disease in the setting of a negative radioiodine diagnostic whole-body scan. A patient was entered in the study if: 1) he/she had non-RAI-avid recurrent/metastatic thyroid carcinoma; 2) specimen corresponded to a lesion identified as PET-avid within 2 years of tissue harvesting. Paraffin tissue was available from 42/70 patients, which are the focus of the current study. We analyzed 55 samples including 19 distant metastases at the following sites: bone (5), lung (5), mediastinal lymph node (3), brain (2), skin (2), chest wall (1) and pelvic soft tissue (1); 22 neck lymph node metastases and 14 recurrences: neck soft tissue (11) and trachea/esophagus (3). The study was approved by the MSKCC Institutional Review Board. We also used a panel of 31 thyroid cancer cell lines, which were genetically fingerprinted by either SNP-CGH or polymorphic short tandem repeat (STR) and verified to be unique (20). All cell lines were grown according to the recommendations of the suppliers and maintained at 5% CO2 at 37°C.
Nucleic Acid Extraction
When necessary, samples were macrodissected to have at least 50% of tumor cells. Genomic DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissues using the PUREGene Genomic DNA purification kit (Gentra, Inc., Minneapolis, MN). DNA from cell lines was extracted using Qiagen DNeasy Blood & Tissue Kit (Qiagen, Maryland, USA) and RNA was isolated using PrepEase™ RNA Spin Kit (USB, Cleveland, OH). DNA and RNA from frozen tissues were isolated by AllPrep DNA/RNA Mini Kit (Qiagen). RNA extraction from FFPE samples was performed using RecoverAll™ Total Nucleic Acid Isolation Kit (Ambion, Austin, TX) (21). DNA/RNA quality was verified by Nanodrop® ND-1000 spectrophotometry followed by PCR with primers specific for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene.
Mass spectrometry genotyping
Selection of Thyroid Cancer Genetic Alterations
We assembled a list of genetic alterations in thyroid cancer by searching the Catalogue of Somatic Mutations in Human Cancer (COSMIC, http:/www.sanger.ac.uk/genetics/CGP/cosmic/) and Pubmed (http://www.ncbi.nlm.nih.gov/sites/entrez/) databases. These included somatic, non-synonymous point mutations and small insertions and deletions in coding regions of selected genes. Genotyping assays, including PCR amplification primers and extension primers were designed using the Sequenom MassARRAY Assay Design 3.1 Software based on sequences obtained from UCSC Genome Browser (http://genome.ucsc.edu). Two or more independent genotyping assays were designed for the most relevant mutations. Overall, we designed 107 genotyping assays to interrogate 111 coding substitutions in 16 genes: BRAF, RET, NRAS, HRAS, KRAS, PIK3CA, AKT1, MET, MAP2K1, IKBKB, PIK3R5, PRKCZ, RHEB, RPS6KA3, RPS6KB1, FRAP1 (Table S1). As the MS assays for codons 12 and 13 of HRAS were not informative, we sequenced all the tumors and cell lines for alterations at these sites.
Genotyping PCR and mass spectrometry
The iPLEX assay is a single-base primer extension assay. First, a PCR reaction amplifying fragments of ∼100 bp with primers bracketing the mutation is conducted in a multiplex reaction for several mutations. Next, extension primers designed immediately adjacent to the mutation site prompt extension by one nucleotide depending on the template sequence. The difference in mass between extended products allows distinction of wild-type and mutant alleles.
Multiplexed PCR was performed in 5 μl containing 0.1 U of HotStart Taq polymerase (Kapa Biosystems, Cape Town, South Africa), 10 ng of genomic DNA, 2.5 pmol of each PCR primer and 2.5 mmol of dNTP. Thermocycling was performed at 95 °C for 15 min followed by 45 cycles of 95 °C for 20 s, 56 °C for 30 s and 72 °C for 30 s. Unincorporated dNTPs were deactivated by incubation with shrimp alkaline phosphatase, followed by heat inactivation. Single-base primer extension was carried out using 5.4 pmol of each extension primer, 50 mmol of the appropriate dNTP/ddNTP combination and 0.5 units of Thermosequenase DNA polymerase (iPlex enzyme). Extension reactions were cycled using a 200-short-cycle program that uses two cycling loops as follows: denaturation at 94° C, annealing at 52° C for 5 seconds and extension at 80° C for 5 seconds. This is repeated for a total of five cycles and then looped back to a 94° C denaturing step for 5 seconds and then a further 5 cycles of annealing and extension. The five annealing and extension steps with the single denaturing step are repeated 40 times, equating to a total of 200 cycles. A final extension is done at 72° C for three minutes and then the sample is cooled to 4° C. After the addition of a cation exchange resin to remove residual salt from the reactions, 7 nl of the purified primer extension reaction were loaded onto a matrix pad (3-hydroxypicoloinic acid) of a SpectroCHIP (Sequenom). SpectroCHIPs were analyzed using a Bruker Biflex III matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometer (SpectroREADER, Sequenom). All results were manually inspected, using the MassARRAY TyperAnalyzer v4.0 software.
To ensure that the samples from patients with multiple tumors were properly categorized, we fingerprinted their genomic DNA using a Sequenom panel that interrogates 42 SNPs. All fingerprinting mismatches were removed from the analysis.
