

Abstract
The different approaches for targeting orally administered drugs to the colon include coating with pH-dependent polymers, design of time-release dosage forms, and the utilization of carriers that are degraded exclusively by colonic bacteria. The aim of the present study was to develop a single unit, site-specific drug formulation allowing targeted drug release in the colon. Matrix tablets were prepared by wet granulation using cross-linked chitosan (ChI) and chondroitin sulfate (ChS) polysaccharides as binder and carrier. ChS was used to form polyelectrolyte complexes (PEC) with ChI, and its potential as a colon-targeted drug carrier was investigated. Indomethacin was used as a model drug. The ChI and ChS PEC was characterized by Fourier transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC) and powder X-ray diffraction studies (XRD). The matrix tablets were tested in vitro for their suitability as colon-specific drug delivery systems. FTIR demonstrated that the PEC forms through an electrostatic interaction between the protonated amine (NH+3) group of ChI with the free carboxylate (COO−) group and sulfate (SO2−4) group of ChS. DSC and XRD indicated that the PEC has different thermal characteristics from ChI or ChS. The dissolution data demonstrates that the dissolution rate of the tablet is dependent upon the concentration of polysaccharide used as binder and matrix and time of cross-linking. The study confirmed that selective delivery of indomethacin to the colon can be achieved using cross-linked ChI and ChS polysaccharides.
Key words: chitosan, chondroitin sulfate, colonic delivery, cross-linking, indomethacin
INTRODUCTION
Colonic drug delivery has gained increased importance not just for the delivery of drugs for the treatment of local diseases of colon but also for its potential for the delivery of proteins and peptides (1). Over the last few years, different approaches have been reported in order to achieve specific colonic drug delivery. Most of the previous literature reports on colonic targeting have focused on the development of a colonic delivery system based on time- and pH-dependent delivery systems as well as systems that utilize bacteria, which colonizes the colon or enzymes produced by these bacteria to affect drug release (2,3). The poor site specificity problem occurs with time release dosage form due to large variation in gastric emptying time (4) and passage across the ileocecal junction. In addition, poor site specificity of pH-dependent system was very well established due to large variation in pH of the gastrointestinal tract (GIT) (5,6). Biodegradable systems formulated using natural polysaccharides are increasingly being developed (7,8). Use of naturally occurring polysaccharides is attracting attention for drug targeting to the colon, since these polymers of monosaccharides are found in abundance, inexpensive, and available in variety of structures with varied properties (9). They can be easily modified chemically and biochemically and are non-toxic, hydrophilic, gel-forming, as well as biodegradable in nature. Conventionally, various polysaccharides are used in the tablet formulations to retard drug release. These have been used either as matrices or as a coating material. For matrices, generally a high concentration of polymer is required. Alternatively, these can be used as binders in tablets (10). Thus, varying the polysaccharides and their concentration affects drug release from the prepared tablet. Based on the above assumption, two different polysaccharides, namely, chitosan (ChI) and a chondroitin sulfate (ChS) were selected for the present study. ChI–ChS complex erodes slowly in phosphate buffer at pH values higher than 6.5, and this behavior leads to suppression of the initial drug release in the upper segments of GIT and controls release in the colon where pH value is in the range of 6.5–7.0 (11).
In the present investigation, for the first time, authors have formulated matrix tablets for colonic delivery using novel cross-linked ChI–ChS PEC. Indomethacin was selected as a model drug because it has good indication for colonic delivery (12,13). Indomethacin is widely used as non-steroidal anti-inflammatory drug. In vitro drug release studies were carried out in simulated colonic fluid with and without rat cecal content, also by using Bacteroides ovatus culture due to its known polysaccharide degradable activity (14).
MATERIALS AND METHODS
Materials
Indomethacin was a generous gift from Micro Lab Limited (Bangalore) India. Chitosan (degree of deacetylation 85%) was obtained from Central Institute of Fisheries, Kochi, India. Chondroitin sulfate was a gift from Markson Pharma., Goa, India. Culture of B. ovatus MTCC 3298 was made available from microbial type culture collection, Chandigarh, India. All other ingredients used were of analytical grade.
