

Abstract
The goal of the present study was to develop and evaluate microsponge-based topical delivery system of mupirocin for sustained release and enhanced drug deposition in the skin. Microsponges containing mupirocin were prepared by an emulsion solvent diffusion method. The effect of formulation and process variables such as internal phase volume and stirring speed on the physical characteristics of microsponges were examined on optimized drug/polymer ratio by 32 factorial design. The optimized microsponges were incorporated into an emulgel base. In vitro drug release, ex vivo drug deposition, and in vivo antibacterial activity of mupirocin-loaded formulations were studied. Developed microsponges were spherical and porous, and there was no interaction between drug and polymer molecules. Emulgels containing microsponges showed desired physical properties. Drug release through cellulose dialysis membrane showed diffusion-controlled release pattern and drug deposition studies using rat abdominal skin exhibited significant retention of active in skin from microsponge-based formulations by 24 h. The optimized formulations were stable and nonirritant to skin as demonstrated by Draize patch test. Microsponges-based emulgel formulations showed prolonged efficacy in mouse surgical wound model infected with S. aureus. Mupirocin was stable in topical emulgel formulations and showed enhanced retention in the skin indicating better potential of the delivery system for treatment of primary and secondary skin infections, such as impetigo, eczema, and atopic dermatitis.
Key words: emulgel, ex vivo drug deposition, microsponges, mouse surgical wound model, mupirocin
INTRODUCTION
Microparticles and nanoparticles have been increasingly investigated to achieve targeted and sustained release of drugs (1). Microsponges are porous, polymeric microspheres that are mostly used for prolonged topical administration. Microsponges are designed to deliver a pharmaceutically active ingredient efficiently at minimum dose and also to enhance stability, reduce side effects, and modify drug release profiles (2). These attributes have been successfully demonstrated in the FDA-approved Retin-A Micro® (0.1% or 0.04% tretinoin) and Carac (0.5% 5-flurouracil) products for acne treatment and actinic keratoses, respectively.
Many of conventional delivery systems require high concentrations of active agents to be incorporated for effective therapy because of their low efficiency as delivery systems. Thus, the need exists for delivery systems to maximize the period of time that an active ingredient is present, either on the skin surface or within the epidermis while minimizing its transdermal penetration into the body. The microsponge-based polymeric microspheres uniquely fulfill such requirements (3).
Microsponges are prepared by several methods utilizing emulsion systems as well as by suspension polymerization in a liquid–liquid system. The most common emulsion system used is oil-in-water (o/w), with the microsponges being produced by the emulsion solvent diffusion (ESD) method (4).
Mupirocin (MP), a topical antibiotic agent is effectively used for treatment of primary and secondary skin infections (5). MP is produced by Pseudomonas fluorescens and has in vitro activity against a range of Gram-positive and some Gram-negative bacteria. However, MP also inhibits the growth of a number of pathogenic fungi in vitro, including a range of dermatophytes and pityrosporum (6). MP inhibits bacterial protein synthesis by binding to the enzyme, isoleucyl-t-RNA synthetase, which prevents the incorporation of isoleucine into proteins (7). Its chemical structure and mechanism of action are unique among antibiotics in clinical use; therefore, cross-resistance between MP and other antibiotics is not a concern (8). MP reaching the systemic circulation is rapidly converted to monic acid, an inactive metabolite that is rapidly excreted in the urine. Penetration into the deeper epidermal and dermal layers is enhanced in traumatized skin and under occlusive dressings. Mupirocin is slowly metabolized by the skin to the antimicrobially inactive metabolite monic acid (6). Therefore, controlling the release of drug will improve the efficacy of formulation and decrease the frequency of application.
The mupirocin 2% ointment with polyethylene glycol applied three times daily has demonstrable activity and is currently marketed. Topical ointment preparations are less acceptable to patients, since ointments have high viscosities leading to inappropriate application to skin lesions, and patients may report garment soiling from greasy residues (9). To enhance patient acceptance and compliance, novel formulations could be developed such as creams, gels, emulgels containing particulate drug carriers like microparticles and nanoparticles. These carriers could protect and release the active in a controlled manner. Benzoyl peroxide microsponges have already been developed for topical treatment of acne and athlete’s foot (10).
Literature search revealed that no study carried out to formulate once a day sustained release medication containing MP. Thus, the aim of the present investigation was to design microsponges as novel carriers for topical delivery of MP. This investigation consisted of preparation, optimization, and evaluation of MP microsponges and incorporation of optimized microsponges in semisolid vehicle base to obtain cosmetically acceptable products. Factorial design or response surface methodology was used to study the effect of factors (formulation and process variables) influencing responses [entrapment efficiency (EE), production yield, and mean particle size] by carrying out limited number of experiments (11). The optimized microsponges were incorporated into an emulgel base that possesses the advantages of both emulsions and gels with exclusively accomplished requirements of vehicle for topical drug delivery systems (12).