PCR products of genomic DNA were sequenced using ABI BigDye Terminator chemistry on an ABI 3730 capillary sequencer (Applied Biosystems).
Vector Ligation and Transformation
DNA samples were amplified by PCR using High-Fidelity Taq DNA Polymerase (Invitrogen, San Diego, CA, USA) and specific primers bracketing the mutation to be analyzed. The purified PCR products were ligated into the pJET1.2/blunt cloning vector using CloneJET PCR Cloning Kit (Fermentas, Vilnius, Lithuania) and the ligation products used to transform DH5α competent bacterial cells (Invitrogen). Positive colonies were screened by colony PCR using High-Fidelity Taq DNA Polymerase (Invitrogen) and analyzed by 2% agarose gel electrophoresis. PCR products of the expected size were sequenced using pJET1.2 sequencing primers.
Screening for RET/PTC and PAX8/PPARγ rearrangements
We used tumor cDNA as template for quantitative PCR to analyze for unbalanced expression of exons 10−11 relative to 12−13 of RET, which flank the rearrangement site in the intron 11. Samples with 12−13 > 10−11 expression were screened for specific RET recombination events using primers bracketing the respective fusion points of RET/PTC1, RET/PTC2 and RET/PTC3 (22). Positive controls were cDNAs from: TPC1 cells (that express RET/PTC1), PCCL3 cells expressing RET/PTC2 and a PTC sample expressing RET/PTC3. We screened for the PAX8-PPARγ fusion by RT-PCR, using primers for all possible transcripts of PAX8/PPARγ (23). cDNA from FTC samples harboring the rearrangement were used as positive controls. GAPDH was used as internal control. PCR products were resolved by 2% agarose gel electrophoresis and selected cases were sequenced. RET rearrangements were analyzed for all cell lines, and in tumors that were wild-type for BRAF or RAS. PAX8-PPARγ was analyzed in all cell lines and primary tumors, and in metastatic lesions that were wild-type for BRAF and RAS.
Statistical methods
Statistical analyses were performed using SPSS 14.0 for Windows. Noncontinuous variables were analyzed using Fischer's exact two-sided test and continuous variables were analyzed using unpaired two-sided T tests. Survival analyses were performed using Kaplan-Meier log-rank tests. Significance was defined as p< 0.05.
RESULTS
Overall detection of thyroid cancer genetic alterations in cell lines and human tissues
We used MS to screen for 111 known mutations in 16 cancer genes. In addition, RET/PTC and PAX8/PPARγ fusion oncogenes were evaluated by quantitative PCR using cDNA as template. We analyzed 31 human thyroid cancer cell lines, 52 primary thyroid cancers (34 PDTC and 18 ATC) and 55 recurrent/metastatic samples from 42 patients with RAIR FDG-PET positive thyroid carcinomas. Altogether we found 122 genetic alterations: these corresponded to 22 of 31 (71%) thyroid cancer cell lines, 26 of 34 (76%) PDTC, 13 of 18 (72%) ATC and 44 of 55 (80%) recurrences/metastases, including RET rearrangements and point mutations in the coding regions of RET, HRAS, KRAS, NRAS, BRAF, PIK3CA, AKT1 and MET. Many of the mutations found by MS (all 9 PIK3CA, 9 AKT1 and a fraction of the RAS- and BRAF-positive tumors and cell lines) were also analyzed by Sanger sequencing to validate the assays. Sequencing and mass spec were 100% concordant for BRAF and RAS mutations. PIK3CA and AKT1 mutations were missed by Sanger sequencing in 11% and 60% of the samples, respectively, indicating that MALDI-TOF genotyping was more sensitive in finding mutations. We subcloned PCR products from AKT1 and PIK3CA-mutated samples missed by the Sanger approach and sequenced about 20 to 30 clones of each, and found that this technique detected as few as 8% (2/24 clones) of mutated cells in a specimen (Figure 1). Many of the alterations found in the cell lines have been previously reported and were useful as positive controls for optimization of our genotyping platform (Table S2). Cell line oncogene mutations not previously reported included: BRAF_T1799A in T235, T238 and KTC2; NRAS_A182G in Hth7 and TT2609-C02; HRAS_C181A in Hth-112, MET_C3029T in ML1 and PIK3CA_G1624A in T238. Twenty one out of 31 (68%) thyroid cell lines have a genetic alteration in the MAPK pathway (NOTE: for simplicity, RET, RAS and BRAF are described as genes encoding MAPK effectors, although RET and RAS also signal through multiple other pathways), with BRAF_T1799A being the most common (11/31, 35%). None of the MAPK alterations overlapped in the cell lines. We also found 3 PIK3CA mutations in cell lines, which overlapped with BRAF mutation in all cases. A MET_C3029T mutation was found in the ML1 follicular thyroid cancer cell line and in one case of ATC. The functional consequence of this alteration in the juxtamembrane domain of MET is controversial, since it has been reported as a somatic mutation conferring tumorigenic advantage (24) and as a germline polymorphism (25). Two mesothelioma cell lines harboring this mutation were more sensitive to the MET inhibitor SU11274 than wild type cells (26). By contrast, we found that growth of ML1 cells was refractory to growth inhibition by SU11274 (IC50 > 10 μM, data not shown).
Figure 1.

