

Abstract
The purpose of this study was to enhance the dissolution of total flavones of Hippophae rhamnoides L. (TFH) by solid dispersions consisting of the drug and a polymeric carrier, poloxamer 188 (PXM). The solvent evaporation method was used to prepare solid dispersions. A 32 full-factorial design approach was used for optimization wherein the amount of solvent (X1) and the drug-to-polymer ratio (X2) were selected as independent variables and the percentage of TFH dissolved in 10 min (Q10) was selected as the dependent variable. Multiple linear regression analysis revealed that a suitable level of X1 and X2 was required for obtaining higher dissolution of TFH from PXM solid dispersions. Solid dispersions were characterized by differential scanning calorimetry, X-ray diffraction, Fourier transform infrared spectroscopy, scanning electron microscopy, and dissolution tests. Characterization studies revealed that solid dispersion of TFH–PXM showed enhancement of TFH dissolution due to the conversion of TFH into a less crystalline and/or amorphous form. In conclusion, dissolution enhancement of TFH was obtained by preparing its solid dispersions in PXM using solvent method.
Key words: poloxamer 188, solid dispersions, solvent method, total flavones of Hippophae rhamnoides L
INTRODUCTION
Total flavones of Hippophae rhamnoides L. (TFH) are extracted from a Chinese herbal medicine, sea buckthorn (1). It is reported that in the high-performance liquid chromatograms of TFH, 12 compounds have been identified, such as quercetin 3-O-glucoside, isorhamnetin 3-O-rutinoside, quercetin, kaempferol, isorhamnetin, etc. (2). With their major constituents including quercetin, isorhamnetin, and kaempferol (3,4), TFH have been demonstrated with most of the bioactive properties of sea buckthorn. Animal and human studies suggest that sea buckthorn flavonoids may have antioxidant, antiulcerogenic, and hepatoprotective actions, which also can scavenge free radicals, lower blood viscosity, lower blood pressure, enhance cardiac function, and suppress platelet aggregation (5–7). TFH have very low and erratic oral bioavailability due to their poor water solubility. Therefore, it is important to introduce effective methods to enhance their dissolution, hence their bioavailability.
Solid dispersion (SD) is defined as the dispersion of one or more active ingredients in inert carriers at solid state prepared by fusion, solvent, or solvent fusion methods (8–10). This system provides the possibility of reducing the particle size of drugs to nearly a molecular level, to transform the drug from the crystalline to the amorphous state, and/or to locally increase the saturation solubility (8). Therefore, preparing solid dispersions of a poorly soluble drug with water-soluble polymers is a promising method for improving the dissolution characteristics and bioavailability of the drug (11). In recent years, the binary and ternary solid dispersions were prepared to enhance the dissolution of poorly soluble drugs to improve oral absorption of these drugs (12–14). Many water-soluble carriers have been employed for preparing solid dispersions, such as polyethylene glycols, polyvinylpyrrolidone (15), mannitol, hydroxypropyl methylcellulose (16), poloxamer (17), etc. In the present paper, poloxamer 188 (PXM) was used. It is a nonionic block copolymer composed of two hydrophilic polyoxyethylene chains connected by a hydrophobic polyoxypropylene chain. It has been used by researchers to increase the aqueous solubility of poorly water-soluble drugs (18–20).
Designing drug delivery formulations with the minimum number of trials is very crucial for pharmaceutical scientists (21). Factorial design is an efficient method of finding the relative significance of number of variables and their interaction based on the response or outcome of the study. Optimization procedure involving factorial designs and analysis of response surfaces is powerful, efficient, and also proven to be a more systematic tool. It has been widely used in the development of various dosage formulations (22–24).
In current study, TFH were selected as a model drug and poloxamer 188 as a polymeric carrier to prepare solid dispersions to enhance the dissolution of TFH. Solid dispersions of PXM–TFH were prepared using solvent evaporation method and studied systematically using an optimization technique. Differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), scanning electron microscopy (SEM), and dissolution tests were employed to characterize the prepared solid dispersions.
MATERIALS AND METHODS
Materials
Total flavones of H. rhamnoides L. powder was obtained from Sichuan Medco Pharmaceutical (Sichuan, China); poloxamer 188 was received as gift samples from BASF, Germany. Tween-80 and ethanol were obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China).