MATERIALS AND METHODS
Materials
MP was procured from Mctony Biotech (Tianjin) Co. Ltd. (Tianjin, China). Ethyl cellulose 46 cp was obtained from Sigma–Aldrich (St. Louis, USA). Polyvinyl alcohol (PVA; MW = approximately 1:25,000), methanol, and acetonitrile were procured from S. D. Fine-Chem Limited (Mumbai, India). Sepigel 305 (polyacrylamide, C13-14 isoparaffin-laureth-7) was purchased from Seppic (Paris, France). All other chemicals and solvents were of analytical reagent grade. Animal studies were performed in accordance with protocols approved by Institutional Animal Ethics Committee (IAEC, C. U. Shah College of Pharmacy, Mumbai, India).
Methods
MP microsponges were prepared by an ESD method. The organic internal phase containing MP and ethyl cellulose in dichloromethane was gradually added into external phase, which contained PVA as emulsifying agent. The mixture was stirred at 1,000–2,000 rpm for 3 h at room temperature to remove dichloromethane from the reaction flask. The formed microsponges were filtered, washed with distilled water, and dried at room temperature. Microsponges were weighed, and production yield (PY) was determined.
Drug Content Analysis
The quantitative determination of MP in microsponges was carried out using a reversed-phase high-performance liquid chromatograph (HPLC) analysis method. The HPLC instrument was equipped with a model series CCPE pump (Tosoh, Tokyo, Japan), with a 20-μL loop injector and a linear model UV absorbance detector. Data were processed using EZChrom™ Chromatography Data System, Version 6.7 (Scientific Software Inc., San Ramon, CA, USA).
Briefly, the chromatographic separations were performed using Hypersil GOLD C18, 5 μm, 250 × 4.6 mm i.d. column, eluted with mobile phase at the flow rate of 0.5 mL/min, and the eluent was monitored using a UV detector at 221 nm. The mobile phase consisting of methanol–water (80:20, v/v) was adjusted to pH 5.0 with ortho-phosphoric acid. The samples were prepared in mobile phase and filtered through 0.2-μm cellulose acetate membrane filter. All the determinations were performed at ambient temperature, and the retention time was found to be 8.2 min for MP. A linear relationship (r2 > 0.9995) was found between the peak area vs. concentration. External standardization by peak area was used for quantitative determination of MP. The developed HPLC method was validated for linearity, accuracy, precision, and specificity as per ICH guidelines.
The actual drug content and EE were calculated as given below:
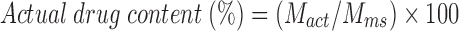 |
1 |
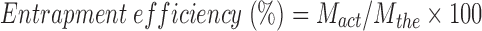 |
2 |
where Mact is the actual amount of MP in weighed quantity of microsponges, Mms is the weighed quantity of microsponges, and Mthe is the theoretical amount of MP in microsponges.
Optimization of Formulation Parameters and Process Variables
Preliminary trials were undertaken to establish the effect of mupirocin/ethylcellulose (MP/EC) ratios on the physical characteristics of microsponges. In all formulations, the total amount of polymer, internal phase volume, PVA concentration, and stirring rate were kept constant.
To optimize the dependent variables such as production yield, EE, and mean particle size of microsponges, a series of formulations were prepared by 32 factorial design using volume of internal phase (X1) and stirring rate (X2) as independent variables.
Differential Scanning Calorimetry
Thermal analysis of MP, ethyl cellulose, and MP-loaded microsponge-based formulations were studied employing differential scanning calorimeter (Mettler Toledo DSC, USA). Samples (5 mg) were accurately weighed into aluminum pans and sealed. All samples were run at a heating rate of 10°C/min over a temperature range 25–300°C in atmosphere of nitrogen.
Scanning Electron Microscopy
The morphology of microsponges was examined using a scanning electron microscope (GEOL 5400, USA) operating at 20 kV. Dried microspheres were coated with gold–palladium alloy for 45 s under an argon atmosphere before observation. SEM photograph was recorded at magnification of ×500.
Particle Size Studies and Porosity Determination
Particle size studies were carried out using laser light scattering technique using Mastersizer 2000 (Malvern Instuments Ltd., Worcestershire, UK). The porous properties were determined by Mercury Intrusion Porosimeter (Autoscan 60, Quantachrome, USA) in the pressure range 0–4,000 kg/cm2.
