

Abstract
Most quantitative trait loci (QTL) studies fail to account for the effect that the maternal genotype may have on an individual’s phenotypes, even though maternal effect QTL have been shown to account for considerable variation in growth and obesity traits in mouse models. Moreover, the fetal programming theory suggests that maternal effects influence an offspring’s adult fitness, although the genetic nature of fetal programming remains unclear. Within this context, our study focused on mapping genomic regions associated with maternal effect QTL by analyzing the phenotypes of chromosomes 2 and 7 subcongenic mice from genetically distinct dams. We analyzed 12 chromosome 2 subcongenic strains that spanned from 70 to 180 Mb with CAST/EiJ donor regions on the background of C57BL/6 J, and 14 chromosome 7 subcongenic strains that spanned from 81 to 111 Mb with BALB/cByJ donor regions on C57BL/6ByJ background. Maternal QTL analyses were performed on the basis of overlapping donor regions between subcongenic strains. We identified several highly significant (P < 5 × 10−4) maternal QTL influencing total body weight, organ weight, and fat pad weights in both sets of subcongenics. These QTL accounted for 1.9-11.7% of the phenotypic variance for growth and obesity and greatly narrowed the genomic regions associated with the maternal genetic effects. These maternal effect QTL controlled phenotypic traits in adult mice, suggesting that maternal influences at early stages of development may permanently affect offspring performance. Identification of maternal effects in our survey of two sets of subcongenic strains, representing approximately 5% of the mouse genome, supports the hypothesis that maternal effects represent significant sources of genetic variation that are largely ignored in genetic studies.
Introduction
Growth and obesity are complex phenotypic traits with a moderate to strong genetic basis in mammals (Bouchard 1991; Mignon-Grasteau et al. 2000), and both traits are linked to health disorders in humans and domestic animals (Bouchard 1991; Brindley and Rolland 1989; Lew and Garfinkel 1979). Mouse models have provided valuable insight into the genetic architecture of growth and obesity through the identification of single-gene mutations (de Magalhães et al. 2005; Horvat and Medrano 2001; Zhang et al. 1994), quantitative trait loci (QTL) (Corva and Medrano 2001; Farber and Medrano 2007; Warden et al. 1995), epistatic interactions between different genomic regions (Brockmann et al. 2000), and quantification of the additive genetic variance (Maes et al. 1997).
Genetic analyses of growth and obesity traits in mice have typically focused on direct genetic effects without taking into account the effects of the maternal genotype. Some studies support the importance of maternal genetic contributions (Jarvis et al. 2005; Wolf et al. 2002), but they only broadly characterized the effects and have not localized associated genomic regions (Jarvis et al. 2005). Recent studies focusing on the identification of QTL for growth and obesity have suggested the presence of maternal effects on chromosomes 2 and 7 in two sets of subcongenic strains (Diament and Warden 2004; Farber and Medrano 2007). In the present study we further investigate each subcongenic set using a QTL fine-mapping approach focused on the overlapping donor regions to identify maternal QTL affecting the phenotypes in adulthood.
The analysis of maternal effects that influence traits in adults has become a major field of research in recent years, characterizing a biological phenomenon known as fetal programming (Schwartz and Morrison 2005). The fetal programming hypothesis proposes that alterations in fetal or early neonatal nutrition and endocrine status result in developmental adaptations that permanently change structure, physiology, and metabolism (Barker 1998), influencing both adult health (Godfrey and Barker 2001) and phenotypic performance (Wu et al. 2006). Fetal programming has been associated with atherosclerosis (Goharkhay et al. 2007), obesity (Yura et al. 2005), cardiovascular disease (Remacle et al. 2004), and fertility (Abbott et al. 2006) in mouse, although little is known about the genetic architecture of these maternal influences. The analysis of the putative genetic mechanisms regulating fetal programming in mice could contribute useful information to the understanding of the genetic basis of growth and obesity in mammals.