Methods
Preparation of Binder Paste
Binder paste of the polysaccharides in various concentrations were prepared by mixing the weighed amount of ChI in a 1% v/v solution of acetic acid in distilled water with magnetic stirrer for 0.5 h so as to enable the ChI to swell. To this mixture, weighed amount of ChS was added slowly. The mixing was carried out for different time intervals such as 12, 18, and 24 h. The cross-linked paste was used as a binder for the powder mixture during wet granulation (Table I).
Table I.
Formulation Composition of Matrix Tablets of Indomethacin
| Sr. | NoName of Ingredient | Quantity/tablet (mg) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Formulation codes | |||||||||||||
| F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 | F10 | F11 | F12 | ||
| 01 | Indomethacin | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 | 75 |
| 02 | ChI | 5.375 | 10.75 | 16.125 | 5.375 | 10.75 | 16.125 | 5.375 | 10.75 | 16.125 | 21.5 | 21.5 | 21.5 |
| 03 | ChS | 5.375 | 10.75 | 16.125 | 5.375 | 10.75 | 16.125 | 5.375 | 10.75 | 16.125 | 21.5 | 21.5 | 21.5 |
| 04 | MCC (Emocel) | 121.375 | 105.2 | 89.125 | 121.375 | 105.25 | 89.125 | 121.375 | 105.25 | 89.125 | 89.125 | 83.75 | 78.375 |
| 05 | Ethyl Cellulose (10% Alcoholic Solution) | 5.375 | 10.75 | 16.125 | – | – | – | – | – | – | – | – | – |
| 06 | Starch paste (10%) | – | – | – | 5.375 | 10.75 | 16.125 | – | – | – | – | – | – |
| 07 | PVP K30 | – | – | – | – | – | – | 5.375 | 10.75 | 16.125 | 5.375 | 10.75 | 16.125 |
| 08 | Magnesium stearate | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 | 2.5 |
| Total weight of tablet | 215 | 215 | 215 | 215 | 215 | 215 | 215 | 215 | 215 | 215 | 215 | 215 | |
ChI chitosan, ChS chondroitin sulfate
Formulation of Granules
All the powdered ingredients were weighed, mixed, and granulated with the binder paste prepared as above. This mixture was thoroughly blended manually and passed through a sieve with a nominal aperture of 1 mm. The granules were prepared and dried in an oven at a temperature between 30°C and 40°C for 4 h. The dried granules were screened, mixed with lubricants, and stored at room temperature for tableting.
Characterization of Granules
Before compression, granules were evaluated for their characteristic parameters. Angle of repose was determined by funnel method. Bulk density and tapped density were determined by cylinder method, and Carr’s index (CI) was calculated using the following equation (15).
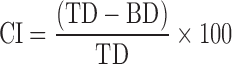 |
1 |
where TD is the tap density and BD is the bulk density
Preparation of Tablets
Tablets weighing 215 mg containing 75 mg of indomethacin were compressed on a ten-station rotary machine (Karnavati Engg., India) using a 7-mm round, concave, plain die-punch.
Characterization of Tablets
The properties of the compressed matrix tablet, such as hardness, friability, weight variation, and content uniformity, were determined using reported procedure (16). Briefly, hardness was determined using Monsanto hardness tester (Campbell Electronics, Mumbai, India), and friability was determined using Roche friability testing apparatus (F. Hoffmann-La Roche Ltd, Basel, Switzerland). Drug content studies were carried out to evaluate the amount of drug present in the prepared tablet.
Determination of Drug Content
Randomly, 20 tablets were selected from each formulation and tested for its drug content. The tablets were finely powdered, and a quantity of powder equivalent to 75 mg of indomethacin was accurately weighed and transferred to 100-ml volumetric flasks containing approximately 50 ml of methanol. The flasks were shaken in a shaking water bath at 37°C for 3 h to solubilize the drug. The volume was made up with methanol and mixed thoroughly. The solutions were filtered through membrane filter paper and analyzed for the content of indomethacin using UV spectrophotometer (1700, Shimadzu, Japan) at 318 nm.
Fourier Transform Infrared Spectroscopy
The electrostatic interaction study of polymers was done by Fourier transform infrared spectroscopy (FTIR) spectroscopic analysis. ChI, ChS, and cross-linked ChI–ChS complex were mixed separately with IR grade KBr in the ratio of 100:1, and corresponding pellets were prepared by applying 10 metric ton of pressure in hydraulic press. The pellets were scanned over a wave range of 4,000–400 cm−1 using FTIR spectrophotometer (8400S, Shimadzu, Japan).