Preparation of Mupirocin-Loaded Emulgel
Oil-in-water emulsion was prepared using Tween-20 and Span-20 (3–4% w/w) as emulsifying agents and liquid paraffin (5% w/w) as an oily phase with stirring at 1,800–2,000 rpm for 20 min. The obtained emulsion was added to sepigel with gentle stirring to get the emulgel base. At this point, either MP (2% w/w) or MP microsponges (equivalent to 2% w/w of MP) were incorporated to get homogenous emulgel-based topical delivery systems.
In Vitro Release Studies
In vitro release studies were carried out using Franz diffusion cells with a receptor compartment volume of 20 mL and an effective diffusion area of 3.14 cm2. Cellulose dialysis membrane (Himedia, Mumbai, India) was soaked in receptor media (phosphate buffer, pH 5.8) for 24 h before experiment. A predetermined amount of emulgel-containing MP microsponges (MS emulgel) was placed on the donor side. The receptor medium was continuously stirred at 600 rpm and thermostated at 32 ± 0.5°C with a circulating jacket. At predetermined time intervals, 2 mL samples were withdrawn from the receiver compartment and replaced with an equal volume of fresh buffer. The collected samples were analyzed by HPLC. The release kinetics of conventional emulgel-containing mupirocin (MP emulgel) and marketed mupirocin ointment formulation (MP ointment) were used for comparison. The drug release data were analyzed to determine the release kinetics (zero-order and first-order) as well as diffusion controlled mechanism (Higuchi model) using linear regression analysis.
Ex Vivo Drug Deposition Studies
Drug deposition study was performed on the excised rat abdominal skin using Franz diffusion cell (13). Epidermal side of the skin was exposed to ambient condition, while dermal side was kept facing the receptor solution. Receptor compartment containing 20 mL phosphate buffer pH 5.8 was thermostated at 32 ± 0.5°C and stirred at 600 rpm. Skin was saturated with diffusion medium for 1 h before the application of sample. A 200-mg of sample was applied on the donor compartment. For determination of drug deposited in the skin, the diffusion cell was dismantled after a period of 4, 8, 16, and 24 h. The skin was carefully removed, and drug present on the skin surface was cleaned with distilled water.
Quantification of Mupirocin in Skin Samples
MP was extracted from skin using a modification of the procedure described by Echevarria et al. (14). Briefly, the skin was cut into small pieces and homogenized with 10 mL methanol by tissue homogenizer. The homogenized sample was subjected to ultrasonication for 10 min for complete extraction of drug. This methanolic extract was centrifuged at 5,000 rpm for 10 min. The supernatant was collected, evaporated, and reconstituted with mobile phase. The sample was filtered through a 0.2-μm membrane filter and analyzed by developed HPLC method as outlined earlier. To determine recovery from skin, the intact skin was spiked with known quantities of the compound and homogenized, extracted, and analyzed as described above.
Primary Skin Irritation Studies
Primary skin irritation studies of the optimized formulations were performed using New Zealand white albino rabbits in accordance with the guidelines of the Consumer Product Safety Commission (15). Formalin and placebo emulgel formulation were used as positive and negative controls, respectively. The scores were recorded as per the Draize patch test.
In Vivo Studies
Swiss albino mice, weighing 18–20 g were used to compare the efficacy of developed MP-loaded formulations with that of marketed MP ointment formulation using mouse surgical wound model infected with Staphylococcus aureus (9). The surgical wound model was constructed as previously described by McRipley et al. (16). Briefly, superficial surgical wounds were produced on the backs of mice by making a longitudinal midline incision 2.3 ± 0.2 cm in length and extending down to the panniculus carnosus. A contaminated suture was inserted through the skin to infect the wound and secured by knotting. Therapy was administered topically 0.1 mL/mouse. Treatment was initiated at 4 h after surgery and continued for further 3 days. On day 5 after surgery, 16 to 20 h after the last topical application, the animals were killed by diethyl ether asphyxiation. A 1- × 2-cm area of skin, including the wound, was excised and homogenized in 1 mL of saline solution. The homogenates were serially diluted, plated on agar plates, and incubated at 37°C. After 24 h, the number of colony forming units (CFU) per wound was counted. Bacterial counts were expressed in terms of mean ± SD.
Stability Studies
The prepared emulgels were packed in aluminum collapsible tubes (5 g) and subjected to stability studies at 5°C, 25°C/60% RH, 30°C/65% RH, and 40°C/75% RH for a period of 3 months. Samples were withdrawn at 15-day time intervals and evaluated for physical appearance, pH, rheological properties, drug content, and drug release profiles.