Preparation of Solid Dispersions
The solid dispersions of THF–PXM were prepared by the solvent evaporation method. Briefly, poloxamer 188 was dissolved in ethanol (80%) under stirring, until a clear solution was obtained, followed by TFH addition and stirring for 45 min. The solvent was recovered by evaporation at 60°C to 70°C under vacuum (Rotavapor, Heidolph, Germany) and used for the preparation of the next SD batch. The recovered solvent was used for the next batch. The resultant solid dispersions were stored in a desiccator at room temperature for 24 h before pulverization and sieving. The yield of solid dispersion was calculated according to the equation shown below:
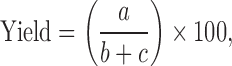 |
1 |
where a is the weight of the solid dispersion sifted through a #120 sieve; b is the weight of THF taken for solid dispersion preparation, and c is the weight of PXM taken for solid dispersion preparation.
Experimental Design
To study all the possible combinations, three-level full-factorial design (32) was constructed and conducted in a fully randomized order. Percent of drug dissolved in 10 min (Q10) and percent yield of SD were selected as dependent variables and studied at three different levels of the selected independent variables (factors). Details of the selected independent variables of the 32 full-factorial design are shown in Table I. The range of a factor was chosen in order to adequately measure its effect on the response variables. This type of design was selected as it provides sufficient degrees of freedom to resolve the main effects as well as the factor interactions. A statistical model incorporating interactive and polynomial terms is used to evaluate the response.
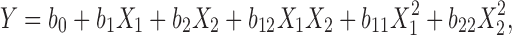 |
2 |
where, Y (Q10, the percentage of TFH dissolved in 10 min) is the dependent variable; b0 is the arithmetic mean response of the nine runs, and bi is the estimated coefficient for the factor Xi. Here, X1 represents the amount of solvent used for SD preparation and X2 is the drug-to-polymer mass ratio. The main effects (X1 and X2) represent the average result of changing one factor at a time from its low to high value. The interaction terms (X1X2) show how the response changes when two factors are simultaneously changed. The polynomial terms (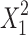 and
and 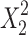 ) are included to investigate nonlinearity. The composition of the factorial design batches SD1 to SD9 are shown in Table I. Two-way analysis of variance (ANOVA) with post hoc Tukey’s test has been applied for data comparison in the factorial design results. Multiple linear regression analysis was used to find out the control factors that affects significantly on response variables. The statistical evaluation of the factorial design results was performed using SPSS 13.0 software (http://www.ts.vcu.edu/faq/software).
) are included to investigate nonlinearity. The composition of the factorial design batches SD1 to SD9 are shown in Table I. Two-way analysis of variance (ANOVA) with post hoc Tukey’s test has been applied for data comparison in the factorial design results. Multiple linear regression analysis was used to find out the control factors that affects significantly on response variables. The statistical evaluation of the factorial design results was performed using SPSS 13.0 software (http://www.ts.vcu.edu/faq/software).
Table I.