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) of ChI, ChS, and cross-linked ChI–ChS complex were performed by using Mettler Toledo DSC 822e instrument. The cell had a nitrogen purge flowing at approximately 40 cm3/min. Sample (5 mg) was scanned in aluminum pan over a temperature range between 75°C to 300°C at a scanning rate of 5°C/min. An indium pan served as reference, and all scans were performed in triplicate. The instrument was calibrated before sample analysis, using an indium standard.
Powder X-Ray Diffraction Studies
Powder X-ray diffraction (XRD) pattern of polymers were obtained using Philips diffractometer (PW3710, Almelo, The Netherland) and Cu-Kα line as a source of radiation, which was operated at the voltage 40 kV and the current 30 mA. All samples were measured in the 2θ angle range between 0° and 80° with a scanning rate of 1°/min and a step size of 0.02°.
Drug Release Studies
The ability of the prepared tablets to retard drug release in the physiological environment of the stomach, small intestine, and the colon was assessed by conducting drug release studies in simulated stomach, small intestine, and colonic fluid, respectively. The changing pH media, method I, USP XXIII, for delayed release tablets was used (10). Dissolution test was conducted in USP I apparatus (TDT 08L, Electrolab, Mumbai, India) at 75 rpm and a temperature of 37 ± 0.5°C. Initial drug release studies were conducted in 900 ml of 0.1 N HCl for 2 h. Then, dissolution media was replaced with phosphate buffer pH 7.4 (900 ml) for 3 h as average small intestine transit time is 3 h and finally in phosphate buffer pH 6.8 up to 24 h. Samples were withdrawn after regular intervals of time and replaced with the same media. The drug release was analyzed spectrophotometrically (UV-1700, Shimadzu, Japan) at a wavelength of 318 nm.
Preparation of Rat Cecal Medium
Male Wistar rats weighing 110–125 g maintained on normal diet were used. Rat cecal content medium at 2% and 4% w/v level was obtained after 7 days of enzyme induction with 1 ml of 2% w/v ChI and ChS dispersion provides the best condition for assessing the susceptibility of ChI and ChS to colonic bacterial degradation (17). Forty-five minutes before the commencement of drug release studies, six rats were killed by spinal traction, the abdomens were opened, and cecum was traced, ligated at both ends, dissected, and immediately transferred into phosphate buffer pH 6.8 previously bubbled with sterile nitrogen. The entire procedure mentioned above was carried out under sterile nitrogen in order to maintain anaerobic conditions.
The susceptibility of ChI and ChS in matrix tablets to enzymatic action of colonic bacteria was assessed by continuing the drug release studies in 100 ml of phosphate buffer pH 6.8 containing 2% and 4% w/v rat cecal content (18). Drug release studies were carried out in USP XXIII dissolution apparatus I (75 rpm, 37 ± 0.5°C) with slight modification. In this modification, beaker (capacity 250 ml) containing 100 ml of dissolution medium was immersed in water containing 1,000 ml vessel, which was, in turn, to water bath apparatus. The tablets were placed in the basket and immersed in the dissolution medium containing rat cecal contents. The experiment was carried out with continuous nitrogen supply into medium to simulate anaerobic environment of the cecum. The drug release studies were carried out for 24 h (usual colonic transit time is 20–30 h), and 1-ml test samples were withdrawn at 1-h interval without prefilter and replaced with 1 ml of fresh phosphate buffer pH 6.8 bubbled with nitrogen. Withdrawn samples were diluted with 1 ml of methanol to ensure the solubility of finely suspended drug particles released due to digestion of polysaccharides by cecal enzymes. The volume was made up to 10 ml with phosphate buffer pH 6.8 and centrifuged, and the supernatant was filtered through bacteria proof filter and filtrate analyzed for indomethacin at 318 nm (18) using UV spectrophotometer (1700, Shimadzu, Japan).
Dissolution Study Using Culture of Bacteroides ovatus
In addition, polysaccharide-prepared matrix tablet with the potential for site-specific delivery to the colon has been evaluated using culture of B. ovatus. The culture was cultivated anaerobically and maintained on nutrient dissolution media (19).