Statistical Analysis
The data obtained from each experiment was subjected to statistical analysis using one-way analysis of variance followed by Bonferroni multiple comparisons test. P < 0.05 was considered to be significant.
RESULTS
Formulation and Optimization of Microsponges
The methods of preparation of microsponges are limited in terms of complexity and cost. Some commercially available microsponges are prepared by suspension polymerization method, but ESD method serves as an alternative for preparing microsponges (2,4). ESD method was found to be an easy, reproducible, rapid technique for the preparation of MP microsponges. Moreover, it had an advantage of avoiding solvent toxicity.
The effect of MP/EC ratio on production yield, EE, and mean particle size is shown in Table I. It is indicated that MP/EC ratio (1.5:1) had the optimum capacity for drug entrapment. With further increase in drug/polymer ratio from 1.5:1 to 2:1, no significant change in EE and production yield was observed. Therefore, further optimization of formulation MP6 was carried out using internal phase volume (X1) and stirring rate (X2) as the independent variables by 32 factorial design (13,17).
Table I.
Optimization of Drug:Polymer Ratio for Preparation of MP Microsponges
| Formulation code | Drug: polymer ratio (by weight) | Theoretical drug content (%) | Actual drug contenta (% ± SD) | Production yielda (% ± SD) | Entrapment efficiencya (% ± SD) | Mean particle sizea (µm ± SD) |
|---|---|---|---|---|---|---|
| MP1 | 0.25:1 | 16.66 | 10.63 ± 1.25 | 67.03 ± 1.36 | 63.81 ± 3.49 | 30.36 ± 4.28 |
| MP2 | 0.5:1 | 28.57 | 20.38 ± 0.57 | 68.11 ± 1.78 | 71.33 ± 2.01 | 32.84 ± 6.35 |
| MP3 | 0.75:1 | 37.50 | 30.66 ± 1.39 | 69.28 ± 2.77 | 81.76 ± 2.34 | 35.28 ± 4.95 |
| MP4 | 1:1 | 44.44 | 38.80 ± 0.80 | 72.41 ± 1.26 | 87.32 ± 1.79 | 38.41 ± 3.34 |
| MP5 | 1.25:1 | 50.00 | 45.55 ± 1.56 | 78.24 ± 3.94 | 91.10 ± 2.43 | 40.32 ± 4.72 |
| MP6 | 1.5:1 | 54.55 | 50.89 ± 0.93 | 81.97 ± 1.98 | 93.29 ± 1.18 | 43.70 ± 3.45 |
| MP7 | 1.75:1 | 58.33 | 53.12 ± 1.12 | 80.43 ± 2.46 | 91.07 ± 1.49 | 47.51 ± 6.83 |
| MP8 | 2:1 | 61.54 | 54.98 ± 1.72 | 79.62 ± 1.85 | 89.34 ± 2.59 | 52.76 ± 5.24 |
aEach observation is the mean ± SD of three determinations
Responses of different batches obtained using factorial design are shown in Table II. A linear model generated for % EE as dependent variable revealed that the internal phase volume and stirring rate had a significant antagonistic influence on % EE without producing any interaction. The regression Eq. 3 for % EE is as follows:
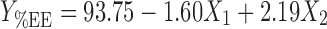 |
3 |
Table II.
Optimization of Internal Phase Volume and Stirring Rate by 32 Factorial Design
| Formulation code | Variable X 1 internal phase volume (mL) | Variable X 2 stirring rate (rpm) | Production yielda (% ± SD) | Entrapment efficiency a (% ± SD) | Mean particle sizea (µm ± SD) |
|---|---|---|---|---|---|
| IS-1 | 5 (−1) | 1,000 (−1) | 88.24 ± 1.13 | 92.18 ± 2.13 | 79.3 ± 6.38 |
| IS-2 | 5 (−1) | 1,500 (0) | 85.93 ± 2.42 | 95.68 ± 2.36 | 65.2 ± 5.24 |
| IS-3 | 5 (−1) | 2,000 (+1) | 83.34 ± 1.68 | 97.28 ± 1.15 | 55.1 ± 4.83 |
| IS-4 | 7.5 (0) | 1,000 (−1) | 82.19 ± 1.46 | 90.86 ± 2.19 | 54.7 ± 5.06 |
| IS-5 | 7.5 (0) | 1,500 (0) | 81.97 ± 1.98 | 93.29 ± 1.18 | 43.7 ± 3.45 |
| IS-6 | 7.5 (0) | 2,000 (+1) | 80.46 ± 2.34 | 95.32 ± 1.68 | 32.8 ± 4.19 |
| IS-7 | 10 (+1) | 1,000 (−1) | 80.12 ± 1.68 | 89.54 ± 1.76 | 28.3 ± 3.64 |
| IS-8 | 10 (+1) | 1,500 (0) | 79.87 ± 1.97 | 92.87 ± 1.63 | 18.2 ± 2.93 |
| IS-9 | 10 (+1) | 2,000 (+1) | 77.46 ± 1.82 | 93.12 ± 1.38 | 12.9 ± 3.14 |
aEach observation is the mean ± SD of three determinations
A linear regression Eq. 4 generated for PY showed the negative influence of both the independent variables with a R2 value of 0.9805.
