

Abstract
Transdermal films of the furosemide were developed employing ethyl cellulose and hydroxypropyl methylcellulose as film formers. The effect of binary mixture of polymers and penetration enhancers on physicochemical parameters including thickness, moisture content, moisture uptake, drug content, drug–polymer interaction, and in vitro permeation was evaluated. In vitro permeation study was conducted using human cadaver skin as penetration barrier in modified Keshary–Chein diffusion cell. In vitro skin permeation study showed that binary mixture, ethyl cellulose (EC)/hydroxypropyl methylcellulose (HPMC), at 8.5:1.5 ratio provided highest flux and also penetration enhancers further enhanced the permeation of drug, while propylene glycol showing higher enhancing effect compared to dimethyl sulfoxide and isopropyl myristate. Different kinetic models, used to interpret the release kinetics and mechanism, indicated that release from all formulations followed apparent zero-order kinetics and non-Fickian diffusion transport except formulation without HPMC which followed Fickian diffusion transport. Stability studies conducted as per International Conference on Harmonization guidelines did not show any degradation of drug. Based on the above observations, it can be reasonably concluded that blend of EC–HPMC polymers and propylene glycol are better suited for the development of transdermal delivery system of furosemide.
Key words: chemical penetration enhancers, ethyl cellulose, furosemide, hydroxypropyl methylcellulose, in vitro skin permeation, transdermal films
INTRODUCTION
Transdermal delivery of drugs provides many advantages over conventional administration including enhanced efficacy, increased safety, greater convenience, and improved patient compliance. This can avoid the “peak and valley” effect of oral or injectable therapy and can enable more effective treatment by delivering drugs at a steady rate into bloodstream over an extended period of time. It also reduces the dosage-related side effects because the amount of drug delivered into the biological system in a very controlled manner and avoids first-pass metabolism (1,2). This route of administration may be particularly significance in infants and children because of their greater surface area to weight ratio (3). The system designs for transdermal patches include matrix, microreservoir, reservoir, adhesive, and membrane-matrix hybrid. Matrix type transdermal patches remain the most popular as they are easy to manufacture (4).
Furosemide (5-(aminosulfonyl)-4-chloro-2-[(2 furanylmethyl) amino] benzoic acid) is potent diuretic agent that induces a powerful diuresis, followed by the loss of sodium, potassium, and chloride into urine, by acting on thick ascending limb of the loop of henle (5). Its usual daily dose for adults is 20–80 mg, while for pediatric use ranges form 1 mg/kg up to a maximum of 40 mg daily. It is commonly used in the treatment of the cardiac and pulmonary disorders in premature infants and neonates. The half-life of furosemide is about 2 h and its bioavailability has been reported to be about 60–70% (6).
Furosemide is administrated peroral or parenterally although its physicochemical and pharmacokinetic characteristics like low molecular weight, lipid solubility, elimination half-life, and low melting point are in agreement with the ideal properties of molecule for effective penetration of the stratum corneum (7). The furosemide containing hydroxypropyl cellulose gel (8) and ethylene-vinyl acetate matrix (9) for transdermal delivery purposes has been reported.
Ethyl cellulose (EC) is regarded as nontoxic, nonallergic, and nonirritating material and has good film forming properties that enable it to form tougher films. However, the water permeability of pure EC is very low. The benefits of EC have been utilized by mixing with other hydrophilic polymers. In the present study, we developed a suitable matrix film for furosemide by employing EC and hydroxypropyl methylcellulose (HPMC) and to study the effect of polymers and chemical penetration enhancers on the physicochemical properties of transdermal film and permeation of drug across human cadaver skin.
MATERIALS AND METHODS
Materials
Furosemide (mol. wt. 330.75, pKa 3.9, logP = 0.74; Hemdeep organic Pvt. Ltd., Ankleshwar, India), ethyl cellulose (45 cps; Colorcon Pvt Ltd., Goa, India), and hydroxy propylmethyl cellulose (15 LV; Aurobindo Pharma Ltd., Hyderabad, India) were received as gift samples. Propylene glycol, isopropyl myristate, and dimethyl sulfoxide (DMSO) were purchased from S. D. Fine Chem. Ltd., Mumbai, India. Di-n-butyl phthalate was purchased from Central Drug House Pvt. Ltd., Mumbai. All other reagents were of analytical grade.