Materials and methods
Mouse husbandry
Two independent sets of subcongenic mouse strains with donor regions on chromosomes 2 and 7 (Figs. 1 and 2) were developed as described by Farber and Medrano (2007) and Warden et al. (1995), and maintained in different vivariums at the University of California-Davis. Note that these were independent mouse populations developed and maintained under different management conditions (e.g., facility, diet; see below). Mice were housed in groups of two to five in polycarbonate cages bedded with a 2:1 mixture of CareFRESH (Absorption Corp., Ferndale, WA) and soft paper chips (Canbrands Intl., Moncton, Canada) and maintained under controlled conditions of temperature (21 ± 2°C), humidity (40–70%), and lighting (14 h light, 10 h dark, lights on at 7 a.m.). Mice were weaned at 3 weeks of age. Chromosome 2 subcongenic strains (and founder stains) were fed with Purina Laboratory Rodent Diet 5008 (Labdiet®, Purina Mills, Inc., St. Louis, MO; 23.5% protein, 6.5% fat, 3.3 kcal/g), whereas chromosome 7 subcongenic mice were fed Research Diets AIN-76A (Research Diets, Inc., New Brunswick, NJ; 20.8% protein from casein, 67.7% carobydrate from sucrose, 11.5% fat from corn oil, 3.9 kcal/g). Food and water were offered ad libitum. All mouse protocols were managed according to the guidelines of the American Association for Accreditation of Laboratory Animal Care (AAALAC; http://www.aaalac.org).
Fig. 1.

Summary of the maternal QTL in mouse chromosome 2. Bottom Locations of the subcongenic strains and boundary markers (red areas represent CAST/EiJ alleles and gray areas represent C57BL6/J alleles; alleles residing between adjacent markers typed as CAST/EiJ and C57BL6/J are undetermined and highlighted in blue; only boundary markers are included). TopP-value plots for maternal QTL affecting body weight (2WK, 3WK, 6WK, and 9WK, i.e., body weight at 2, 3, 6, and 9 weeks of age, respectively), organ (LW liver weight, SW spleen weight, KW kidneys weight, HW heart weight, TW testis weight), and fat pads weight (GFP gonadal fat pads, RFP retroperitoneal fat pad, MFP mesenteric fat pad, FFP femoral fat pads, TFP total fat pads weight). The dotted horizontal line represents the significance threshold under Bonferroni’s correction (α = 0.05; P = 1.62 × 10-4)
Fig. 2.

Summary of the maternal QTL in mouse chromosome 7. Bottom Locations of the subcongenic strains and boundary markers (red areas represent B6.C-TyrcH1bHbbd/By alleles and gray areas represent C57BL/6ByJ alleles; alleles residing between adjacent markers typed as B6.C-TyrcH1bHbbd/By and C57BL/6ByJ are undetermined and highlighted in blue; only boundary markers are included). TopP-value plots for maternal QTL affecting body size (BL body length, BW body weight, CW carcass weight) and fat pads weight (GFP gonadal fat pads, RFP retroperitoneal fat pad, MFP mesenteric fat pad, FFP femoral fat pads, TFP total fat pads weight). The spotted horizontal line highlights the significance threshold under Bonferroni’s correction (α = 0.05; P = 4.46 × 10-4)
Genotyping
During the development of chromosome 2 subcongenic strains, mice were genotyped for 36 microsatellite loci (D2Mit329, D2Mit93, D2Mit94, D2Mit160, D2Mit476, D2Mit15, D2Mit439, D2Mit130, D2Mit389, D2Mit43, D2Mit477, D2Mit185, D2Mit58, D2Mit207, D2Mit101, D2Mit420, D2Mit445, D2Mit17, D2Mit224, D2Mit107, D2Mit488, D2Mit223, D2Mit136, D2Mit490, D2Mit212, D2Mit194, D2Ucd22, D2Mit260, D2Mit286, D2Mit409, D2Mit262, D2Mit196, D2Mit342, D2Mit456, D2Mit213, and D2Mit148; Table 1) and the nonagouti locus. DNA samples were isolated from 1.0-mm tail clips and genotyped following standard PCR protocols as described by Farber et al. (2006). Chromosome 7 subcongenic strains were genotyped by standard PCR protocols for ten microsatellite loci (D7Mit318, D7Mit90, D7Mit62, D7Mit301, D7Mit373, D7Mit321, D7Mit354, D7Mit353, D7Mit96, and D7Mit37; Table 1) after extracting DNA samples from kidney or toe (Diament and Warden 2004). Marker D7Mit37 is placed as the most distal marker by the Ensembl genome browser (Ensembl; http://www.ensembl.org) and by the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov), whereas Celera (http:www.celera.com) assumes two copies, one placed in the distal location and the other one placed between markers D7Mit373 and D7Mit321. Our analyses were performed on the basis of D7Mit37 placed at the most distal position.