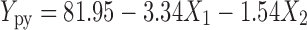 |
4 |
A polynomial regression Eq. 5 revealed that the negative influence of both the independent variables with positive interaction on mean particle size of the microsponges.
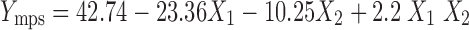 |
5 |
Results of multiple regression analysis of all the parameters investigated are summarized in Table III. Response surface methodology study indicated that formulation IS-8 was better in terms of EE, production yield, and particle size. Thus, optimized microsponges batch (IS-8) was subjected to further characterization studies and incorporated into an emulgel base.
Table III.
Summary of Regression Analysis Results for Measured Responses
| Parameters | Coefficients | ||||||
|---|---|---|---|---|---|---|---|
| β 0 | β 1 | β 2 | β 11 | β 22 | β 12 | r 2 | |
| Entrapment efficiency | 93.75 | −1.60 | 2.19 | 0.28 | −0.89 | −0.38 | 0.9871 |
| Production yield | 81.95 | −3.34 | −1.54 | 0.953 | −0.621 | 0.56 | 0.9805 |
| Mean particle size | 42.74 | −23.36 | −10.25 | −0.56 | 1.48 | 2.2 | 0.9990 |
Characterization of Microsponges
The thermal behavior of MP, ethyl cellulose, and optimized MP microsponge formulations is shown in thermograms (Fig. 1). MP presented a sharp endothermic peak at 63.4°C corresponding to its melting point, while at that temperature, optimized MP microsponges presented a broad endothermic peak with the loss of its sharp appearance. SEM of MP-loaded microsponges as shown in Fig. 2 revealed that the microsponges were uniform, spherical in shape, and porous in nature. No intact MP crystals were seen visually. The result obtained from porosimetry experiments of different microsponges formulations is depicted in Table IV. The porosity of microsponges formulations was found to increase with increasing the drug amount.
Fig. 1.

DSC thermograms of a ethylcellulose, b MP, and c MP-loaded microsponges (1.5:1)
Fig. 2.

SEM of MP loaded microsponges under ×500
Table IV.
The Effect of Drug/Polymer Ratio on Porosity of Microsponges Formulations
| Formulation code | Total intrusion volumea (mL g−1) | Total pore areaa (m2 g−1) | Average pore diametera (µm) | Bulk densitya (g mL−1) | True densitya (g mL−1) | Porositya (%) |
|---|---|---|---|---|---|---|
| MP1 | 1.02 | 20.21 | 0.20 | 0.29 | 0.42 | 30.64 |
| MP2 | 1.08 | 25.63 | 0.17 | 0.32 | 0.48 | 32.14 |
| MP3 | 1.14 | 29.84 | 0.15 | 0.33 | 0.56 | 39.73 |
| MP4 | 1.21 | 34.73 | 0.14 | 0.41 | 0.69 | 41.26 |
| MP5 | 1.25 | 42.15 | 0.12 | 0.43 | 0.80 | 46.81 |
| MP6 | 1.26 | 53.46 | 0.09 | 0.39 | 0.83 | 53.44 |
| MP7 | 1.31 | 56.71 | 0.09 | 0.44 | 1.05 | 58.23 |
| MP8 | 1.33 | 62.15 | 0.08 | 0.43 | 1.18 | 63.71 |
aEach observation is the mean of three determinations
Characterization of Emulgels
The developed emulgel formulations were white, viscous preparations with smooth, and homogeneous appearance. They were thixotropic, easily spreadable, and had good aesthetic appeal. The pH and viscosity values of prepared emulgels were found to be in the range of 5.5–6.0 and 7500–9000 cps, respectively. The drug content was 99 ± 0.5%. The drug content uniformity test for all the emulgels indicated that the drug was uniformly dispersed in emulgel formulations.
In Vitro Drug Release Studies
The influence of composition and vehicle on release profile of different formulations was investigated using a cellulose dialysis membrane. In vitro release profiles of MP from different formulations shown in Fig. 3 indicated that MP ointment and MP emulgel released the drug within 4 and 10 h, respectively, while MS emulgel was able to show a sustained release up to period of 24 h. Drug release from emulgels was found to be diffusion-controlled mechanism with R2 value ranging from 0.9738 to 0.9790. The steady state flux (Jss) and permeability coefficient (Kp) were calculated, and the results are tabulated in Table V.