Mass spectrometry (left panel) and Sanger sequencing (right panel) traces of representative samples with NRAS, AKT1 and PIK3CA mutations. Note that NRAS mutant and wild-type peaks are of comparable size, which is also reflected in the sequencing trace. By contrast, the mutant peaks in AKT1 and PIK3CA were small, and missed by Sanger sequencing. Subcloning and sequencing detected 8 of 27 (29%) and 2 of 24 (8%) clones to be mutated for AKT1 and PIK3CA, respectively. wt, wild type; mut, mutant; UEP, unextended primer.
We also screened for an activating mutation in exon 2 of MEK1 (K57N), recently reported in 1% of lung cancers and shown to confer sensitivity to MEK inhibitors. None of the cell lines, primary or metastatic tumors had this mutation (27). We also screened 13 thyroid cancer cell lines and 36 primary or metastatic PDTC for mutations at 11 additional sites in exons 11 and 15 of BRAF as well as 26 KIT alterations previously found in other cancers. No mutations were found, suggesting that these particular alterations are absent or uncommon in thyroid cancer.
Genotype of primary PDTC and ATC
The genetic alterations found in primary PDTC and ATC are shown in Figure 2 and Table S3. Eight of 18 ATC were derived from PTC (4 TCV and 4 WD), whereas 10 were of unknown origin. PDTC derivation was as follows: 23/34 PTC (2 TCV and 21 WD), 7/34 FTC, 3/34 Hurthle cell carcinomas (HCC) and 1 mixed HCC/PTC (Table S3). MAPK changes were present in 25 of 34 (74%) PDTCs and 12 of 18 (67%) ATCs. We found H-K-N-RAS mutations to be much more prevalent (44%) in PDTC than BRAF mutations (12%; p=0.002). Moreover, patients with RAS-positive PDTC had a median survival of 6.6 years compared to 3.3 years for BRAF (p=0.08). Accordingly, BRAF-mutated PDTC were associated with extrathyroidal extension (ETE, p=0.04), whereas RAS mutations were robustly associated with absence of ETE (p=0.002), consistent with an opposing role of these oncogenes on key prognostic variables in PDTC. Although ATC patients have a rapidly fatal outcome irrespective of tumor genotype, BRAF mutations were more common in this aggressive histotype (44%) than in PDTC (12%; p=0.02). Six of 34 (17%) PDTC had unbalanced expression of RET exons 10−11 vs 12−13. One of these cases had a RET/PTC3 rearrangement, whereas the others probably carry RET rearrangements other than RET/PTC1, 2 or 3 (Figure S1). RET positive tumors were not associated with differences in survival compared to other groups, although they were associated with ETE (p=0.01). Five cases of RET/PTC-positive PDTC were derived from PTC and one from a HCC. None of the ATC had RET rearrangements and none of the PDTC or ATC had PAX8/PPARγ fusion oncogenes.
Figure 2.

A. Mutational frequency of BRAF, RET/PTC, NRAS, HRAS, KRAS, AKT1, PIK3CA in (A) 18 primary ATC, (B) 34 primary PDTC and (C) 23 RAIR FDG-PET positive PDTC. RAS is significant more prevalent than BRAF in primary PDTC (p=0.002) and BRAF is more prevalent than RAS in RAIR PET positive PDTC (p=0.04). RET/PTC rearrangements were analyzed in cases that were wild-type for BRAF and RAS.
Histology and genotype of recurrent and metastatic RAIR thyroid cancers
There were 55 recurrent/metastatic samples from 42 patients with RAIR disease (Table 1). Their histology was as follows: PDTC: 32/55 (58%); TCV-PTC: 12/55 (22%); WD-PTC: 7/55 (13%); HCC: 3/55 (5%) and ATC: 1/55 (2%). Overall 33 of 42 (79%) patients and 44 of 55 (80%) samples had at least one mutation. BRAF mutations were the most frequent, being found alone or associated with another mutation in 62% of the samples, followed by AKT1 (16%), RAS (13%), PIK3CA (5%) and RET/PTC (4%). The mutation frequency in recurrent/metastatic samples according to histologic subtype is shown in Table 1 and Table S4. BRAF mutations were the most common genetic alteration in PDTC (15/32, 47%) and PTC (18/19, 95%), the latter including both WD-PTC and TCV-PTC. No differences in genotype were found when comparing recurrences or nodal metastases against distant metastases.
Table 1.
Histotype and genotype of 55 samples from 42 patients with RAIR recurrent/metastatic thyroid carcinomas:
| Histotype | n | BRAF | BRAF-AKT1 | BRAF-PIK3CA | BRAF-NRAS | BRAF Total | NRAS | HRAS | RET/PTC | AKT1 | Unknown |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WD-PTC | 7 (13%) | 4 (57%) | - | 1 (14%) | 1 (14%) | 6 (85%) | 1 (14%) | - | - | - | - |
| TCV-PTC | 12 (22%) | 10 (83%) | 2 (17%) | - | - | 12 (100%) | - | - | - | - | - |
| HCC | 3 (5%) | - | - | - | - | - | - | - | - | 1 (33%) | 2 (67%) |
| PDTC | 32 (58%) | 9 (28%) | 5 (16%) | 1 (3%) | - | 15 (47%) | 3 (9%) | 2 (6%) | 2 (6%) | 1 (3%) | 9 (28%) |
| ATC | 1 (2%) | - | - | 1 (100%) | - | 1 (100%) | - | - | - | - | - |
| All types | 55 (100%) | 23 (42%) | 7 (13%) | 3 (5%) | 1 (2%) | 34 (62%) | 4 (7%) | 2 (4%) | 2 (4%) | 2 (4%) | 11 (20%) |
RAIR: Radioactive iodine refractory; WD: Well-differentiated; PTC: Papillary thyroid carcinoma; TCV: Tall cell variant; HCC: Hurthle cell carcinoma; PDTC: Poorly differentiated thyroid carcinoma; ATC: Anaplastic thyroid carcinoma.