Table 1.
Microsatellite markers typed in the chromosome 2 and chromosome 7 subcongenic strains
| Marker | Mba | Marker | Mba |
|---|---|---|---|
| Chromosome 2 | D2Mit212 | 141.8 | |
| D2Mit329 | 74.9 | D2Mit194 | 143.8 |
| D2Mit93 | 76.7 | D2Ucd22 | 146.0 |
| D2Mit94 | 80.0 | D2Mit260 | 149.0b |
| D2Mit160 | 84.8 | D2Mit286 | 154.3 |
| D2Mit476 | 86.3 | Nonagouti | 154.8 |
| D2Mit15 | 91.9 | D2Mit409 | 158.3 |
| D2Mit439 | 92.0 | D2Mit262 | 155.7 |
| D2Mit130 | 97.4 | D2Mit196 | 160.3 |
| D2Mit389 | 103.1 | D2Mit342 | 164.7 |
| D2Mit43 | 104.0 | D2Mit456 | 168.8 |
| D2Mit477 | 104.4 | D2Mit213 | 174.4 |
| D2Mit185 | 105.3 | D2Mit148 | 178.5 |
| D2Mit58 | 108.1 | ||
| D2Mit207 | 112.0 | Chromosome 7 | |
| D2Mit101 | 114.9 | D7Mit318 | 81.1 |
| D2Mit420 | 118.1b | D7Mit90 | 87.5 |
| D2Mit445 | 121.1 | D7Mit62 | 91.8 |
| D2Mit17 | 122.6b | D7Mit301 | 98.7 |
| D2Mit224 | 129.1 | D7Mit373 | 103.2 |
| D2Mit107 | 133.1 | D7Mit321 | 105.1 |
| D2Mit488 | 133.1 | D7Mit354 | 106.1 |
| D2Mit223 | 134.7b | D7Mit353 | 106.7 |
| D2Mit136 | 136.2 | D7Mit96 | 107.7 |
| D2Mit490 | 138.7 | D7Mit37 | 111.0 |
aMb according to Ensembl genome browser (http://www.ensembl.org) April 2007 assembly (Hubbard et al. 2002)
bMb according to UCSC Genome Bioinformatics (http://genome.ucsc.edu) July 2007 assembly (Karolchik et al. 2003)
Subcongenic F2 crosses
For mouse chromosome 2, seven subcongenic strains were developed from the B6.CAST-(D2Mit329-D2Mit457)N(6) congenic strain (B62D) (Farber and Medrano 2007; Farber et al. 2006). Five additional subcongenic strains were generated on the basis of the HG.CAST-(D2Mit329-D2Mit457)N(6) congenic line (HG2D) (R. J. Gularte, personal communication), following the procedures detailed by Farber and Medrano (2007). Both B62D and HG2D possessed identical CAST/EiJ donor regions introgressed on C57BL6/J (B6) and C57BL/6 J-hg/hg (HG) (Corva and Medrano 2000) backgrounds, respectively. Note that the HG strain is isogenic to B6, except for the high growth locus, a 500-kb deletion on mouse chromosome 10 (Horvat and Medrano 2001). As described by Farber and Medrano (2007), seven B6 × (B62D × B6) males with narrow (approximately 20 Mb) overlapping donor regions were selected and backcrossed to B6 females to generate large numbers of F1 heterozygous mice of both sexes. Subcongenic F2 populations were created by intermating F1 heterozygous mice derived from each of the seven founders. In a similar way, the F2 HG2D subcongenic populations were generated from five HG × (HG2D × HG) males with narrow overlapping donor regions mated with HG females and the subsequent mating of heterozygous F1 individuals (R. J. Gularte, personal communication). See Table 2 and Fig. 1 for a detailed characterization of F2 populations and donor regions.
Table 2.