Methods
Preparation of Transdermal Film
Transdermal films of furosemide (4.52 mg/cm2) containing different ratio of EC and HPMC were prepared on mercury surface. The required amount of drug and polymers were dissolved in methanol-dichloromethane (1:1) solvent system. Di-n-butyl phthalate (15% w/w of polymer) was used as plasticizer. Propylene glycol (PG), isopropyl myristate (IPM), and DMSO were added to the polymer–drug solution. The resultant homogeneous solution was poured into a glass ring placed on mercury substrate. The films were dried for a period of 24 h, and the rate of evaporation was controlled by inverting funnel over the Petri dish. The dry films were wrapped in aluminum foil and kept in desiccators until used. The composition of different films is given in Table I.
Table I.
Composition of Furosemide Transdermal Films
| Formulation | Drug (% w/w) | EC (% w/w) | HPMC (% w/w) | Penetration enhancer (10 % w/w of polymer) | ||
|---|---|---|---|---|---|---|
| PG | DMSO | IPM | ||||
| H | 26.22 | 100 | 00 | – | – | – |
| H1 | 26.22 | 95 | 05 | – | – | – |
| H2 | 26.22 | 90 | 10 | – | – | – |
| H3 | 26.22 | 85 | 15 | – | – | – |
| H4 | 24.64 | 95 | 05 | √ | – | – |
| H5 | 24.64 | 90 | 10 | √ | – | – |
| H6 | 24.64 | 85 | 15 | √ | – | – |
| H7 | 24.64 | 95 | 05 | – | √ | – |
| H8 | 24.64 | 90 | 10 | – | √ | – |
| H9 | 24.64 | 85 | 15 | – | √ | – |
| H10 | 24.64 | 95 | 05 | – | – | √ |
| H11 | 24.64 | 90 | 10 | – | – | √ |
| H12 | 24.64 | 85 | 15 | – | – | √ |
In all formulations, plasticizer at a level of 15% w/w of polymer is added
% w/w weight of drug to total weight of dry film, EC ethyl cellulose, HPMC hydroxypropyl methylcellulose, PG propylene glycol, DMSO dimethyl sulfoxide, IPM isopropyl myristate
Thickness
The thickness of film before and after the permeation study was determined using micrometer gauge (Mitoyoto, Japan). Film was measured at different places and mean value was calculated.
Drug Content Analysis
The uniformity of drug distribution was determined by taking known area of the films at different places of the film. The films were dissolved in 2 ml of casting solvent and subsequently diluted with phosphate buffer pH 8.0. After appropriate dilution, solutions were analyzed spectrophotometrically (UV Pharmaspec-1700, Shimadzu, Japan) for furosemide at 229 nm using solution of films prepared without drug as reference to neglect the absorption of components of the formulation if any.
Moisture Content
The prepared films were weighed individually and kept in desiccators containing activated silica at room temperature (30°C) for 24 h till a constant weight was attained. The percentage of moisture content was calculated as the difference between initial and final weight with respect to final weight (10).
Moisture Uptake
A weighed film kept in desiccator at room temperature (30°C) for 24 h was taken out and exposed to 84% relative humidity (RH) in a stability chamber (Lab Care, Mumbai, India) until a constant weight of film was obtained. The percentage moisture uptake was calculated as the difference between final and initial weight with respect to initial weight (10).
In Vitro Skin Permeation Study
Permeation studies were performed for different formulations across cadaver skin in modified Keshary–Chein diffusion cell at 32 ± 0.5°C. The diameter of the donor compartment cell was 2 cm, providing 3.14 cm2 effective constant area. The films with area 2.83 cm2 were applied to the skin using adhesive tape (cellophane) as backing layer. The phosphate buffer pH 8.0 (70 ml) was used as receptor compartment medium to ensure sink conditions and stability of the drug (11). This whole assembly was kept on a magnetic stirrer and the solution was stirred continuously using a magnetic bead. The samples were withdrawn at different time interval and replaced with equal volume of diffusion medium. Samples were analyzed spectrophotometrically at 229 nm. To ascertain whether the components of the skin or other excipients of the film interfere in the drug analysis, blank experiment (films without drug) was run using skin as barrier membrane using phosphate buffer pH 8.0. When the solution was analyzed at 229 nm for any interfering constituents, the released constituents were amounting to an average of 0.06 ± 0.02%.