Histology and genotype of FDG-PET positive recurrent/metastatic lesions
Table 2 shows the histotype and mutational analysis of the 35 recurrent/metastatic RAIR tumors that were also FDG-PET positive. BRAF was the most frequently mutated gene, being present in 19/35 samples (54%) followed by RAS (11%). BRAF mutations were detected in all 9 (100%) FDG-PET positive recurrent/metastatic PTC samples and in 9 of 23 (39%) FDG-PET positive PDTC specimens. All 13 (100%) recurrent/metastatic FDG-PET positive carcinomas containing significant amount of tall cells (TCV-PTC and some PDTC) harbored BRAF mutations, whereas only 6 of 22 (27%) FDG-PET positive lesions without tall cell features displayed BRAF mutations (p=0.00002).
Table 2.
Histotype and genotype of 35 RAIR FDG-PET positive recurrent/metastatic cases
| Histotype | n | BRAF | BRAF-AKTl | BRAF-PIK3CA | BRAF-NRAS | BRAF Total | NRAS | HRAS | RET/PTC | AKT1 | Unknown |
|---|---|---|---|---|---|---|---|---|---|---|---|
| WD-PTC | 4 (11%) | 3 (75%) | - | - | 1 (25%) | 4 (100%) | - | - | - | - | - |
| TCV-PTC | 5 (14%) | 5 (100%) | - | - | - | 5 (100%) | - | - | - | - | - |
| HCC | 2 (6%) | - | - | - | - | - | - | - | - | 1 (50%) | 1 (50%) |
| PDTC | 23 (66%) | 6 (26%) | 3 (13%) | - | - | 9 (39%) | 2 (9%) | 1 (4%) | 2 (9%) | 1 (4%) | 8 (35%) |
| ATC | 1 (3%) | - | - | 1 (100%) | - | 1 (100%) | - | - | - | - | - |
| All types | 35 (100%) | 14 (40%) | 3 (8%) | 1 (3%) | 1 (3%) | 19 (54%) | 2 (6%) | 1 (3%) | 2 (6%) | 2 (6%) | 9 (26%) |
RAIR: Radioactive iodine refractory, FDG-PET: 18F-fluorodeoxyglucose-positron emission tomography; WD: Well-differentiated; PTC: Papillary thyroid carcinoma; TCV: Tall cell variant; HCC: Hurthle cell carcinoma; PDTC: Poorly differentiated thyroid carcinoma; ATC: Anaplastic thyroid carcinoma.
There was a reciprocal relationship in BRAF and RAS mutation frequency between primary PDTC and FDG-PET positive PDTC. In primary PDTC RAS>BRAF (44% vs 12%; p=0.002); whereas BRAF > RAS in FGD-PET positive PDTC (39% vs 13%; p=0.04) (Figure 2). One RET/PTC2 and one RET/PTC3 were found among the PET-positive PDTC. These were the only RET alterations found in all 55 RAIR recurrent/metastatic samples.
Histopathologic and molecular correlations in patients with multiple tumor specimens
Twelve patients had multiple specimens genotyped (Table 3). For mutations of genes encoding MAPK effectors, 9 of 10 (90%) patients had concordant genotype between the different samples. One patient had the same RAS mutation in two metastatic sites. BRAF mutations were present in 9 patients. In 8 of these (89%), the BRAF mutations were present in all tumor sites tested (Figure 3). Four of the 12 patients with multiple samples also had specimens of the primary tumor. All four had a BRAF mutation in primary tumors and at all metastatic sites. As opposed to the concordance of MAPK genetic alterations between samples of individual patients, mutations of genes encoding PI3K pathway effectors (PIK3CA and AKT1) were discordant in 5 of 6 patients.
Table 3.