Summary of the subcongenic F2 crosses and their congenic regions
| N | Males | Females | Litters | Boundary microsatellites of the congenic region | ||
|---|---|---|---|---|---|---|
| Chromosome 2 subcongenics | ||||||
| B62D-1 | 87 | 87 | 0 | 15 | D2Mit329 | D2Mit476 |
| B62D-2 | 128 | 128 | 0 | 19 | D2Mit94 | D2Mit101 |
| HG2D-1 | 363 | 159 | 204 | 52 | D2Mit43 | D2Mit103 |
| B62D-3 | 157 | 157 | 0 | 25 | D2Mit477 | D2Mit103 |
| B62D-4 | 145 | 145 | 0 | 24 | D2Mit207 | D2Mit17 |
| HG2D-2 | 233 | 95 | 138 | 27 | D2Mit420 | D2Mit490 |
| B62D-5 | 111 | 111 | 0 | 21 | D2Mit445 | D2Mit107 |
| B62D-6 | 118 | 118 | 0 | 17 | D2Mit107 | D2Mit194 |
| B62D-7 | 129 | 129 | 0 | 22 | D2Mit490 | D2Mit409 |
| HG2D-3 | 371 | 189 | 182 | 47 | D2Mit194 | D2Mit262 |
| HG2D-4 | 336 | 166 | 170 | 38 | D2Mit286 | D2Mit213 |
| HG2D-5 | 377 | 170 | 207 | 50 | D2Mit456 | D2Mit148 |
| Overall | 2555 | 1654 | 901 | 357 | ||
| Chromosome 7 subcongenics | ||||||
| B6.C-H1 | 51 | 51 | 0 | 31 | D7Mit318 | D7Mit37 |
| B6.C-C | 76 | 76 | 0 | 44 | D7Mit318 | D7Mit318 |
| B6.C-D | 24 | 24 | 0 | 14 | D7Mit18 | D7Mit90 |
| B6.C-M | 36 | 36 | 0 | 22 | D7Mit90 | D7Mit90 |
| B6.C-N | 86 | 86 | 0 | 51 | D7Mit90 | D7Mit62 |
| B6.C-L | 33 | 33 | 0 | 19 | D7Mit62 | D7Mit301 |
| B6.C-A | 47 | 47 | 0 | 31 | D7Mit 301 | D7Mit301 |
| B6.C-B | 29 | 29 | 0 | 20 | D7Mit301 | D7Mit37 |
| B6.C-I | 88 | 88 | 0 | 44 | D7Mit373 | D7Mit373 |
| B6.C-F | 123 | 123 | 0 | 71 | D7Mit373 | D7Mit37 |
| B6.C-H | 48 | 48 | 0 | 26 | D7Mit37 | D7Mit37 |
| B6.C-J | 57 | 57 | 0 | 36 | D7Mit37 | D7Mit37 |
| B6.C-K | 29 | 29 | 0 | 14 | D7Mit37 | D7Mit37 |
| B6.C-E | 22 | 22 | 0 | 13 | D7Mit321 | D7Mit321 |
| B6.C-G | 31 | 31 | 0 | 14 | D7Mit354 | D7Mit354 |
| Overall | 780 | 780 | 0 | 450 | ||
Subcongenic strains for mouse chromosome 7 were developed from the BALB/cByJ (B6.C-H1) congenic strain (The Jackson Laboratory, Bar Harbor, ME, USA), a commercially available strain retaining the H1 histocompatibility antigen allele of the BALB/cBy strain on the background of the C57BL/6ByJ (B6By) strain. Fourteen subcongenic strains were generated by backcrossing B6By × B6.C-H1 mice to B6By mice. Then, each recombinant mouse containing only a part of the full congenic region was backcrossed again to B6By and genotyped to produce F1 progeny with identical recombinant donor region boundaries. These heterozygous progeny were sib-mated to produce F2 littermate mice of all three genotype classes (Diament and Warden 2004). The genomic intervals captured by each subcongenic strain are summarized in Fig. 2 and Table 2.
Recording of phenotypic data
Genotypes were available for mice in all generations (F0, F1, and F2) and phenotypic information was collected from all F2 mice from each subcongenic. Chromosome 2 subcongenic male and female mice were weighed at 2 (2WK), 3 (3WK), 6 (6WK), and 9 (9WK) weeks of age. At sacrifice (9WK ± 5 days), mice were weighed, anesthetized under isofluorane, sacrificed by decapitation, and exsanguinated. The weight of liver (LW), spleen (SW), kidneys (KW), heart (HW), and testis (TW) were registered. Gonadal fat pads (GFP; adipose tissue surrounding the gonads and other reproductive organs), retroperitoneal fat pad (RFP; fat deposit behind each kidney along the lumbar muscles), mesenteric fat pad (MFP; adipose tissue surrounding the gastrointestinal tract from the gastroesophageal sphincter to the end of the rectum), and femoral fat pads (FFP; subcutaneous adipose deposit from the outer thigh) were dissected following Johnson and Hirsch (1972). Total fat pad weight (TFP) was calculated as the sum of GFP, RFP, MFP, and FFP. At 16–18 weeks of age, chromosome 7 subcongenic male mice were anesthetized with isofluorane and measurements of body length (BL) and weight (BW) were taken. Animals were sacrificed by cervical dislocation. After sacrifice, carcass weight (CW) was registered and gonadal, retroperitoneal, mesenteric, and femoral fat pads were dissected and weighed as described by Warden et al. (1995). See Tables 2 and 3 for a detailed summary of available mice and phenotypic records.