Stability Studies
The stability studies were conducted according to International Conference on Harmonization guidelines by storing the TD films at 40 ± 2°C/75% RH in stability chamber (Lab Care, Mumbai, India) for 6 months. The samples were withdrawn after 6 months and analyzed for drug content in a UV spectrophotometer.
Statistical Analysis of Data
The results were analyzed by one-way analysis of variance with Tukey post t test using Graph Pad Prism software-5 version (Graph Pad software Inc., San Diego, CA, USA).
RESULT AND DISCUSSION
When all the dried films were subjected to physical examination, films appeared to be translucent suggesting that the drug was not completely solubilized rather dispersed/suspended in the matrix. In preliminary studies, the EC films were found to be brittle and di-n-butyl phthalate was used as plasticizer to reduce the brittleness of the films. The studies revealed that addition of di-n-butyl phthalate at 15% w/w of polymer produces smooth, uniform, and flexible films. Hence, further studies were carried out using plasticizer at 15% w/w level in the films.
Thickness
Thickness of films varied between 0.083 and 0.117 mm (Table II) suggesting that formulation variables used in the study did not produce any significant effect (p > 0.05) on the thickness of films.
Table II.
Drug Content and Physicochemical Parameters of Films
| Formulation | Drug content (mg/cm2) | %Moisture uptake ± SD, n = 4 | %Moisture content ± SD, n = 4 | Thickness before permeation study (mm) ± SD, n = 4 | Thickness after permeation study (mm) ± SD, n = 4 |
|---|---|---|---|---|---|
| H | 4.44 ± 0.04 | 02.27 ± 0.06 | 01.27 ± 0.09 | 0.083 ± 0.006 | 0.0845 ± 0.002 |
| H1 | 4.45 ± 0.03 | 05.52 ± 0.08 | 03.27 ± 0.08 | 0.097 ± 0.012 | 0.099 ± 0.004 |
| H2 | 4.48 ± 0.06 | 08.50 ± 0.02 | 05.77 ± 0.15 | 0.107 ± 0.032 | 0.105 ± 0.001 |
| H3 | 4.49 ± 0.05 | 13.60 ± 0.02 | 09.29 ± 0.43 | 0.103 ± 0.021 | 0.105 ± 0.006 |
| H4 | 4.45 ± 0.03 | 06.06 ± 0.09 | 03.52 ± 0.06 | 0.110 ± 0.020 | 0.112 ± 0.004 |
| H5 | 4.47 ± 0.03 | 09.62 ± 0.05 | 06.04 ± 0.09 | 0.103 ± 0.015 | 0.107 ± 0.004 |
| H6 | 4.44 ± 0.01 | 14.00 ± 0.07 | 09.79 ± 0.07 | 0.110 ± 0.010 | 0.112 ± 0.003 |
| H7 | 4.44 ± 0.04 | 05.75 ± 0.20 | 04.53 ± 016 | 0.107 ± 0.021 | 0.108 ± 0.001 |
| H8 | 4.43 ± 0.04 | 08.68 ± 0.18 | 06.80 ± 0.12 | 0.117 ± 0.015 | 0.120 ± 0.002 |
| H9 | 4.51 ± 0.07 | 13.82 ± 0.09 | 11.33 ± 0.07 | 0.103 ± 0.012 | 0.106 ± 0.001 |
| H10 | 4.45 ± 0.01 | 05.97 ± 0.03 | 03.77 ± 0.05 | 0.110 ± 0.026 | 0.112 ± 0.004 |
| H11 | 4.45 ± 0.04 | 09.25 ± 0.08 | 06.29 ± 0.09 | 0.110 ± 0.010 | 0.112 ± 0.003 |
| H12 | 4.51 ± 0.06 | 13.89 ± 0.42 | 10.05 ± 0.03 | 0.117 ± 0.015 | 0.120 ± 0.001 |
SD standard deviation
Drug Content Analysis
The drug content of all the formulations (Table II) was ≥4.43 mg/cm2 with a low standard deviation (≤0.07). The results of drug content analysis have shown that the method employed to prepare films in this study was capable of giving films with uniform drug distribution with an insignificant batch variability (p > 0.001).