Composition of Factorial Design Batches (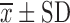 , n = 3)
, n = 3)
| Batch code | Variable levels in coded form | % Q 10 ± SD | % Yield ± SD | |
|---|---|---|---|---|
| X 1 a | X 2 b | |||
| SD1 | −1 | −1 | 68.98 ± 3.28 | 81 ± 0.78 |
| SD2 | −1 | 0 | 80.25 ± 1.18 | 86 ± 1.23 |
| SD3 | −1 | +1 | 85.03 ± 1.97 | 84 ± 0.94 |
| SD4 | 0 | −1 | 70.54 ± 3.20 | 85 ± 1.53 |
| SD5 | 0 | 0 | 95.14 ± 0.95 | 88 ± 0.64 |
| SD6 | 0 | +1 | 92.06 ± 1.53 | 85 ± 0.95 |
| SD7 | +1 | −1 | 71.28 ± 2.15 | 89 ± 1.72 |
| SD8 | +1 | 0 | 87.33 ± 1.65 | 81 ± 0.89 |
| SD9 | +1 | +1 | 88.98 ± 2.17 | 89 ± 1.16 |
Values represent the mean ± SD of three experiments
X 1 the amount of solvent, X 2 the drug-to-polymer mass ratio, Q 10 the percentage of TFH dissolved in 10 min
aActual values: −1 = 150 mL; 0 = 200 mL; +1 = 250 mL
bActual values:−1 = 1:2 (1.5:3 g); 0 = 1:4 (1.5:6 g); +1 = 1:6 (1.5:9 g)
Characterization
In Vitro Dissolution Studies
Dissolution tests were performed with a dissolution apparatus (RCZ-8A, Precise Apparatus of Tianjin University Co., Ltd., China) using the paddle method according to US Pharmacopeia XXIX (25). Samples of original TFH, physical mixtures of TFH and polymeric carrier, and various solid dispersions equivalent to 40 mg of TFH were added to 900-mL deionized water with 0.5% Tween-80 to satisfy the sink condition. Paddle rotation speed was set at 100 rpm and the temperature was maintained at 37 ± 0.5°C. At predetermined intervals (5, 10, 20, 30, 45, 60, 90, and 120 min), 5-mL sample was withdrawn from each vessel, filtered with a 0.45-μm membrane filter. The absorbance of each sample was analyzed spectrophotometrically at 374 nm (model UV-1601 UV-Visible spectrophotometer, Shimadzu). The same volume of fresh medium was replaced after sampling. The percentage of TFH dissolved was calculated using a regression equation generated from the standard curve (r2 = 0.9999). Our previous tests have confirmed that there was no change in the λmax of TFH despite the presence of poloxamer 188 dissolved in the dissolution medium. All tests were performed in triplicates.
Fourier Transform Infrared Spectroscopy
A Nicolet Nexus FTIR 670 spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used for FTIR analysis. The samples were ground and mixed thoroughly with KBr, an infrared-transparent matrix. Sample disks were prepared by compressing the mixtures. The scans were executed from 450 to 4,400 cm−1.
Differential Scanning Calorimetry
DSC was performed by a Perkin-Elmer DSC7 differential scanning calorimeter with a Pyris Series Workstation (Perkin-Elmer, Waltham, MA, USA). The accurately weighed sample was placed in an aluminum pan and an empty aluminum pan was used as reference. The heating rate of the DSC run was 10°C/min starting from 40°C to 280°C. Liquid nitrogen was pumped at 20 mL/min through the system for cooling purpose.
X-Ray Diffraction Studies
Vacuum grease was applied over a glass slide to stick the sample. About 100 mg of sample was sprinkled over it to make a layer having a thickness of ∼0.5 mm. All the experiments were performed on an X-ray diffraction (X’Pert PRO, PANalytical, The Netherlands) having a sensitivity of 0.1 mg. The samples were exposed to CuKα radiation under 40 kV and 40 mA over the 2θ range from 0° to 40° in increments of 0.5°/min every 0.033°.
Scanning Electron Microscopy Analysis
SEM was used to study the surface morphology of TFH, poloxamer 188, and the solid dispersion of TFH with poloxamer 188. The samples were mounted on a brass stage using adhesive carbon tape and placed in a low-humidity chamber prior to analysis. Samples were coated with gold–palladium, and microscopy was performed using a PHILIPS environmental scanning electron microscope (XL 30 ESEM, PHILIPS Inc.) operating at an excitation voltage of 20 kV.
RESULTS AND DISCUSSION
TFH was found to be 23.37% and 50.88% dissolved in 5 min and in 2 h, respectively, explaining the need for improvement of its dissolution characteristics. Therefore, a solid dispersion technique using poloxamer 188 was employed for dissolution enhancement of TFH in the present investigation.
Preliminary investigations of the process parameters revealed that factors X1 (solvent amount) and X2 (the drug-to-polymer mass ratio) highly influenced the rate of in vitro dissolution (Table II) and, hence, were used for subsequent systematic studies. The Q10 for the nine batches (SD1 to SD9) showed a wide variation from 68.98 to 95.14 (Table I). The data clearly suggested that X1 and X2 strongly influence the Q10. All the batches of factorial design exhibited yield greater than 80% (Table I).
Table II.