Table 3.
Number of phenotypic records and summary statistics for the analyzed traits
| Trait | n | Mean ± SE |
|---|---|---|
| Chromosome 2 subcongenics | ||
| Weight at 2 weeks of age (g) | 2541 | 7.467 ± 0.025 |
| Weight at 3 weeks of age (g) | 2553 | 10.688 ± 0.035 |
| Weight at 6 weeks of age (g) | 2544 | 25.741 ± 0.085 |
| Weight at 9 weeks of age (g) | 2548 | 30.200 ± 0.111 |
| Liver weight (g) | 2531 | 1.339 ± 0.005 |
| Spleen weight (g) | 2527 | 0.098 ± 0.001 |
| Kidneys weight (g) | 2532 | 0.316 ± 0.001 |
| Heart weight (g) | 2526 | 0.149 ± 0.001 |
| Testis weight (g) | 1636 | 0.130 ± 0.002 |
| Gonadal fat pads weight (g) | 2527 | 0.392 ± 0.002 |
| Retroperitoneal fat pad weight (g) | 2527 | 0.079 ± 0.001 |
| Mesenteric fat pad weight (g) | 2527 | 0.230 ± 0.001 |
| Femoral fat pads weight (g) | 2527 | 0.345 ± 0.002 |
| Total fat pads weight (g) | 2525 | 1.045 ± 0.005 |
| Chromosome 7 subcongenics | ||
| Body length (cm) | 775 | 9.822 ± 0.010 |
| Body weight (g) | 780 | 26.593 ± 0.105 |
| Gonadal fat pads weight (g) | 780 | 0.556 ± 0.011 |
| Retroperitoneal fat pad weight (g) | 774 | 0.195 ± 0.005 |
| Mesenteric fat pad weight (g) | 779 | 0.171 ± 0.004 |
| Femoral fat pads weight (g) | 780 | 0.311 ± 0.006 |
| Total fat pads weight (g) | 780 | 1.233 ± 0.024 |
| Carcass weight (g) | 766 | 24.871 ± 0.102 |
Statistical analyses
As shown in Figs. 1 and 2, the donor regions of subcongenic strains with donor regions on the same chromosome partially overlapped. This property allowed us to define 22 overlapping regions (OR) on chromosome 2 and 15 OR on chromosome 7 (see Figs. 1 and 2). Note that OR were sequentially numbered from 2.1 to 2.22 in chromosome 2 (Fig. 1) and from 7.1 to 7.15 in chromosome 7 (Fig. 2). Although previous analyses performed on these data modeled maternal genetic contributions on the broad basis of each subcongenic strain (Diament and Warden 2004; Farber and Medrano 2007), the OR approach allowed fine-mapping (~10 Mb) of maternal QTL by examining differences between background/background and donor/background F1 females (mothers of the F2 population) at each genomic location. As was initially applied by Clee et al. (2006) on a direct QTL, this analysis extends the method for QTL mapping by allowing for strain differences at each OR. The specific genetic architecture of our subcongenic strains was used to test differences in offspring phenotypic performance in relation to the maternal genetic background within each overlapping region. For chromosome 2, 22 analyses focused on each overlapping region were performed for 14 phenotypic traits (Fig. 1) using the following mixed model,
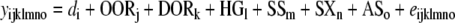 |
where yijklmno and eijklmno were the phenotypic record and residual term belonging to the oth mouse, respectively. This model accounts for the random environmental effect of each dam (di), the genotype of the offspring in the overlapping chromosomal regions (background/background, background/donor, or donor/donor; OORj), dam genotype in the chromosomal overlapping region (background/donor or background/background; DORk), genotype for the high-growth locus (high growth or wild type; HGl), subcongenic strain (12 strains; SSm), sex (male or female; SXn) and age at sacrifice (ASo) as continuous effect. Preliminary analyses of fat pad weights also accounted for weight at sacrifice, although this factor was dropped from the operational model because it did not reach statistical significance when compared with age at sacrifice. On chromosome 2, maternal influences were captured by di (genetic and environmental influences related to the