Moisture Content and Moisture Uptake
Moisture content and moisture uptake studies provide information regarding stability of the formulation (4). The results revealed that the moisture content and moisture uptake were found to increase with increasing concentration of hydrophilic polymer (HPMC). The presence of penetrations enhancers PG and IPM did not show any major changes in moisture content and moisture uptake values. In case of DMSO, slight increment in both parameters was observed (Table II). This may be due to the water affinity of DMSO (12). The small moisture content in the formulation helps them to remain stable and from being a completely dried and brittle films (13), and low moisture uptake protects the material from microbial contamination and bulkiness of the films (10). Thus, the results of physicochemical studies conducted on different polymeric films containing furosemide favored the combination of these polymers for preparation of transdermal films.
In Vitro Skin Permeation Study
An in vitro skin permeation study is predictive of in vivo performance (4) and also valuable and necessary for studying the rate and mechanisms of percutaneous absorption of drugs (14). In the present study, in vitro permeation study was carried out using human cadaver skin as penetration barrier. The cumulative amount of drug permeated per centimeter squared was plotted against time and the steady state permeation flux was calculated from slope of linear portion of the curve. The control (H) containing furosemide in polymeric matrix of ethyl cellulose film showed a flux of 5.78 ± 1.25 mcg/cm2 h and release of 63.92 ± 2.20 mcg/cm2 in 8 h (Table III). A few of workers have used penetration enhancers in HPMC gels (8) and plasticizers in ethylene-vinyl acetate matrix (9) to improve the permeation of furosemide. In our work, the concentration of HPMC, a hydrophilic polymer, was used as a variable. It was observed that as the concentration of HPMC was increased in the formulations (H1, H2, and H3), the mean cumulative amounts of drug permeated (90.22, 209.64, 263.94 mcg/cm2) and flux (8.38, 22.05, 30.08 mcg/cm2/h) increased substantially (Table III; Fig. 1). The hydrophilic nature of HPMC seems to have contributed toward increase in penetration of the solvent molecules into the polymeric matrix and disturbed the compactness of polymeric matrix resulting in faster release.
Table III.
Permeation Parameters
| Formulation | Drug permeated (8 h) mcg/cm2 ± SD, n = 4 | Flux (mcg/cm2/h) ± SD, n = 4 | Enhancement factor (E) |
|---|---|---|---|
| H | 63.92 ± 2.20 | 5.78 ± 1.25 | 1 |
| H1 | 90.22 ± 1.52 | 8.38 ± 2.11 | 1.44 |
| H2 | 209.64 ±1.04 | 22.05 ± 1.17 | 3.81 |
| H3 | 263.94 ± 2.86 | 28.85 ± 3.14 | 4.99 |
| H4 | 204.61 ± 1.44 | 19.42 ± 1.30 | 3.35 |
| H5 | 283.08 ± 1.35 | 27.19 ± 1.72 | 4.70 |
| H6 | 428.87 ± 1.72 | 45.09 ± 2.86 | 7.80 |
| H7 | 125.39 ± 1.89 | 12.83 ± 2.45 | 2.21 |
| H8 | 231.85 ± 2.34 | 23.74 ± 1.89 | 4.10 |
| H9 | 299.89 ± 2.18 | 30.08 ± 1.83 | 5.20 |
| H10 | 171.15 ± 2.72 | 17.18 ± 1.24 | 2.97 |
| H11 | 239.60 ± 1.64 | 23.95 ± 1.68 | 4.14 |
| H12 | 403.11 ± 3.56 | 36.91 ± 3.50 | 6.38 |
SD standard deviation
Fig. 1.

Effect of HPMC concentrations on the permeation of furosemide from EC film
Further, to enhance the permeation of drug for achieving the desired therapeutic plasma level, one of the approaches makes use of penetration enhancers. In the present study, formulations H1, H2, and H3 were selected to evaluate the effect of enhancers like PG, IPM, and DMSO on in vitro permeation of drug through the human cadaver skin. Figures 2, 3, and 4 show that addition of enhancers further increased (p < 0.05) the values of the amount of drug permeated and flux compared to their counterpart without enhancers. Among penetration enhancers used, PG has shown the highest amounts of drug permeated and flux. The calculated enhancement factor (15) for PG was found to be higher (3.35, 4.70, and 7.80) compared to IPM and DMSO at same composition (Table III) and the relative order of the release rates for the enhancers was PG > IPM > DMSO. The higher enhancement capacity of PG may be attributed to its cosolvent and enhancer effects. As cosolvent, it may solubilize the drug and as penetration enhancer, it may affect the intracellular lipids (16) or protein (17) and thus increase the partitioning of the drug in favor of stratum corneum.