Factors in Preliminary Experiments and the Results of One-Way ANOVA (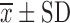 , n = 3)
, n = 3)
| Factor | Level | % Q 10 ± SD | P value | F | Fcrit |
|---|---|---|---|---|---|
| The drug-to-polymer mass ratio | 1:2 | 72.94 ± 0.11 | 3.04E−08 | 111.07 | 3.48 |
| 1:3 | 86.69 ± 1.09 | ||||
| 1:4 | 92.21 ± 1.23 | ||||
| 1:5 | 87.34 ± 4.24 | ||||
| 1:6 | 89.35 ± 0.83 | ||||
| Stirring time | 25 min | 92.21 ± 1.23 | 0.055 | 4.88 | 5.14 |
| 45 min | 92.67 ± 0.97 | ||||
| 60 min | 89.14 ± 1.64 | ||||
| Alcohol content | 60% | 87.76 ± 2.97 | 0.056 | 4.85 | 5.14 |
| 80% | 92.21 ± 1.23 | ||||
| 100% | 88.75 ± 2.02 | ||||
| Solvent amount | 150 mL | 83.17 ± 0.56 | 2.42E−05 | 27.04 | 3.48 |
| 200 mL | 88.11 ± 2.85 | ||||
| 250 mL | 92.21 ± 1.23 | ||||
| 300 mL | 90.41 ± 0.77 | ||||
| 350 mL | 84.28 ± 2.92 |
Values represent the mean ± SD of three experiments. The drug-to-polymer mass ratio: 1:2 means 1.5:3 g, 1:3 means 1.5:4.5 g, 1:4 means 1.5:6 g, 1:5 means 1.5:7.5 g, 1:6 means 1.5: 9 g; solvent amount means 1:4 (1.5:6 g) in 150, 200, 250, 300, 350, respectively. The statistical evaluation of the results in preliminary experiments was performed using SPSS 13.0
Data comparison in Table I was performed by two-way ANOVA with post hoc Tukey’s test since they are normally distributed and the variances of the different treatments are equal. The results revealed that both X1 and X2 and the interaction term X1 × X2 strongly influenced Q10 (P < 0.001). When factors X1 and X2 are considered separately, a statistically significant difference was observed between the levels used (P < 0.001), with level 2 (i.e., coded value 0, Table I) performing better for both factors. Therefore, level 2 was selected for further investigation.
Multivariable linear regression was performed to evaluate the relationship obtained between the response and independent variables. The fitted equation relating the response Q10 to the transformed factors is shown in Table III. It is obvious from the results that X2 (the drug-to-polymer mass ratio) showed the largest positive effect, whereas the term X1 (the amount of solvent) showed insignificant positive effect on Q10. The quadratic term of X2 presented slight negative effect and the quadratic term of X1 exhibited slight positive effect on percentage of drug dissolved. However, when both X1 and X2 were changed simultaneously, a statistically significant negative effect on Q10 was observed (P < 0.05). This may be probably attributed to the different effect exhibited separately by X1 and X2 on Q10. Accordingly, the interaction between these two factors needs to be further investigated in future work.
Table III.
Results of Regression Analysis
| Coefficient response Q 10 | b 0 | b 1 | b 2 | b 11 | b 22 | b 12 |
|---|---|---|---|---|---|---|
| FM | 91.310 | 2.222 | 9.212 | 0.413* | −5.605* | −8.095 |
| RM | 87.573 | 2.222 | 9.212 | – | – | −8.095 |
FM full model, RM reduced model
*P = 0.05, response is insignificant at this level
The significance levels of the coefficients b11 and b22 were found to be P = 0.846 and 0.134, respectively, so they were omitted from the full model to generate a reduced model. The results of the regression analysis are shown in Table III. The coefficients b0, b1, b2, and b12 were found to be significant at P < 0.05; hence, they were retained in the reduced model. The reduced model was tested in portions to determine whether the coefficients b11 and b22 significantly affect the prediction of Q10. The results of the model testing are shown in Table IV. The critical value of F for α = 0.05 is equal to 9.28 (df = 3, 3). Since the calculated value (F = 9.689) in FM and the calculated value (F = 10.249) in RM are more than the critical value (F = 9.28), it may be concluded that the interaction terms b11 and b22 did not contribute significantly to the prediction of Q10 and should be omitted from the full model to generate the reduced model.