Fig. 3.

Comparative in vitro release profiles of MP from different formulations. Each value represents mean ± SD (n = 6)
Table V.
Permeation Characteristics of MP from Different Formulations Across Cellulose Dialysis Membrane
| Parameters | MP ointment | MP emulgel | MS emulgel |
|---|---|---|---|
| Fluxa (J, μg cm−2 h−1) | 151.42 ± 4.01 | 84.79 ± 1.7 | 49.89 ± 1.25 |
| Permeability coefficienta (P, 10−3 cm h−1) | 7.57 ± 0.20 | 4.24 ± 0.08 | 2.49 ± 0.06 |
| Zero order (R 2) | 0.6735 | 0.9021 | 0.9175 |
| First order (R 2) | 0.9113 | 0.9557 | 0.9640 |
| Higuchi (R 2) | 0.8957 | 0.9790 | 0.9738 |
Mean ± SD (n = 3)
aSignificant difference between all formulations (P < 0.05)
Ex Vivo Drug Deposition Studies
Amount of MP deposited in excised rat abdominal skin from different formulations at different time intervals has been shown in Fig. 4. The amount of MP deposited in skin was higher from the MS emulgel (204.4 ± 6.9 µg/cm2) as compared to that with the MP emulgel (87.57 ± 9.7 µg/cm2) or MP ointment (37.03 ± 7.6 µg/cm2) at the end of 24 h. This indicates that microsponges improved the drug residence in skin.
Fig. 4.

Amount of MP deposited in rat skin from different formulations. Each value represents mean ± SD (n = 3)
Primary Skin Irritation Studies
The scores for erythema and edema were totaled for intact and abraded skin for all rabbits at 24 and 72 h. The primary irritation index was calculated based on the sum of the scored reactions divided by 24 (two scoring intervals multiplied by two test parameters multiplied by six rabbits). The developed formulation showed no erythema or edema on the intact and abraded rabbit skin. The primary irritation index of the formulation was calculated to be 0.00. Thus, the formulations found to be nonirritant to the rabbit skin.
Stability Studies
The developed MP-loaded formulations were found to be stable upon storage for 3 months. No change was observed in their physical appearance, pH, rheological properties, drug content, and drug release profiles.
In Vivo Studies
MP ointment (three times a day), MP emulgel (twice in a day) and MS emulgel (once a day) administered topically demonstrated significant efficacy (P < 0.001) compared with untreated as well as placebo control groups (Fig. 5). There were no significant differences between untreated and placebo controls (P > 0.05).
Fig. 5.

a Mouse surgical wound infected with S. aureus. b Comparative efficacy of MP-loaded formulations against an experimental surgical wound infection in mice caused by S. aureus. Each value represents mean ± SD (n = 6)
MS and MP emulgel formulations reduced mean bacterial counts to 3.37 ± 0.49 and 3.50 ± 0.71 log10 CFU/wound, respectively. The efficacy of developed MS emulgel formulations applied once a day was not significantly different from that of conventional MP ointment applied three times a day (P > 0.05).
DISCUSSION
Factorial design (32) approach was used to optimize formulation and process variables in preparation of microsponges. The higher drug entrapment efficiencies obtained at high drug/polymer ratios can be explained through the fact that more amount of drug was present per unit polymer. The reason for increased production yield at high MP/EC ratios could be due to the reduced diffusion rate of dichloromethane from concentrated solutions into aqueous phase. This provides more time for the droplet formation and may improve the yield of microparticles (2). The viscosity of the polymer solution at high MP/EC ratio was responsible for formation of larger particles.
The data shown in Table II was subjected to multiple regression analysis, and results were fitted in a statistical model, Y = β0 + β1X1 + β2X2 + β11X1X1 + β22X2X2 + β12X1X2 incorporating interactive and polynomial terms to evaluate the responses, where Y is the dependent variable, β0 is the arithmetic mean response of the nine runs, and βi is the estimated coefficient for the factor Xi (17).
The negative influence of internal phase volume and positive influence of stirring rate on % EE were attributed to the lower concentration of drug in higher volume of dichloromethane and decreased drug loss at higher stirring rate, respectively.
The negative influence of both the independent variables as shown by Eq. 4 may be attributed to the decreased concentration of MP at high level of internal phase volume. It was also observed that at the higher stirring rates employed, due to the turbulence created within the external phase, polymer adhered to the paddle and PY decreased (10).