Fig. 2.

Effect of PG, IPM, and DMSO on the permeation of furosemide from EC/HPMC (9.5:0.5) film
Fig. 3.

Effect of PG, IPM, and DMSO on the permeation of furosemide from EC/HPMC (9:1) film
Fig. 4.

Effect of PG, IPM, and DMSO on the permeation of furosemide from EC/HPMC (8.5:1.5) film
Release Kinetics and Mechanism of Drug Release
Different kinetic models (zero-order, first-order (18), and Korsmeyer et al. expression (18,19)) were applied to interpret the drug release kinetics and to know the mechanism of drug release from these matrix systems with the help of Eqs. 1, 2, and 3.
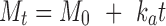 |
1 |
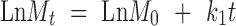 |
2 |
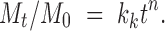 |
3 |
In these equations, Mt is the cumulative amount of drug released at any specified time point and M0 is the dose of the drug incorporated in the delivery system. k0, k1, and kk are rate constants for zero-order, first-order, and Korsmeyer’s model, respectively, and n is the diffusional release exponent indicative of the operating release mechanism. The correlation coefficient values (R2) are presented in Table IV.
Table IV.
Permeation Kinetics Data from Different Transdermal Films
| Formulation | Zero-order kinetics | First-order kinetics | Korsmeyer et al. model | |
|---|---|---|---|---|
| R 2 (±SD, n = 4) | R 2 (±SD, n = 4) | R 2 (±SD, n = 4) | n (±SD, n = 4) | |
| H | 0.9271 ± 0.0127 000.015 | 0.9304 ± 0.0124 | 0.9935 ± 0.0040 | 0.49 ± 0.01 |
| H1 | 0.9486 ± 0.0188 | 0.9521 ± 0.0161 | 0.9727 ± 0.0152 | 0.51 ± 0.01 |
| H2 | 0.9651 ± 0.0125 | 0.9723 ± 0.0202 | 0.9984 ± 0.0009 | 0.64 ± 0.01 |
| H3 | 0.9583 ± 0.0315 | 0.9680 ± 0.0153 | 0.9966 ± 0.0007 | 0.66 ± 0.03 |
| H4 | 0.9591 ± 0.0224 | 0.9655 ± 0.0189 | 0.9728 ± 0.0138 | 0.54 ± 0.02 |
| H5 | 0.9568 ± 0.0158 | 0.9669 ± 0.0175 | 0.9910 ± 0.0051 | 0.56 ± 0.04 |
| H6 | 0.9830 ± 0.0090 | 0.9856 ± 0.0083 | 0.9761 ± 0.0168 | 0.65 ± 0.05 |
| H7 | 0.9638 ± 0.0182 | 0.9681 ± 0.0134 | 0.9887 ± 0.0082 | 0.60 ± 0.04 |
| H8 | 0.9775 ± 0.0160 | 0.9804 ± 0.0093 | 0.9851 ± 0.0095 | 0.66 ± 0.02 |
| H9 | 0.9674 ± 0.0126 | 0.9732 ± 0.0156 | 0.9790 ± 0.0157 | 0.66 ± 0.03 |
| H10 | 0.9695 ± 0.0230 | 0.9744 ± 0.0119 | 0.9841 ± 0.0127 | 0.59 ± 0.01 |
| H11 | 0.9665 ± 0.0112 | 0.9735 ± 0.0111 | 0.9895 ± 0.0075 | 0.61 ± 0.06 |
| H12 | 0.9810 ± 0.0097 | 0.9879 ± 0.0080 | 0.9926 ± 0.0005 | 0.67 ± 0.01 |
SD standard deviation
The in vitro permeation profiles of all formulation (Figs. 1, 2, 3, and 4) obtained by plotting cumulative amount of drug permeated against time shows a similar pattern of drug permeation having initial faster (burst) release followed by slower release. Hence, the in vitro permeation data neither fit into zero-order (R2 = 0.9271 to 0.9830) nor first-order (R2 = 0.9304 to 0.9879) kinetics completely. When the polymeric layer is placed in contact with the skin, the drug compound migrates through the polymer, partitions across the polymer/skin interface, and then migrates into skin (20). The initial faster