After a 5-h dissolution study, the phosphate buffer pH 7.4 was replaced with nutrient medium containing culture of B. ovatus (109 CFU/ml). All of the material was then transferred to a sterile jar of dissolution apparatus. Sterile nitrogen was bubbled through vessels to maintain anaerobic condition. Samples (1 ml; after each interval) were withdrawn periodically and diluted with phosphate buffer pH 6.8. Each sample was subsequently centrifuged at 2,000 rpm for 5 min; the absorbance of resulting supernatant was measured at 318 nm.
Kinetic Analysis of Dissolution Data
To study the mechanism of drug release from the matrix tablets, the release data were fitted to zero-order, first-order, and Higuchi equations (20). The dissolution data was also fitted to the well-known exponential equation (Korsmeyer equation), which is often used to describe the drug release behavior from polymeric systems (21).
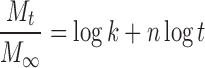 |
2 |
where Mt is the amount of drug release at time t; M∞ is the amount of drug release after infinite time; k is a release rate constant incorporating structural and geometric characteristics of the tablet; and n is the diffusional exponent indicative of the mechanism of drug release.
Statistical Analysis
The cumulative percent of indomethacin released from cross-linked ChI–ChS matrix tablets (n = 3) in the dissolution medium at 24 h with and without rat cecal contents (control study) was compared, and the statistical significance was tested by using Student’s t test. A value of P < 0.05 was considered statistically significant.
RESULTS
Characterization of Granules
The granules for matrix tablet were prepared according to the composition given in Table I and characterized with respect to angle of repose, bulk density, tapped density, Carr’s index, and total drug content (Table II). Physical characterization of granules was found to be in acceptable range (15).
Table II.
Characterization of Granules and Matrix Tablets of Indomethacin
| Parameters | Formulation Code | |||||
|---|---|---|---|---|---|---|
| F7 | F8 | F9 | F10 | F11 | F12 | |
| Granules | ||||||
| Angle of repose (deg) | 28 ± 1.1 | 25 ± 0.8 | 27 ± 0.9 | 22 ± 0.6 | 28 ± 1.2 | 27 ± 0.9 |
| Bulk Density (g/mL) | 0.68 | 0.57 | 0.46 | 0.53 | 0.43 | 0.41 |
| Tap Density (g/mL) | 0.78 | 0.65 | 0.53 | 0.61 | 0.50 | 0.48 |
| Carr’s Index | 13.0 ± 0.5 | 12.3 ± 0.5 | 13.2 ± 1.1 | 13.1 ± 0.6 | 14.0 ± 1.2 | 14.6 ± 1.1 |
| Total drug content (%) | 98.0 ± 3.6 | 98.5 ± 3.9 | 99.0 ± 3.7 | 98.2 ± 4.1 | 98.7 ± 3.8 | 99.1 ± 3.7 |
| Tablets | ||||||
| Weight variation (%) | ± 4.0 | ± 3.0 | ± 2.0 | ± 3.0 | ± 2.0 | ± 4.0 |
| Friability (%) | 0.24 | 0.32 | 0.11 | 0.22 | 0.19 | 0.17 |
| Hardness (kg/cm2) | 5.5 ± 0.1 | 5.7 ± 0.2 | 6.1 ± 0.4 | 5.7 ± 0.3 | 5.6 ± 0.4 | 6.3 ± 0.2 |
| Content uniformity (%) | 98.3 ± 4.3 | 98.7 ± 4.0 | 99.0 ± 0.5 | 98.4 ± 4.3 | 98.8 ± 4.0 | 99.3 ± 3.5 |
*All values represent mean ± SD (n = 3)
Characterization of Tablets
Weight variation, friability, hardness, and content uniformity (98–101%) falls within the limits of Indian Pharmacopoeia (16).
Fourier Transform Infrared Spectroscopy
The IR spectra of ChI (A), ChS (B) and cross-linked ChI–ChS complex (C), respectively, are shown in Fig. 1. IR spectra of ChI (A) confirm the presence of –OH and N–H stretching vibration at 3,435 cm−1 in which the –OH stretching vibration are overlapped by N–H stretching; the absorption of C–H stretching of methyl or methylene group of ChI is at 2,920 cm−1; the peak at 1,648 cm−1 corresponds to the amide bonds; the peak at 1,599 cm−1 corresponds to the symmetrical stretch vibration of amino group; the peak at 1,384 cm−1 corresponds to stretching vibrations of C=N bond; the stretch vibrations of C–O are found at 1,079 and 1,030 cm−1.
Fig. 1.