Histotype and genotype of 12 patients with multiple tumor specimens
| Patient # | Age, sex | Primary | Rec/met #1 | Rec/met #2 | Rec/met #3 | BRAF/RAS | PIK3CA/AKT1 |
|---|---|---|---|---|---|---|---|
| 1 | 65, F | PDTC BRAF1799T>A | PDTC BRAF1799T>A | PDTC BRAF1799T>A | NA | Concordant | NA |
| 2 | 78, F | PDTC BRAF1799T>A | PDTC BRAF1799T>A | PDTC BRAF1799T>A | NA | Concordant | NA |
| 3 | 49, M | TCV PTC BRAF1799T>A | TCV PTC BRAF1799T>A | TCV PTC BRAF1799T>A | NA | Concordant | NA |
| 4 | 85, F | TCV PTC BRAF1799T>A | PDTC BRAF1799T>A | TCV PTC BRAF1799T>A | NA | Concordant | NA |
| 5 | 61, M | NA | HCC AKT149G>A PDTC | HCC No mutations PDTC | NA | NA | Discordant |
| 6 | 81, F | NA | BRAF1799T>A AKT149G>A | BRAF1799T>A AKT149G>A | NA | Concordant | Concordant |
| 7 | 67, F | NA | PDTC NRAS182A>G | PDTC NRAS182A>G | NA | Concordant | NA |
| 8 | 65, F | NA | PDTC No mutations | PDTC No mutations | NA | NA | NA |
| 9 | 58, M | NA | PDTC BRAF1799T>A PIK3CA1624G>A ¶ | TCV PTC BRAF1799T>A | NA | Concordant | Discordant |
| 10 | 49, F | NA | TCV PTC BRAF1799T>A | TCV PTC BRAF1799T>A AKT149G>A | NA | Concordant | Discordant |
| 11 | 25, M | NA | WD PTC BRAF1799T>A PIK3CA1624G>A | TCV PTC BRAF1799T>A | NA | Concordant | Discordant |
| 12 | 25, F | NA | PDTC HRAS37G>T | PDTC RET/PTC2 | PDTC BRAF1799T>A AKT149G>A | Discordant | Discordant |
RAIR: Radioactive iodine refractory; WD: Well-differentiated; PTC: Papillary thyroid carcinoma; TCV: Tall cell variant; HCC: Hurthle cell carcinoma; PDTC: Poorly differentiated thyroid carcinoma; NA: Not applicable.
Figure 3.
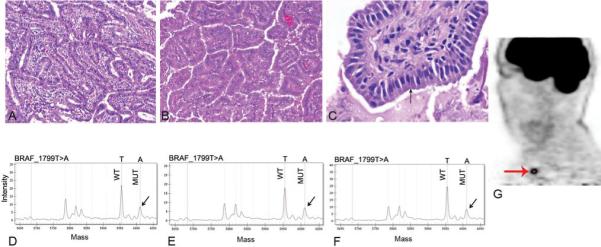
Genotype of multiple tumor sites in a patient with radioactive iodine refractory, FDG-PET positive thyroid carcinoma. A: Histology of primary tumor showing a papillary thyroid carcinoma (PTC), tall cell variant (TCV). B: Histology of metastatic TCV-PTC to lung 15 months after diagnosis C: Histology of metastatic TCV-PTC to right supraclavicular lymph node that developed 10 years after removal of primary tumor. This high power view shows the tumor cells (arrow) to be tall (their height at least twice their width) with strong eosinophilic cytoplasm. D, E, F: Mass spectrometry traces for BRAF mutation from primary tumor (A), first (B) and second (C) recurrence, respectively. Note the mutant BRAF_T1799A peak (arrow). G: FDG-PET scan from the second recurrence showing PET positive lesion (arrow) in the right supraclavicular area corresponding to specimen C.
The PIK3CA and AKT1 oncogenes in advanced thyroid cancer
Altogether, 9/55 (16%) recurrent/metastatic samples from 42 patients with RAIR FDG-PET positive thyroid carcinomas had an AKT1_G49A mutation (Table S4). None of the primary PDTC or ATC, or the cell lines harbored this mutation. We found AKT1 mutations in 6/32 (19%) PDTC, 1/3 (33%) HCC and 2 of 12 (17%) TCV-PTC. All but 2 (7/9) had a concomitant BRAF mutation. This is the first report of AKT1 mutations in thyroid cancer, and we therefore expanded the analysis to other possible mutation sites and additional samples. The entire exon 1 of AKT1 was sequenced in all cancer cell lines, PDTC and ATC as well as 13 follicular adenomas, 12 PTC and 3 FTC. No further mutations were found. We found 9 samples with PIK3CA alterations: 3 thyroid cancer cell lines, 2 primary PDTC, 1 primary ATC and 3 recurrent/metastatic lesions from patients with RAIR disease: 1 ATC, 1 PDTC and 1 WD-PTC. PIK3CA mutations were found concomitantly with BRAF mutations in 8/9 cases. Altogether, 15/18 (83%) cases with mutations in PIK3CA or AKT1 also had a BRAF mutation. We also designed Sequenom assays for other putative oncoproteins in the PI3K pathway discovered as part of the systematic resequencing of other cancer genomes: RHEB_E139K, RPS6KA3_I416V, RPS6KB1_G289E, PIK3R5_R28C, PRKCZ_S514F, IKBKB_A360S, IKBKB_Q611, FRAP1_P2476L, FRAP1_S2215Y (28, 29). No samples had mutations at any of these sites.
DISCUSSION
The feasibility of using high throughput MALDI-TOF MS to genotype patient tumor samples was recently demonstrated in a large survey of multiple tumor types (15). There are few genotyping studies of advanced thyroid cancer, and in particular no comprehensive survey of mutations in primary and metastatic cancers that are RAIR, and/or FDG-PET positive, yet these are the tumors most likely to require treatment with kinase inhibitors. These advanced cancers often have features that alter the sensitivity of mutation detection, such as ploidy changes or infiltration with stromal or immune cells (17). We found that sequence alterations present in only 8% of cells were detectable by MS, but missed by Sanger sequencing. Whereas BRAF/RAS mutations were generally detected by both methods, PIK3CA/AKT1 mutations were often missed by Sanger sequencing, possibly because the latter occur later in tumor progression, when the cancer cells are heavily intermingled with stromal cells. Alternatively, the PIK3CA or AKT1 mutations may be subclonal. This approach allowed us to screen for a large number of genetic alterations simultaneously with enhanced sensitivity, yielding a more comprehensive view of the oncogenic abnormalities in advanced forms of the disease.