release may be attributed to the rapid diffusion of the drug immediate to the surface of the film. Thus, rapid depletion of the surface drug and consequent increase in mean diffusional path length might have caused latter slower release. In addition, latter slower release of the drug from the formulations (except H) can also be accounted for the increase in diffusion path length due to the swelling of hydrated HPMC. The hypothesis was further confirmed by the increase (p < 0.01) in thickness of the films after the permeation study. This assumption was further confirmed by fitting the release data into Eq. 3. The formulation H showed strong linearity with R2 value 0.9935 with an “n” value of 0.49. It indicates that diffusion is the mechanism of drug release from formulation H. However, when HPMC loaded formulations plotted with Eq. 3, irrespective of drug concentration showed high linearity (R2 = 0.9727 to 0.9984) with a comparatively higher slope (n) values (p < 0.01, except H1) ranging from 0.51 to 0.66 (Table IV). It indicates that drug release was leaning toward diffusion and swelling coupled mechanism—so called anomalous diffusion. Presence of swellable polymer (HPMC) in the matrix might be responsible for the drug release controlled by more than one process.
Stability Study
Table V shows drug content of the formulations before and after stability study. These formulations were stored at 40 ± 2°C/75% RH in stability chamber (Lab Care, Mumbai, India) for 6 months. After 6 months, visual examination of the dispersed did not show any changes in particle size. Drug content of the patches after stability studies was 4.39 to 4.46 mg/cm2 and did not show any significant variations. These result indicates that drug remain stable after stability studies.
Table V.
Stability Study Data of Films
| Formulation | Drug content(mg/cm2) | |
|---|---|---|
| Before (±SD), n = 4 | After 6 months (±SD), n = 4 | |
| H | 4.44 ± 0.04 | 4.43 ± 0.00 |
| H1 | 4.45 ± 0.03 | 4.43 ± 0.08 |
| H2 | 4.48 ± 0.06 | 4.46 ± 0.03 |
| H3 | 4.49 ± 0.05 | 4.43 ± 0.00 |
| H4 | 4.45 ± 0.03 | 4.39 ± 0.02 |
| H5 | 4.47 ± 0.03 | 4.43 ± 0.09 |
| H6 | 4.44 ± 0.01 | 4.39 ± 0.01 |
| H7 | 4.44 ± 0.04 | 4.40 ± 0.01 |
| H8 | 4.43 ± 0.04 | 4.39 ± 0.04 |
| H9 | 4.51 ± 0.07 | 4.44 ± 0.10 |
| H10 | 4.45 ± 0.01 | 4.40 ± 0.03 |
| H11 | 4.45 ± 0.04 | 4.43 ± 0.01 |
| H12 | 4.51 ± 0.06 | 4.45 ± 0.03 |
SD standard deviation
CONCLUSION
The furosemide transdermal films using EC/HPMC polymer blend were prepared. Among the penetration enhancers, propylene glycol, dimethyl sulfoxide, and isopropyl myristate used, the highest permeation rates were noticed with propylene glycol. Incorporation of HPMC enhanced the flux of the drug and also was responsible for the swelling coupled diffusion controlled drug release.
Acknowledgment
Authors are thankful to Hemdeep Organic Pvt. Ltd, Colorcon Pvt Ltd., and Aurobindo Pharma Ltd. for providing furosemide, ethyl cellulose, and hydroxypropyl methyl cellulose, respectively, as gift samples.