FTIR spectra’s of a ChI, b ChS, and c cross-linked ChI–ChS. ChI chitosan, ChS chondroitin sulfate, ChI–ChS chitosan–chondroitin sulfate polyelectrolyte complex
The IR spectra of ChS (B) confirm the presence of –OH and N–H stretching vibration at 3,436 cm−1 in which the –OH stretching vibration is overlapped by N–H stretching; the absorption of C–H stretching of methyl or methylene group is at 2,923 cm−1; the stretch vibrations of C–O are found at 1,039 cm−1; the peak at 1,636 cm−1 corresponds to the amide bands; the bands at 1,413 and 1,380 cm−1 are due to the coupling of the C–O stretch vibration and O–H variable-angle vibration and indicates the existence of the free carboxyl group; the peak at 1,253 cm−1 corresponds to the stretching vibrations of S=O bond (SO2−4), which is the characteristic absorption peak of ChS.
In the IR spectra of cross-linked ChI–ChS complex (C), the peak at 1,648 cm−1 (CONH2) in the spectrum of ChI shifts to 1,642 cm−1, whereas the peak of amino group disappeared, which indicates cross-linking of amino group of ChI with ChS. The peak at 1,253 cm−1 (S=O) turns weaker and shifts to 1,250 cm−1, which indicates cross-linking of sulfate group of ChS with ChI. The coupling peak of the C–O stretch vibrations and O–H variable-angle vibration turns weaker and divide into two peaks (the peak at 1,413 cm−1 divide into 1,411 and 1,402 cm−1, the peak at 1,380 cm−1 divide into 1,383 and 1,378 cm−1), which indicates that one part of free carboxyl group reacted with ChI.
Differential Scanning Calorimetry
It is evident from Fig. 2a that ChI powder exhibited one endothermic and exothermic transition each at 52.90°C and 253.11°C, respectively. The DSC thermogram of ChS revealed one endotherm at 94.24°C and one exotherm at 211.89°C in Fig. 2b. However, cross linked ChI–ChS exhibited a large endotherm at 75.30°C followed by another endotherm at 185.32°C (Fig. 2c).
Fig. 2.

DSC thermograms of a ChI, b ChS, and c cross-linked ChI–ChS. ChI Chitosan, ChS chondroitin sulfate, ChI–ChS chitosan–chondroitin sulfate polyelectrolyte complex
Powder X-Ray Diffraction Studies
XRD patterns of ChI, ChS, and cross-linked ChI–ChS is shown in Fig. 3. The characteristic peaks appeared in the XRD of ChI (Fig. 3a) and ChS (Fig. 3b) at different angles. ChI and ChS is crystalline in the solid state, but its diffractogram exhibits well-defined peaks at 2θ = 9.4° and 19.1° for ChI (Fig. 3a) and 46.2° for ChS (Fig. 3b). The complexes (Fig. 3c) give diffractograms showing only the amorphous part, and the intensity of two peaks mentioned above, which is characteristic of ChI and ChS, is much lower than the peaks in individual XRD pattern of ChI and ChS.
Fig. 3.

Powder X-ray diffraction patterns of a ChI, b ChS, and c cross-linked ChI–ChS. ChI chitosan, ChS chondroitin sulfate, ChI–ChS chitosan–chondroitin sulfate polyelectrolyte complex
In Vitro Drug Release Studies
Formulations F1 to F6 were disintegrated within 45 min in 0.1 N HCl and hence were not studied further. Studies using cross-linked ChI and ChS complex as a binder showed that, at a concentration of 5% (F7) the drug release in the initial 5 h was not more than 10%. It indicates that cross-linked polymers are capable of preventing drug from being released completely in the physiological environment of stomach and small intestine. Increasing the concentration of ChI and ChS complex in the formulation up to 10% in F8 and up to 15% in F9 further retards the drug release as compared to F7. At the end of 5 h, the drug release was found to be 6% in F8 and to 4% in F9.
Effects of cross-linking time of ChI and ChS on the percentage drug release at different time periods from the formulation F7 to F12 without rat cecal content are shown in Fig. 4. Results indicate that, as the degree of cross-linking time of ChI and ChS increases, drug release from matrix tablet decreases. Drug released after 24 h cross-linking from formulation F7 to F12 was found to 57.12%, 55.1%, 45.64%, 54.0%, 52.12%, and 51.9%, respectively, in the absence of rat cecal contents (control study). Hence, F9 formulation was selected for further study, as it shows only 45.64% release after 24 h. On the other hand, in the presence of 2% and 4% w/v rat cecal content, drug release was found to be 69.84% and 99.5%, respectively, from F9 formulation (Fig. 5).