The histological definition of PDTC is controversial, and the few genetic studies of this disease have shown conflicting information. Our analysis shows that PDTC with BRAF or RAS mutations have distinct biological and clinical behavior. BRAF mutations predict for poor outcome in WD-PTC (30), and we now demonstrate that this also applies to PDTC. A previous analysis of RAS in PDTC showed an association of this oncogene with poor prognosis and aggressive behavior (31). Although this paper did not genotype cancers for BRAF, this is inconsistent with our observation. This study found a high prevalence of KRAS mutations in PDTC, while we found mostly mutations in NRAS. We are confident about the mutation calls, since we had three assays interrogating NRAS_Q61R, and most KRAS assays for mutations in codons 12 and 13 were validated in colorectal tumors with positive controls for these alterations. The overall predilection for NRAS, as opposed to KRAS, mutations in thyroid cancer is also consistent with the COSMIC database. The differential outcomes of patients harboring cancers with BRAF and NRAS mutations has been also seen in melanomas (32). Although RAS mutations in PDTC indicate a better prognosis, these patients still have a median survival of 6.6 years (1). Currently, there are no effective therapies for tumors harboring oncogenic RAS mutants. A combination of MEK and PI3K/AKT/mTOR pathway inhibitors is more effective than the respective monotherapy in mouse models of RAS-driven cancers (33-35), raising expectations that this may also be the case in humans.
BRAF mutations were present in 62% of RAIR recurrent/metastatic thyroid carcinomas, and in 54% of these tumors that were also FDG-PET positive. The difference in BRAF positivity between RAIR tumors and thyroid carcinomas in general is even more marked when comparing these 2 groups by histotype. Indeed, 100% of FDG-PET positive RAIR PTCs were BRAF positive, compared to 45% of PTC in general. This is consistent with a previous study of 13 RAIR PTC cases, in which 10 out of 13 (77%) PTC harbored BRAF mutations (36). The evidence for causality is supported by the fact that conditional activation of BRAFV600E downregulates expression of the sodium iodide symporter (NIS) in thyroid cells in vitro (37). Moreover, human thyroid cancers with BRAF mutations show greater reduction of NIS mRNA compared to tumors with other mutations or with no identifiable genetic changes (38). This report is the first to demonstrate an inordinately high prevalence of BRAF mutations in tumors with functional evidence of loss of RAI avidity. In PDTC, RAIR FDG-PET positive tumors harbor BRAF mutations in 39% of the samples, whereas BRAF is significantly less frequent (12%) in primary PDTC, which more commonly presented with RAS mutations (44%). The high frequency of BRAF mutation in RAIR FDG-PET positive tumors makes BRAF an attractive target for therapy to induce cell death and/or to restore RAI uptake. The putative success of such therapeutic strategies requires that all metastatic or recurrent tumors in the same patient harbor the same genetic defect. BRAF mutation is an early event in thyroid cancer pathogenesis (reviewed in (39)), and likely to be required for the viability of all subclones of the primary tumor. However, genetic heterogeneity between different primary tumors in multifocal PTC has been reported (40, 41), as well as between primary tumors and regional lymph node metastases (42). By contrast we found that 8/9 patients with BRAF-positive cancers had the same BRAF mutation in all tumor sites tested. Primary tumors of 4/9 of these patients were available, and in all of them the BRAF mutation found in the primary tumor was also present in all metastases. This supports the evidence that BRAF is an early event in thyroid carcinogenesis, and increases the likelihood that these tumors may be addicted to the oncoprotein.
Most cancers with RET/PTC rearrangements are WD-PTC, which are usually not associated with aggressive behavior. Accordingly, we found only two recurrent/metastatic RAIR tumors harboring RET/PTC oncogenes. Although rare, this information could be of clinical value as treatment selection becomes more individualized, particularly since multikinase inhibitors with potent activity on RET kinase are entering the clinic (43).
By contrast to BRAF, for which primary and metastatic lesions were highly concordant, mutations in PIK3CA or AKT1 were frequently discordant between metastatic samples of the same individuals, consistent with a late acquisition of these oncogeness during tumor progression. Mutations of AKT1 and PIK3CA were mutually exclusive. This is the first report of AKT1 mutations in this disease. The AKT1G49A mutation was first detected in breast, colon, ovarian and lung cancers. It constitutively activates AKT signaling, and induces leukemia in mice (13). AKT1 activation is associated with tumor invasion in papillary and follicular subtypes of thyroid cancer (44, 45). Moreover, PI3K-AKT activation has been proposed to play an important role in the development of FTC metastasis in TRßPV/PV mice, which have homozygous inactivating mutations of the thyroid hormone receptor β (44).
PIK3CA/AKT1 mutations almost invariably coexisted with BRAF mutations in our study, pointing to possible cooperativity of co-activation of MAPK and PI3K in disease progression. The PIK3CA alterations found are within the known hotspots, which have been shown to induce AKT phosphorylation and possess strong oncogenic potential (46). We did not find concomitant alterations in RAS and PIK3CA. This association has been reported in a subset of anaplastic cancers (9), but based on the current analysis are likely to be comparatively infrequent.