Table IV.
Calculations for Testing the Model in Portions
| Regression | df | SS | MS | R | P | F |
|---|---|---|---|---|---|---|
| FM | 5 | 733.314 | 146.663 | 0.970 | 0.045 | 9.689 |
| RM | 3 | 669.802 | 223.267 | 0.927 | 0.014 | 10.249 |
| Error | ||||||
| FM | 3 | 45.411 | 15.137 | |||
| RM | 5 | 108.924 | 21.785 | |||
df degree of freedom, SS sum of squares, MS mean of squares, R regression coefficient, FM full model, RM reduced model
Batch SD5 (1:4 ratio) and SD6 (1:6 ratio) exhibited the largest Q10 values, i.e., 95.14% and 92.06%, respectively. Considering lower drug-to-polymer mass ratio and higher percentage drug dissolved simultaneously, batch SD5 may be a promising formulation batch for dissolution enhancement of TFH. Hence, this batch was selected for subsequent physicochemical characterization.
The dissolution profiles of optimized formulation (1:4 ratio, 200 mL), physical mixture (1:4 ratio), and pure drug are shown in Fig. 1. It is clear from the figure that the dissolution rate of pure drug and physical mixture is very low as compared to the optimized formulation. It is also observed from the dissolution profile of optimized formulation that the total quantity of the drug present in the solid dispersion gets dissolved within 20 min.
Fig. 1.

Dissolution profiles of optimized formulation, physical mixture, and pure drug (n = 3)
The solid dispersion of best batch SD5 was evaluated for physical characterization via FTIR, DSC, XRD, and SEM. TFH and pure poloxamer 188 were also run as control. The samples used for the study were freshly prepared (48 h in advance) and preserved in desiccator before use.
The FTIR spectrum of TFH, poloxamer 188, and solid dispersion are shown in Fig. 2. The characteristic peaks of TFH can be found at 2,926, 1,654, and 1,617 cm−1 due to stretching of C–H, C=O, and C=C groups. The poloxamer 188 exhibits characteristic peaks at 3,447, 2,886, and 1,112 cm−1 due to stretching of O–H, C–H, and C–O groups. The peak at 1,654 and 1,617 cm−1 of the C=O and C=C is the important characteristics of TFH. The characteristic carbonyl stretching band of TFH was inconspicuous in FTIR spectra of solid dispersion. Furthermore, the peak at 2,926 cm−1 of C–H in TFH is shifted to a lower frequency of 2,877 cm−1 in the solid dispersion. The lowering of shift to 49 cm−1 and the disappearance of the peak at 1,654 and 1,617 cm−1 in the solid dispersion could be attributed to the Van der Waals interactions and/or intermolecular hydrogen bonding between the drug and polymers, which may result in dissolution enhancement of TFH.
Fig. 2.

FITR spectra of TFH, poloxamer 188, and FITR spectra of solid dispersion (SD5)
Figure 3 shows the DSC curve of TFH, poloxamer 188, and solid dispersion. The TFH, poloxamer 188, and solid dispersion show endothermic peaks at 245.500°C, 54.666°C, and 51.500°C, respectively. The endothermic peak corresponding to melting of TFH was absent in the DSC thermogram of solid dispersion which could be attributable to the transformation of TFH into amorphous form in the solid dispersion or just simply because of lack of crystalline structure of TFH in the solid dispersion.
Fig. 3.

DSC curves of TFH, poloxamer 188, and solid dispersion (SD5)
XRD patterns of TFH, poloxamer 188, and solid dispersion of TFH and poloxamer 188 are shown in Fig. 4. For TFH, very sharp characteristic peaks at diffraction angle (2θ) of 9.02°, 9.78°, 13.35°, and 25.82° were observed indicating the presence of crystalline lattice of the drug, whereas, for pure poloxamer 188, the characteristic peaks were observed at 19.17° and 23.33°. For the prepared solid dispersion, however, characteristic peaks for the crystalline drug disappeared and only exhibited the characteristic peaks of those of poloxamer 188 indicating the formation of amorphous drug within crystalline polymer matrix. It is well known that, at amorphous state, drug possesses higher Gibbs energy than its more stable crystalline state exhibiting a higher dissolution rate. This can be the reason why the dissolution rates of total flavones in solid dispersions of TFH and poloxamer 188 were higher than those of drug themselves.