Fig. 4.

Effect of cross-linking time of ChI and ChS on the percentage of drug released at different time periods from matrix tablets from the formulations F7 to F12 without rat cecal content (a 12 h cross-linking; b 18 h cross-linking, and c 24 h cross-linking). ChI chitosan, ChS chondroitin sulfate. All results were calculated as mean ± 3SD
Fig. 5.

Percent cumulative release from optimized (F9) formulation with 2% and 4% w/v rat cecal content. All results were calculated as mean ± 3SD
Pure colonic bacteria B. ovatus selected for its known polysaccharide degradable activity (14). The results of dissolution experiment using B. ovatus MTCC 3298 showed that drug is rapidly released after predetermined lag time from matrix tablet, as shown in Fig. 6. Result of dissolution study with culture of B. ovatus provides the best alternative method to dissolution study with 4% w/v rat cecal content.
Fig. 6.

Percent cumulative release from optimized (F9) formulation in presence of culture of bacteroides ovatus (109 CFU/ml); all results were calculated as mean ± 3SD
Kinetic Analysis of Dissolution Data
Drug release data of F9 formulation in presence of rat cecal content showed good fit into the first order equation (r2 = 0.90), partially fitted in zero-order equation (r2 = 0.88) and into the Higuchi equation (r2 = 0.85) and show high linearity with Korsmeyer equation r2 = 0.91. Value of release exponent n determined for F9 in the presence of rat cecal content is 0.72, indicating combined effect of diffusion and predominately erosion mechanisms for controlled drug release.
Statistical Analysis
The cumulative percent of indomethacin released from cross-linked ChI–ChS matrix tablets (n = 3) in the dissolution medium at 24 h with and without rat cecal contents (control study) were compared statistically using Student’s t test. An increase in the amount of indomethacin released was found significant (p < 0.05) from F9 with rat cecal contents when compared to the in vitro release without rat cecal contents.
DISCUSSION
Angle of repose was less than 30° for all the batches of granules, indicating satisfactory flow behavior. Other parameters for granules were also determined and found to be in acceptable range (15). The weight variation and friability was less than 4% and 0.4%, respectively. Good drug content uniformity was found among different batches of the tablets (Table II). FTIR behavior reflects the interaction between the amino group of ChI with the sulfate and free carboxyl group of ChS. PEC was obtained after the interaction between the ChI and ChS. From DSC studies, the endothermic peak (189°C) can be assigned to the interaction between ChI and ChS. It was observed that the XRD of cross-linked ChI–ChS PEC shows absence of characteristic peaks of ChI and ChS, and intensity of peaks in cross-linked ChI–ChS PEC was also reduced.
The purpose of colon targeted drug delivery system is not only to prevent the drug from being release in the physiological environment of the stomach and intestine but also to release the drug in the colon after enzymatic degradation of polysaccharides from matrix tablets by colonic bacteria. Hence, the ability of the polymers used in the formulations to retain the integrity of tablet in upper GI tract was assessed by conducting drug release studies in 0.1 N HCl for 2 h and phosphate buffer pH 7.4 for 3 h (condition mimicking mouth to the colon transit).
The decrease in release rate on increasing the concentration of ChI and ChS can be explained on the basis that, as the concentration of binder in the system is increased, hardness, porosity, and capillary sizes are reduced. This reduces the wicking of water into the tablet, which reduces the disintegration and dissolution processes. These tablets formulated using ChI did not show any swelling in basic environments, so drug release due to swelling and polymer erosion was minimized. This explains why the rate of drug release was not as high as in the case of other swellable gums.
The presence of rat cecal contents or B. ovatus culture in the dissolution medium resulted in improved drug release at different periods when compared to control, indicating that polysaccharides are degraded by colonic bacterial species. It appears that several contaminants, grown in the dissolution medium, might have broken down the matrix tablets. In fact, a bioreactor (with a relevant culture medium) was used by earlier workers to test the colon-specific delivery systems (14). However, due to several limitations, alternative methods, like rat cecal content medium or a dissolution medium containing a specific enzyme, were developed.