dam) and DORk (genetic effects restricted to the analyzed chromosomal overlapping region) effects, whereas direct genetic effects were inferred by OORj (loci located in the analyzed chromosomal overlapping region), HGl (high-growth genotype), and SSm (genetic background of each subcongenic strain) or accumulated in the residual term (eijklmno). The separation of maternal and direct genetic sources of variation was accomplished by including the two factors DORk and OORj in the model; both of this factors could be partially correlated. The same operational model was used to analyze chromosome 7 subcongenic strains but without the HGl term, since the high-growth mutation was not present in B6.C-H1 mice. For both chromosomes 2 and 7 subcongenic strains, an independent analysis was sequentially performed on each chromosomal overlapping region (note that overlapping regions 7.12 and 7.14 were contributed by the same subcongenic strains with donor genotype: B6.C-H1, B6.C-B, B6.C-F, B6.C-J, and B6.C-K; Fig. 2). Inference was focused on DORk in order to scan maternal QTL with effects on growth and obesity traits in mice. Although di could also capurate some genetic variability due to mutations (Casellas and Medrano 2008) in the background genome, its main effect must be due to environmental sources of variation and without additional contributions to the maternal QTL.
When the QTL scan detected two or more significant peaks for a given trait, the appropriate DORk effects associated with the minimum P-value locations were jointly analyzed in the same model (with the remaining systematic and random sources of variation defined above) to determine their statistical relevance in this population of congenic mice. All analyses were performed with the Mixed procedure of SAS 8.2 (SAS Institute, Inc., Cary, NC). Given that these analyses were performed within a multiple-testing scheme, Bonferroni’s correction was applied (α = 0.05). Threshold for statistical significance was placed at P = 1.62 × 10-4 (chromosome 2) and P = 4.46 × 10-4 (chromosome 7).
Results
Maternal QTLs for growth and organ weight in mouse chromosomes 2 and 7
A significant QTL was detected for 9WK (P = 1.23 × 10−5; Fig. 1) within the chromosomal region flanked by markers D2Mit477 and D2Mit207 (OR 2.5). The remaining growth traits (2WK, 3WK, and 6WK) had similar P-value profiles but failed to reach statistical significance after correcting for multiple testing. The peak additive effect was negative (−0.828 ± 0.233 g), indicating that maternal CAST/EiJ alleles reduced adult weight at 9 weeks in the offspring.
All organ weights showed significant maternal QTL on mouse chromosome 2 with the exception of heart (P > 0.23; Fig. 1). Two maternal QTL were detected for KW between D2Mit329 and D2Mit94 (P = 8.55 × 10−8; OR 2.1) and between D2Mit107 and D2Mit194 (P = 1.12 × 10−6; OR 2.13–2.15), with positive and negative effects for CAST/EiJ alleles on organ weight, respectively (Table 4). In a similar way, two maternal QTL with opposite effects were obtained for TW between D2Mit477 and D2Mit101 (P = 6.32 × 10−7; OR 2.5–2.6) and between D2Mit107 and D2Mit194 (P = 6.96 × 10−8; OR 2.13–2.15). For both KW and TW, the two maternal QTL were corroborated under joint analyses with P ≤ 7.21 × 10−5 for all cases. The two remaining organs (LW and SW) showed a unique maternal QTL between markers D2Mit477 and D2Mit101 (P = 3.50 × 10−7 and P = 1.46 × 10−7, respectively; OR 2.5 and 2.6), although slightly differing in the location of the P-value peak (between D2Mit477 and D2Mit207 for LW and between D2Mit207 and D2Mit101 for SW; Table 4).
Table 4.