Thus, the genotype of primary thyroid cancers for BRAF is likely to give an accurate account of the status of this oncogene at metastatic sites. This may not be the case for PIK3CA or AKT1. This introduces significant caveats to the use of primary cancer specimens for genetic analysis, particularly if patients will be segregated for specific therapies based on this information. This is worth noting in light of the fact that there are many experimental compounds targeting effectors in the PI3K-AKT-mTOR pathway at various stages of clinical development.
In summary, we show that PDTC with BRAF and RAS have distinct biological and clinical behaviors. BRAF mutations are highly prevalent in FDG-PET positive RAIR metastatic thyroid cancers, thus placing this oncoprotein as a prime therapeutic target in the advanced forms of this disease. Mutations of PIK3CA and AKT1, the latter not previously described in this disease, are comparatively frequent in advanced thyroid cancers, particularly in metastatic or recurrent lesions. BRAF mutations are concordant between primary and metastatic specimens, yet this is not the case for PIK3CA or AKT1, which has implications for our understanding of the sequence of events in thyroid cancer pathogenesis, and on how we may apply this information for patient management.
Acknowledgements
This work was supported by NIH grant CA50706, the Margot Rosenberg Pulitzer Foundation, and a Byrne award from MSKCC (to JAF). J.C. Ricarte-Filho was supported by a research grant from CAPES (BEX-0644/07-2). The MSKCC Sequenom facility is supported by the Anbinder Fund.
References
- 1.Rivera M, Ghossein RA, Schoder H, Gomez D, Larson SM, Tuttle RM. Histopathologic characterization of radioactive iodine-refractory fluorodeoxyglucose-positron emission tomography-positive thyroid carcinoma. Cancer. 2008;113(1):48–56. doi: 10.1002/cncr.23515. [DOI] [PubMed] [Google Scholar]
- 2.Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer research. 2003;63(7):1454–7. [PubMed] [Google Scholar]
- 3.Soares P, Trovisco V, Rocha AS, et al. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene. 2003;22(29):4578–80. doi: 10.1038/sj.onc.1206706. [DOI] [PubMed] [Google Scholar]
- 4.Frattini M, Ferrario C, Bressan P, et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene. 2004;23(44):7436–40. doi: 10.1038/sj.onc.1207980. [DOI] [PubMed] [Google Scholar]
- 5.Nikiforova MN, Lynch RA, Biddinger PW, et al. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. The Journal of clinical endocrinology and metabolism. 2003;88(5):2318–26. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- 6.Santoro M, Papotti M, Chiappetta G, et al. RET activation and clinicopathologic features in poorly differentiated thyroid tumors. The Journal of clinical endocrinology and metabolism. 2002;87(1):370–9. doi: 10.1210/jcem.87.1.8174. [DOI] [PubMed] [Google Scholar]
- 7.Hou P, Liu D, Shan Y, et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13(4):1161–70. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 8.Santarpia L, El-Naggar AK, Cote GJ, Myers JN, Sherman SI. Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. The Journal of clinical endocrinology and metabolism. 2008;93(1):278–84. doi: 10.1210/jc.2007-1076. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Rostan G, Costa AM, Pereira-Castro I, et al. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer research. 2005;65(22):10199–207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 10.Paes JE, Ringel MD. Dysregulation of the phosphatidylinositol 3-kinase pathway in thyroid neoplasia. Endocrinol Metab Clin North Am. 2008;37(2):375–87, viii-ix. doi: 10.1016/j.ecl.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research. 2008;68(15):6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bleeker FE, Felicioni L, Buttitta F, et al. AKT1(E17K) in human solid tumours. Oncogene. 2008 doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- 13.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Hou P, Ji M, et al. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. The Journal of clinical endocrinology and metabolism. 2008;93(8):3106–16. doi: 10.1210/jc.2008-0273. [DOI] [PubMed] [Google Scholar]
- 15.Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nature genetics. 2007;39(3):347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 16.Vivante A, Amariglio N, Koren-Michowitz M, et al. High-throughput, sensitive and quantitative assay for the detection of BCR-ABL kinase domain mutations. Leukemia. 2007;21(6):1318–21. doi: 10.1038/sj.leu.2404635. [DOI] [PubMed] [Google Scholar]
- 17.Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocrine-related cancer. 2008;15(4):1069–74. doi: 10.1677/ERC-08-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiltzik D, Carlson DL, Tuttle RM, et al. Poorly differentiated thyroid carcinomas defined on the basis of mitosis and necrosis: a clinicopathologic study of 58 patients. Cancer. 2006;106(6):1286–95. doi: 10.1002/cncr.21739. [DOI] [PubMed] [Google Scholar]
- 19.Volante M, Collini P, Nikiforov YE, et al. Poorly differentiated thyroid carcinoma: the Turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. The American journal of surgical pathology. 2007;31(8):1256–64. doi: 10.1097/PAS.0b013e3180309e6a. [DOI] [PubMed] [Google Scholar]