Fig. 4.

X-ray diffraction spectra of TFH, poloxamer 188, physical mixture of TFH and poloxamer 188, solid dispersion (SD5)
The scanning electron micrographs of TFH, poloxamer 188, and TFH–poloxamer 188 SDs revealed an unformed sheet with particles size less than 5 μm for TFH (Fig. 5a), spherical particles with smooth surface for poloxamer 188 (Fig. 5b), and a smooth irregular-shaped mixed mass for SDs (Fig. 5c). The particle size of SDs is much bigger than TFH but similar to that of poloxamer 188 and the surface of SDs is slightly rougher compared with poloxamer 188, suggesting that the surface morphology of SDs is much similar to the surface morphology of poloxamer 188.
Fig. 5.

SEM of THF a, poloxamer 188 b, and TFH–poloxamer 188 SDs (SD5) particles c
CONCLUSIONS
The results of the experimental study confirmed that the factors X1, X2 and X1 × X2 significantly influence the dissolution rate of TFH. The application of experimental design techniques for optimization of formulation helps in reaching the optimum point in the shortest time with minimum efforts. Characterization studies revealed that solid dispersion of TFH–poloxamer 188 showed enhancement of TFH dissolution probably due to the conversion of TFH into a less crystalline and/or an amorphous form. In conclusion, dissolution enhancement of TFH was obtained by preparing its solid dispersions in PXM using solvent method.
Acknowledgment
This work was supported by the Shanghai City College Scientific Research Fund for Choosing and Cultivating Excellent Youth Teacher of China (szy-07034). The authors are grateful to Vice-professor Yue Su for her instruction on the statistics analysis, Ms. Fuyuan Ye for her FTIR and X-ray recording, and Mr. Jun Li for his DSC recording.
References
- 1.Lachman J, Pivec V, Hubacek J, Rehakova V. Flavonoid substances in the fruit s of sea buckthorn (Hippophae rhamnoides) Sci Agric Bohem. 1985;3:169–82. [Google Scholar]
- 2.Chen C, Zhang H, Xiao W, Yong ZP, Bai N. High-performance liquid chromatographic fingerprint analysis for different origins of sea buckthorn berries. J Chromatogr A. 2007;1154:250–9. doi: 10.1016/j.chroma.2007.03.097. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, Cui H. Simultaneous determination of quercetin, kaempferol, and isorhamnetin in phytopharmaceuticals of Hippophae rhamnoides L. by high-performance liquid chromatography with chemiluminescence detection. J Sep Sci. 2005;28:1171–8. doi: 10.1002/jssc.200500055. [DOI] [PubMed] [Google Scholar]
- 4.Zu Y, Li C, Fu Y, Zhao C. Simultaneous determination of catechin, rutin, quercetin kaempferol and isorhamnetin in the extract of sea buckthorn(Hippophae rhamnoides L.) leaves by RP-HPLC with DAD. J Pharm Biomed Anal. 2006;41:714–9. doi: 10.1016/j.jpba.2005.04.052. [DOI] [PubMed] [Google Scholar]
- 5.Suomela JP, Ahotupa M, Yang B, Vasankari T, Kallio H. Absorption of flavonols derived from sea buckthorn (Hippophae rhamnoides L.) and their effect on emerging risk factors for cardiovascular disease in humans. J Agric Food Chem. 2006;54:7364–9. doi: 10.1021/jf061889r. [DOI] [PubMed] [Google Scholar]
- 6.Pang X, Zhao J, Zhang W, Zhuang X, Wang J, Xu R, et al. Antihypertensive effect of total flavones extracted from seed residues of Hippophae rhamnoides L. in sucrose-fed rats. J Ethnopharmacol. 2008;117:325–31. doi: 10.1016/j.jep.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Cheng J, Kondo K, Suzuki Y, Ikeda Y, Meng X, Umemura K. Inhibitory effects of total flavones of Hippophae rhamnoides L. on thrombosis in mouse femoral artery and in vitro platelet aggregation. Life Sci. 2003;72:2263–71. doi: 10.1016/S0024-3205(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 8.Damian F, Blaton N, Naesens L, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur J Pharm Sci. 2000;10:311–22. doi: 10.1016/S0928-0987(00)00084-1. [DOI] [PubMed] [Google Scholar]