The negative influence of internal phase volume on mean particle size indicated that increasing the internal phase volume decreased the particle size of microsponges. Particle sizes of microsponges were directly proportional to apparent viscosity of internal phase. When the internal phase with lower viscosity was poured into continuous phase, the globules of the formed emulsion could easily divide into smaller droplets and mean particle size decreased. The negative correlation of stirring rate with mean particle size may be ascribed to the high shearing effect of stirring blades (18).
The data shown in Table III clearly indicate that the output responses were strongly dependent on the selected independent variables. The high values of correlation coefficient indicated a good fit (i.e., good agreement between the dependent and independent variables).
Differential scanning calorimetry (DSC) provides information on the physical properties of the sample and its crystalline or amorphous nature and demonstrates a possible interaction between drug and other compounds in microsponges. A broad endothermic peak for MP microsponges, as shown in Fig. 1, indicated a significant reduction in MP crystallinity during the microsponge formation without producing any interaction between drug and polymer.
Both SEM and porosity data show the presence of pores on the surface of microsponges. SEM not only shows the authentic presence of pores but also gives additional information regarding 3-D structure of the microcarriers. Undoubtedly, porosity can be established as in-process control for routine estimation of presence of pores for well-characterized microsponge preparations.
To know the mechanism of drug release from these formulations, the data were treated according to zero-order (cumulative amount of drug released vs. time), first-order (log cumulative percentage of drug remaining vs. time), Higuchi’s (cumulative percentage of drug released vs. square root of time) model equations. The in vitro release profiles of MP from emulgels could be best expressed by Higuchi’s, as the plots of percent cumulative drug release vs. square root of time were found to be linear with R2 values ranging from 0.9738 to 0.9790.
The cumulative amount of drug permeated per unit area was plotted against time, and the slope of the linear portion of the plot was estimated as steady state flux (JSS). The permeability coefficient (Kp) was calculated as Kp = Jss/C0, where C0 is the initial concentration of drug in donor compartment.
The slower flux of MP from MS emulgel (49.89 ± 1.25 μg cm−2 h−1) indicated the controlled release of entrapped drug from polymeric carrier systems (10).
Effective topical drug therapy requires sufficient amount of drug uptake into skin over a particular period of time for maximal pharmacological activity. The amount of MP deposited in skin from the MS emulgel was higher as compared to that with the MP emulgel or MP ointment at the end of 24 h. This indicates microsponges improved the drug residence in skin. This result is in agreement with previous studies, reporting that use of particulate drug carriers like microparticles and nanoparticles improved the drug residence in the skin without increasing transdermal transport (19). The high concentration of drug in the skin after application of microparticles could be explained by the occlusive effect, since microparticles produced a film on the skin surface, which reduced the transepidermal water loss and favored the drug penetration into the skin. De-Jalon et al. (1) reported that acyclovir-loaded PLGA microparticles deposited higher amount of drug in basal epidermis. The localization and quantification of particulate drug carriers are therefore of substantial interest in furthering our understanding of their mechanism of action.
Mouse surgical wound model infected with S. aureus has been used to compare the efficacy of developed formulations with that of marketed MP ointment. The outcome measure of the efficacy was expressed in terms of mean number of bacteria remaining in the treated wounds (log10 CFU/wound).
Although the developed MP-loaded formulations were as effective as marketed MP ointment formulations, they exhibited benefits like reduction in frequency of application, improved patient compliance, and good aesthetic appeal. The in vivo efficacy results were also supported by ex vivo drug deposition studies, which demonstrated that higher amount of MP was deposited in skin from microsponge-based emulgel formulations, and therapeutic drug concentration were maintained up to 24 h.
CONCLUSION
Microsponge-based novel delivery system has been developed to provide once a day sustained release medication for topical delivery of mupirocin. The formulations showed enhanced retention of drug in skin, indicating better potential of delivery system as compared with marketed mupirocin ointment and conventional mupirocin emulgel. On the grounds of efficacy and improved patient compliance due to reduced frequency of application, microsponge-based emulgel formulations will have significantly better role in treatment of primary and secondary skin infections.
Acknowledgment
The authors are grateful to the Indian Institute of Technology (IIT), Bombay, India for help in performing characterization studies.