- 20.Schweppe RE, Klopper JP, Korch C, et al. DNA Profiling Analysis of 40 Human Thyroid Cancer Cell Lines Reveals Cross-Contamination Resulting in Cell Line Redundancy and Misidentification. The Journal of clinical endocrinology and metabolism. 2008 doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Smyth P, Cahill S, et al. Improved RNA quality and TaqMan Pre-amplification method (PreAmp) to enhance expression analysis from formalin fixed paraffin embedded (FFPE) materials. BMC Biotechnol. 2008;8:10. doi: 10.1186/1472-6750-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imkamp F, von Wasielewski R, Musholt TJ, Musholt PB. Rearrangement analysis in archival thyroid tissues: punching microdissection and artificial RET/PTC 1−12 transcripts. The Journal of surgical research. 2007;143(2):350–63. doi: 10.1016/j.jss.2006.10.033. [DOI] [PubMed] [Google Scholar]
- 23.Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. The American journal of surgical pathology. 2002;26(8):1016–23. doi: 10.1097/00000478-200208000-00006. [DOI] [PubMed] [Google Scholar]
- 24.Ma PC, Kijima T, Maulik G, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer research. 2003;63(19):6272–81. [PubMed] [Google Scholar]
- 25.Schmidt L, Junker K, Nakaigawa N, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18(14):2343–50. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- 26.Jagadeeswaran R, Ma PC, Seiwert TY, et al. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer research. 2006;66(1):352–61. doi: 10.1158/0008-5472.CAN-04-4567. [DOI] [PubMed] [Google Scholar]
- 27.Marks JL, Gong Y, Chitale D, et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer research. 2008;68(14):5524–8. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 30.Xing M. BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev. 2007;28(7):742–62. doi: 10.1210/er.2007-0007. [DOI] [PubMed] [Google Scholar]
- 31.Garcia-Rostan G, Zhao H, Camp RL, et al. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J Clin Oncol. 2003;21(17):3226–35. doi: 10.1200/JCO.2003.10.130. [DOI] [PubMed] [Google Scholar]
- 32.Ugurel S, Thirumaran RK, Bloethner S, et al. B-RAF and N-RAS mutations are preserved during short time in vitro propagation and differentially impact prognosis. PLoS ONE. 2007;2(2):e236. doi: 10.1371/journal.pone.0000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Downward J. Targeting RAS and PI3K in lung cancer. Nature medicine. 2008;14(12):1315–6. doi: 10.1038/nm1208-1315. [DOI] [PubMed] [Google Scholar]
- 34.Bedogni B, Welford SM, Kwan AC, Ranger-Moore J, Saboda K, Powell MB. Inhibition of phosphatidylinositol-3-kinase and mitogen-activated protein kinase kinase 1/2 prevents melanoma development and promotes melanoma regression in the transgenic TPRas mouse model. Molecular cancer therapeutics. 2006;5(12):3071–7. doi: 10.1158/1535-7163.MCT-06-0269. [DOI] [PubMed] [Google Scholar]
- 35.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nature medicine. 2008;14(12):1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mian C, Barollo S, Pennelli G, et al. Molecular characteristics in papillary thyroid cancers (PTCs) with no 131I uptake. Clinical endocrinology. 2008;68(1):108–16. doi: 10.1111/j.1365-2265.2007.03008.x. [DOI] [PubMed] [Google Scholar]
- 37.Mitsutake N, Knauf JA, Mitsutake S, Mesa C, Jr., Zhang L, Fagin JA. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer research. 2005;65(6):2465–73. doi: 10.1158/0008-5472.CAN-04-3314. [DOI] [PubMed] [Google Scholar]
- 38.Durante C, Puxeddu E, Ferretti E, et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. The Journal of clinical endocrinology and metabolism. 2007;92(7):2840–3. doi: 10.1210/jc.2006-2707. [DOI] [PubMed] [Google Scholar]
- 39.Fagin JA. Challenging dogma in thyroid cancer molecular genetics--role of RET/PTC and BRAF in tumor initiation. The Journal of clinical endocrinology and metabolism. 2004;89(9):4264–6. doi: 10.1210/jc.2004-1426. [DOI] [PubMed] [Google Scholar]
- 40.Zhu Z, Ciampi R, Nikiforova MN, Gandhi M, Nikiforov YE. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: effects of the detection methods and genetic heterogeneity. The Journal of clinical endocrinology and metabolism. 2006;91(9):3603–10. doi: 10.1210/jc.2006-1006. [DOI] [PubMed] [Google Scholar]
- 41.Giannini R, Ugolini C, Lupi C, et al. The heterogeneous distribution of BRAF mutation supports the independent clonal origin of distinct tumor foci in multifocal papillary thyroid carcinoma. The Journal of clinical endocrinology and metabolism. 2007;92(9):3511–6. doi: 10.1210/jc.2007-0594. [DOI] [PubMed] [Google Scholar]
- 42.Oler G, Ebina KN, Michaluart P, Jr., Kimura ET, Cerutti J. Investigation of BRAF mutation in a series of papillary thyroid carcinoma and matched-lymph node metastasis reveals a new mutation in metastasis. Clinical endocrinology. 2005;62(4):509–11. doi: 10.1111/j.1365-2265.2005.02235.x. [DOI] [PubMed] [Google Scholar]
- 43.Santoro M, Carlomagno F. Drug insight: Small-molecule inhibitors of protein kinases in the treatment of thyroid cancer. Nature clinical practice. 2006;2(1):42–52. doi: 10.1038/ncpendmet0073. [DOI] [PubMed] [Google Scholar]
- 44.Kim CS, Vasko VV, Kato Y, et al. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146(10):4456–63. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 45.Vasko V, Saji M, Hardy E, et al. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. Journal of medical genetics. 2004;41(3):161–70. doi: 10.1136/jmg.2003.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104(13):5569–74. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]