- 9.Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60:1281–302. doi: 10.1002/jps.2600600902. [DOI] [PubMed] [Google Scholar]
- 10.Ford JL. The current status of solid dispersions. Pharm Acta Helv. 1986;61:69–88. [PubMed] [Google Scholar]
- 11.Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272:1–10. doi: 10.1016/j.ijpharm.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Wang X. Improved dissolution of oleanolic acid with ternary solid dispersions. AAPS PharmSci Tech. 2007;8(4):E1–5. doi: 10.1208/pt0804080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cirri M, Maestrelli F, Corti G, Mura P. Fast-dissolving tablets of glyburide based on ternary solid dispersions with PEG 6000 and surfactants. Drug Deliv. 2007;14:247–55. doi: 10.1080/10717540601067802. [DOI] [PubMed] [Google Scholar]
- 14.Kwon SH, Kim SY, Ha KW, Kang MJ, et al. Pharmaceutical evaluation of genistein-loaded pluronic micelles for oral delivery. Arch Pharm Res. 2007;30:1138–43. doi: 10.1007/BF02980249. [DOI] [PubMed] [Google Scholar]
- 15.Abdul-Fattah AM, Bhargava HN. Preparation and in vitro evaluation of solid dispersions of halofantrine. Int J Pharm. 2002;235:17–33. doi: 10.1016/S0378-5173(01)00941-3. [DOI] [PubMed] [Google Scholar]
- 16.Zheng X, Yang R, Tang X, Zheng L. Part I: characterization of solid dispersions of nimodipine prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2007;33:791–802. doi: 10.1080/03639040601050213. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Zhang GGZ, Neilly J, Marsh K, Mawhinney D, Sanzgiri YD. Enhancing the bioavailability of ABT-963 using solid dispersion containing Pluronic F-68. Int J Pharm. 2004;286:69–80. doi: 10.1016/j.ijpharm.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Passerini N, Albertini B, Gonzalez-Rodriguez ML, Cavallari C, Rodriguez L. Preparation and characterization of ibuprofen–poloxamer 188 granules obtained by melt granulation. Eur J Pharm Sci. 2002;15:71–8. doi: 10.1016/S0928-0987(01)00210-X. [DOI] [PubMed] [Google Scholar]
- 19.Kwon GS. Polymeric micelles for delivery of poorly water-soluble compounds. Crit Rev Ther Drug Carrier Syst. 2003;20:357–403. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i5.20. [DOI] [PubMed] [Google Scholar]
- 20.Yong CS, Yang CH, Rhee JD, et al. Enhanced rectal bioavailability of ibuprofen in rats by poloxamer 188 and menthol. Int J Pharm. 2004;269:169–76. doi: 10.1016/j.ijpharm.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Hamed E, Sakr A. Application of multiple response optimization technique to extended release formulations design. J Control Rel. 2001;73:329–38. doi: 10.1016/S0168-3659(01)00356-X. [DOI] [PubMed] [Google Scholar]
- 22.Rhee Y-S, Chang S-Y, Park C-W, et al. Optimization of ibuprofen gel formulations using experimental design technique for enhanced transdermal penetration. Int J Pharm. 2008;364:14–20. doi: 10.1016/j.ijpharm.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 23.Patel VF, Patel NM. Statistical evaluation of influence of viscosity and content of polymer on dipyridamole release from floating matrix tablets: a technical note. AAPS PharmSciTech. 2007;8(Sup 3):E1–5. doi: 10.1208/pt0803069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang YT, Tsai TR, Cheng CJ, et al. Formulation design of an HPMC-based sustained tablet for pyridostigmine bromide as a highly hygroscopic model drug and its in vivo/in vitro dissolution properties. Drug Dev Ind Pharm. 2007;33:1183–91. doi: 10.1080/03639040701377334. [DOI] [PubMed] [Google Scholar]
- 25.USP . The United States Pharmacopeia (USP) XXIX. NF XXIV. Rockville: US Pharmacopeial Convention; 2006. p. 2673. [Google Scholar]