Thus, systems formulated using ChI as a binder have been found to protect majority of drug release during the usual upper GIT transit time of 5 h. However, there have been a number of reports where ChI has been found to be digested by the microflora of the colon (8,22,23,25). On similar lines, once these matrix tablets reaches the colon, ChI and ChS shall be broken down by the microflora of the colon, and the total amount of drug shall be released from the dosage form. These tablets can also tolerate variation in upper GIT transit time, since the rate of drug release before arrival into the colon remains retarded. From the results, it seems that the complex, i.e., cross-linked ChI–ChS is more susceptible for bacterial degradation and may release the drug completely in the colon when compared with drug release in the absence of microbial flora.
However, it has been reported that when water solubility of the drug is low, as is in the case of indomethacin, the possibility of release by diffusion is practically zero, and release takes place by surface erosion (24,25). The kinetic data obtained from in vitro drug release studies in presence of rat cecal content indicates that the drug release was followed by mixed order transport, i.e., release of the drug from tablets was by more than one mechanism. The P value found to be less than 0.05 denotes significant difference at 24 h in the amount of indomethacin released from F9 with rat cecal contents when compared to the in vitro release without rat cecal contents.
CONCLUSION
Based on the results of the present investigation, it can be concluded that polyelectrolyte complex of ChI–ChS might assist in improving drug delivery to the colon.
Acknowledgments
Authors are thankful to the Principal, R.C. Patel College of Pharmacy for providing facilities to carry out the research work. The authors acknowledges Micro Lab Limited, Bangalore, India, Central Institute of Fisheries, Kochi, and Markson Pharma., Goa, India, for providing gift samples of indomethacin, chitosan, and chondroitin sulfate, respectively.
References
- 1.Gupta K, Beckert TE, Price JC. A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. AAPS PharmSciTech. 2001;213:83–91. doi: 10.1016/s0378-5173(00)00649-9. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez M, Vila-Jato JL, Torres D. Design of a new multiparticulate system for potential site-specific and controlled drug delivery to the colonic region. J Control Release. 1998;55:67–77. doi: 10.1016/S0168-3659(98)00029-7. [DOI] [PubMed] [Google Scholar]
- 3.Khan MZ, Prebeg Z, Kurjakovic N. A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers. I. Manipulation of drug release using Eudragit L100-55 and Eudragit S100 combinations. J Control Release. 1999;58:215–222. doi: 10.1016/S0168-3659(98)00151-5. [DOI] [PubMed] [Google Scholar]
- 4.Davis SS, Hardy JG, Taylor MJ, Stockwell A, Whalley DR, Wilson CG. The in vivo evaluation of an osmotic device (Osmet) using gscintigraphy. J Pharm Pharmacol. 1984;36:740–742. doi: 10.1111/j.2042-7158.1984.tb04862.x. [DOI] [PubMed] [Google Scholar]
- 5.Ashford M, Fell JT, Attwood D, Sharma HL, Woodhead PJ. An in vivo investigation into the suitability of pH dependent polymers for colonic targeting. Int J Pharm. 1993;95:193–199. doi: 10.1016/0378-5173(93)90406-6. [DOI] [Google Scholar]
- 6.Evans DF, Pye G, Bramely R, Clark AG, Dysoa TJ. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988;29:1035–1041. doi: 10.1136/gut.29.8.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinha VR, Kumria R. Colonic drug delivery: prodrug approach. Pharm Res. 2001;18:557–564. doi: 10.1023/A:1011033121528. [DOI] [PubMed] [Google Scholar]
- 8.Sinha VR, Kumria R. Polysaccharides for colon specific drug delivery. Int J Pharm. 2001;224:19–38. doi: 10.1016/S0378-5173(01)00720-7. [DOI] [PubMed] [Google Scholar]