References
- 1.Cleary GW. Medical applications of controlled release. In: Langer DL, editor. Transdermal controlled release systems. Boca Raton FL: CRC; 1984. pp. 203–251. [Google Scholar]
- 2.Kydonieus AF. Controlled release technologies. Boca Raton, FL: CRC; 1987. [Google Scholar]
- 3.Sintov AC, Krymberk I, Gavrilov V, Gorodischer R. Transdermal delivery of paracetamol for paediatric use: effect of vehicle formulation on the percutaneous penetration. J Pharm Pharamcol. 2003;55:911–919. doi: 10.1211/0022357021486. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee B, Mahapatra S, Gupta R, Patra B, Tiwari A, Arora P. A comparison of povidone-ethylcellilose and povidone-eidragit transdermal dexamethasone matrix patches based on in vitro skin permeation. Eur J Pharm Biopharm. 2005;59:475–483. doi: 10.1016/j.ejpb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Giebisch G. The use of a diuretic agent as a probeto investigate site and mechanism of ion transport process. Arzneim Forsch/Drug Res. 1985;35:336–342. [PubMed] [Google Scholar]
- 6.Micromedex® Healthcare Series for Windows, Micromedex Thompson Healthcare. 2001;vol. 109.
- 7.Barry BW. Is transdermal drug delivery research still important today? Drug Discov Today. 2001;6:967–971. doi: 10.1016/S1359-6446(01)01938-9. [DOI] [PubMed] [Google Scholar]
- 8.Agyralides GG, Dallas PP, Rekkas DM. Development and in vitro evaluation of furosemide transdermal formulation using experimental design techniques. Int J Pharm. 2004;281:35–43. doi: 10.1016/j.ijpharm.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Cho CW, Choi JS, Shin SC. Controlled release of furosemide from ethylene vinyl acetate matrix. Int J Pharm. 2005;299:127–133. doi: 10.1016/j.ijpharm.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Arora P, Mukherjee B. Design, development, physicochemical, and in vitro and in vivo evaluation of transdermal patches containing diclofenac diethyl ammonium salt. J Pharm Sci. 2002;91:2076–2089. doi: 10.1002/jps.10200. [DOI] [PubMed] [Google Scholar]
- 11.Bundgaard H, Norgaard T, Nielsen NM. Photodegradation and hydrolysis of furosemide and furosemide esters in aqueous solutions. Int J Pharm. 1988;42:217–224. doi: 10.1016/0378-5173(88)90178-0. [DOI] [Google Scholar]
- 12.Rowe RC, Sheskery PJ, Weller PJ. Hand book of pharmaceutical excipient, Fourth Edition. London: Pharmaceutical; 2003. [Google Scholar]
- 13.Mutalik S, Udupa N. Glibenclamide transdermal patches: physicochemical, pharmacodynamic, and pharmacokinetic evaluations. J Pharm Sci. 2004;93:1577–1594. doi: 10.1002/jps.20058. [DOI] [PubMed] [Google Scholar]
- 14.Singh P, Roberts MS. Skin permeability and local tissue concentration of nonsteriodal anti-inflammatory drugs after topical application. J Pharmacol Expt Ther. 1994;268:144–151. [PubMed] [Google Scholar]
- 15.Yu JW, Chien T, Chien YW. Transdermal dual controlled delivery of testosterone and estradiol. I. Impact of system design. Drug Dev Ind Pharm. 1991;17:1883–1904. doi: 10.3109/03639049109048057. [DOI] [Google Scholar]
- 16.Bouwstra JA, De Vries MA, Gooris GS, Brass W, Brussee J, Ponec M. Thermodynamic and structural aspects of the skin barrier. J Control Rel. 1991;15:209–220. doi: 10.1016/0168-3659(91)90112-Q. [DOI] [Google Scholar]
- 17.Goodman M, Barry BW. Action of penetration enhancers on human stratum corneum as assessed by differential scanning calorimetry. In: Bronaugh RL, Maibach H, editors. Percutaneous absorption: mechanisms–methodology–drug delivery. New York: Dekker; 1989. pp. 567–593. [Google Scholar]
- 18.Khatun M, Islam SMA, Akter P, Quadir MA, Reza S. Md. Controlled release of naproxen sodium from Eudragit RS 100 transdermal film. Dhaka Univ J Pharm Sci. 2004;3:1–2. [Google Scholar]
- 19.Kormeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [Google Scholar]
- 20.Kurnik RT, Potts RO. Modeling of diffusion and crystal dissolution in controlled release system. J Control Rel. 1997;45:257–264. doi: 10.1016/S0168-3659(96)01575-1. [DOI] [Google Scholar]