Abbreviations
- CFU
colony forming units
- EE
entrapment efficiency
- ESD
emulsion solvent difusion
- JSS
steady-state flux
- KP
permeability coefficient
- MP emulgel
emulgel containing mupirocin
- MP
mupirocin
- MP ointment
marketed mupirocin ointment formulation
- MP/EC
mupirocin/ethylcellulose
- MS emulgel
emulgel containing mupirocin-loaded microsponges
- PVA
polyvinyl alcohol
- PY
production yield
References
- 1.De-Jalon EG, Blanco-Prieto MJ, Ygartua P, Santoyo S. Topical application of acyclovir-loaded microparticles: quantification of the drug in porcine skin layers. J Control Release. 2001a;75:191–197. doi: 10.1016/S0168-3659(01)00395-9. [DOI] [PubMed] [Google Scholar]
- 2.Orlu M, Cevher E, Araman A. Design and evaluation of colon specific drug delivery system containing flubiprofen microsponges. Int J Pharm. 2006;318:103–117. doi: 10.1016/j.ijpharm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Nacht S, Katz M. The microsponge: a novel topical programmable delivery system. In: Osborne DW, Amann AH, editors. Topical drug delivery formulations. New York: Marcel Dekker; 1990. pp. 299–325. [Google Scholar]
- 4.Comoglu T, Gonul N, Baykara T. Preparation and in vitro evaluation of modified release ketoprofen microsponges. Il Farmaco. 2003;58:101–106. doi: 10.1016/S0014-827X(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 5.Pappa KA. The clinical development of mupirocin. J Am Acad Dermatol. 1990;22:873–879. doi: 10.1016/0190-9622(90)70116-Y. [DOI] [PubMed] [Google Scholar]
- 6.Ward A, Campoli-Richards DM. Mupirocin: review of its antibacterial activity, pharmacokinetic properties and therapeutic use. Drugs. 1986;32:425–444. doi: 10.2165/00003495-198632050-00002. [DOI] [PubMed] [Google Scholar]
- 7.Conly JM, Johnston BL. Mupirocin- are we in danger of losing it? Can J Infect Dis. 2002;13:157–159. doi: 10.1155/2002/692581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parenti MA, Hatfield SM, Leyden JJ. Mupirocin: a topical antibiotic with a unique structure and mechanism of action. Clin Pharm. 1987;6:761–769. [PubMed] [Google Scholar]
- 9.Gisby J, Bryant J. Efficacy of a new cream formulation of mupirocin: comparison with oral and topical agents in experimental skin infections. Antimicrob Agents Chemother. 2000;44:255–260. doi: 10.1128/AAC.44.2.255-260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jelvehgari M, Siahi-Shabad MR, Azarmi S, Martin GP, Nokhodchi A. The microsponge delivery system of benzoyl peroxide: preparation, characterization and release studies. Int J Pharm. 2006;308:124–132. doi: 10.1016/j.ijpharm.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 11.Kincl M, Turk S, Vrecer F. Application of experimental design methodology in development and optimization of drug release method. Int J Pharm. 2005;291:39–49. doi: 10.1016/j.ijpharm.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 12.Mohamed MI. Optimization of chlorphenesin emulgel formulation. AAPS J. 2004;6:1–7. doi: 10.1208/aapsj060326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padamwar MN, Pokharkar VB. Development of vitamin loaded topical liposomal formulation using factorial design approach: drug deposition and stability. Int J Pharm. 2006;320:37–44. doi: 10.1016/j.ijpharm.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 14.Echevarria L, Blanco-Prieto MJ, Campanero MA, Santoyo S, Ygartua P. Development and validation of a liquid chromatographic method for in vitro mupirocin quantification in both skin layers and percutaneous penetration studies. J Chromatogr B. 2003;796:233–241. doi: 10.1016/j.jchromb.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Thomas J, Schloemer B. Primary skin irritation test in the rabbit of water jel burn dressing. Accessed 11/03/06 from http://waterjel.com/public/SkinIrritationTest.pdf.
- 16.Mcripley RJ, Whitney RR. Characterization and quantitation of experimental surgical wound infections used to evaluate topical antibacterial agents. Antimicrob Agents Chemother. 1976;10:38–44. doi: 10.1128/aac.10.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shivkumar HN, Desai BG, Deshmukh G. Design and optimization of diclofenac sodium controlled release solid dispersion by response surface methodology. Ind J Pharm Sci. 2008;70:22–30. doi: 10.4103/0250-474X.40327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Re MI, Biscans B. Preparation of microspheres of ketoprofen with acrylic polymers by a quasi-emulsion solvent diffusion method. Powder Technol. 1999;101:120–133. doi: 10.1016/S0032-5910(98)00163-6. [DOI] [Google Scholar]
- 19.Alvarez-Roman R, Naik A, Kalia YN, Guy RH, Fessi H. Enhancement of topical delivery from biodegradable nanoparticles. Pharm Res. 2004;21:1818–1824. doi: 10.1023/B:PHAM.0000045235.86197.ef. [DOI] [PubMed] [Google Scholar]