- 9.Hovgaard L, Brondsted H. Current applications of polysaccharides in colon targeting. Crit Rev Ther Drug Carrier Syst. 1996;13:185–223. doi: 10.1615/critrevtherdrugcarriersyst.v13.i3-4.10. [DOI] [PubMed] [Google Scholar]
- 10.Sinha VR, Kumria R. Binders for Colon specific drug delivery: an in vitro evaluation. Int J Pharm. 2002;249:23–31. doi: 10.1016/S0378-5173(02)00398-8. [DOI] [PubMed] [Google Scholar]
- 11.Tapia C, Escobar Z, Costa E, Hagar J, Valenzuela F, Basualto C, Gai MN, Pedram MY. Comparative studies on polyelectrolyte complex and mixtures of chitosan-carageenan as prolonged diltiazem clorhydrate release systems. Eur J Pharm Biopharm. 2004;57:65–75. doi: 10.1016/S0939-6411(03)00153-X. [DOI] [PubMed] [Google Scholar]
- 12.Hull MA, Gardner H, Hawcroft G. Activity of the non-steroidal anti-inflammatory drug indomethacin against colorectal cancer. Cancer Treat Rev. 2003;29:309–320. doi: 10.1016/S0305-7372(03)00014-8. [DOI] [PubMed] [Google Scholar]
- 13.Kapitanovic S, Cacev T, Antica M, Kralj M, Cavric G, Pavelic K, Spaventi R. Effect of indomethacin on E-cadherin and β-catenin expression in HT-29 colon cancer cells. Exp Mol Pathol. 2006;80:91–96. doi: 10.1016/j.yexmp.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Chourasia MK, Jain SK. Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Sci. 2003;6(1):33–66. [PubMed] [Google Scholar]
- 15.Aulton ME, Wells TI, editors. The science of dosage form design. London, UK: Churchill Livingstone; 1988. [Google Scholar]
- 16.Government of India Ministry of Health and Family Welfare . The pharmacopoeia of India. Delhi, India: Controller of Publication; 2007. [Google Scholar]
- 17.Krishnaiah YSR, Bhaskar Reddy PR, Satyanarayana V, Karthikeyan RS. Studies on the development of oral colon targeted drug delivery systems for metronidazole in the treatment of amoebiasis. Int J Pharm. 2002;236:43–55. doi: 10.1016/S0378-5173(02)00006-6. [DOI] [PubMed] [Google Scholar]
- 18.Krishnaiah SR, Satyanarayana S, Rama Prasad YV, Narasimha Rao S. Evaluation of gaur gum as a compression coat for drug targeting to colon. Int J Pharm. 1998;171:137–146. doi: 10.1016/S0378-5173(98)00172-0. [DOI] [Google Scholar]
- 19.Macfarlane GT, Hay S, Macfarlane S, Gibson GR. Effect of different carbohydrates on growth, polysaccharidase and glycosidase production by Bacteroides ovatus, in batch and continuous culture. J Appl Bacteriol. 1990;68(2):179–187. doi: 10.1111/j.1365-2672.1990.tb02564.x. [DOI] [PubMed] [Google Scholar]
- 20.Higuchi T. Mechanism of sustained-action medication: theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. doi: 10.1002/jps.2600521210. [DOI] [PubMed] [Google Scholar]
- 21.Korsmeyer RW, Gurny R, Docler E, Buri P, Peppas NA. Mechanism of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15:25–35. doi: 10.1016/0378-5173(83)90064-9. [DOI] [Google Scholar]
- 22.Tozaki H, Komoike J, Tada C, Maruyama T, Terabe A, Suzuki T, Yamamoto A, Muranishi S. Chitosan capsules for colon specific drug delivery: improvement of insulin absorption from the rat colon. J Pharm Sci. 1997;86:1016–1021. doi: 10.1021/js970018g. [DOI] [PubMed] [Google Scholar]
- 23.Tozaki H, Fujita T, Odoriba T, Terabe A, Suzuki T, Tanaka C, Okabe S, Muranishiand SS, Yamamoto A. Colon specific delivery of R68070, a new thromboxane synthase inhibitor, using chitosan capsules: therapeutic effect against 2, 4, 6,-trinitrobenzene sulphonic acid-induced ulcerative colitis in rats. Life Sci. 1999;64:1155–1162. doi: 10.1016/S0024-3205(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 24.Lapidus H, Lordi NG. Some factors affecting the release of a water-soluble drug from a compressed hydrophilic matrix. J Pharm Sci. 1966;55:840–843. doi: 10.1002/jps.2600550818. [DOI] [PubMed] [Google Scholar]
- 25.Nigalaye AG, Adusumilli P, Bolto S. Investigation of prolonged drug release from matrix formulations of chitosan. Drug Dev Ind Pharm. 1990;16:449–467. doi: 10.3109/03639049009114897. [DOI] [Google Scholar]