Maternal QTL in mouse chromosomes 2 and 7 (single-QTL analysis)
| Traita | Boundary microsatellites | P valueb | QTL effectb,c | Phenotypic varianced (%) |
|---|---|---|---|---|
| Chromosome 2 | ||||
| 9WK (g) | D2Mit185 to D2Mit207 | 1.23 × 10−5 | −0.828 ± 0.233 | 1.9 |
| LW (g) | D2Mit185 to D2Mit101 | 3.50 × 10−7 | −0.065 ± 0.012 | 2.5 |
| SW (g) | D2Mit185 to D2Mit101 | 1.46 × 10−7 | −0.006 ± 0.001 | 1.3 |
| KW (g) | D2Mit329 to D2Mit94 | 8.55 × 10−8 | 0.036 ± 0.006 | 11.7 |
| KW (g) | D2Mit107 to D2Mit194 | 1.12 × 10−6 | −0.022 ± 0.004 | 9.7 |
| TW (g) | D2Mit185 to D2Mit101 | 6.32 × 10−7 | 0.010 ± 0.001 | 5.4 |
| TW (g) | D2Mit107 to D2Mit194 | 6.96 × 10−8 | −0.020 ± 0.003 | 3.1 |
| GFP (g) | D2Mit185 to D2Mit101 | 1.62 × 10−5 | 0.038 ± 0.008 | 7.2 |
| RFP (g) | D2Mit185 to D2Mit101 | 2.81 × 10−5 | 0.011 ± 0.002 | 2.1 |
| FFP (g) | D2Mit185 to D2Mit101 | 5.81 × 10−7 | 0.027 ± 0.005 | 3.7 |
| TFP (g) | D2Mit185 to D2Mit101 | 6.83 × 10−6 | 0.087 ± 0.019 | 7.2 |
| Chromosome 7 | ||||
| BL (cm) | D7Mit62 to D7Mit301 | 1.34 × 10−5 | −0.126 ± 0.039 | 3.3 |
| BW (g) | D7Mit90 to D7Mit373 | 1.90 × 10−8 | −2.176 ± 0.484 | 8.9 |
| CW (g) | D7Mit373 to D7Mit321 | 2.32 × 10−5 | −1.083 ± 0.354 | 2.4 |
| GFP (g) | D7Mit90 to D7Mit62 | 1.44 × 10−7 | −0.171 ± 0.042 | 7.6 |
| GFP (g) | D7Mit301 to D7Mit373 | 2.34 × 10−9 | −0.238 ± 0.049 | 9.7 |
| RFP (g) | D7Mit90 to D7Mit62 | 2.07 × 10−7 | −0.075 ± 0.013 | 5.0 |
| RFP (g) | D7Mit301 to D7Mit373 | 1.79 × 10−7 | −0.093 ± 0.022 | 7.2 |
| MFP (g) | D7Mit90 to D7Mit90 | 1.62 × 10−5 | −0.044 ± 0.014 | 2.7 |
| MFP (g) | D7Mit301 to D7Mit301 | 9.67 × 10−5 | −0.043 ± 0.015 | 2.5 |
| FFP (g) | D7Mit90 to D7Mit62 | 8.58 × 10−7 | −0.093 ± 0.024 | 6.1 |
| FFP (g) | D7Mit301 to D7Mit373 | 2.17 × 10−6 | −0.110 ± 0.029 | 6.9 |
| TFP (g) | D7Mit90 to D7Mit62 | 2.78 × 10−7 | −0.380 ± 0.097 | 2.5 |
| TFP (g) | D7Mit301 to D7Mit373 | 4.89 × 10−8 | −0.496 ± 0.114 | 2.7 |
a9WK Weight at 9 weeks of age, BL Body length, BW Body weight, CW Carcass weight, LW Liver weight, SW Spleen weight, KW Kidneys weight, TW Testes weight, GFP Gonadal fat pads weight, RFW Retroperitoneal fad pads weight, MFW Mesenteric fat pad weight, FFP Femoral fad pads weight, TFP Total fad pads weight
bWhen a QTL spanned two or more overlapping regions, estimates were focused on the region with smaller P value
cEstimates as the difference between donor and background genetic environments
dPercentage of the phenotypic variance accounted for by the maternal QTL
On chromosome 7, BW showed a two-peak profile (Fig. 2); however, only the maternal QTL placed between markers D7Mit301 and D7Mit373 (OR 7.7–7.9) remained significant after the joint analysis of both QTL (P = 4.15 × 10−7). Maternal QTL for BL and CW were detected between D7Mit62 and D7Mit301 (P = 1.34 × 10−5; OR 7.5–7.7) and between D7Mit373 and D7Mit321 (P = 2.32 × 10−5; OR 7.9–7.11), respectively. All three maternal QTL implied a reduction in body weight and length linked with B6.C-H1 alleles (Table 4).
The maternal QTL accounted for a significant percentage of the phenotypic variance, between 1.9% (9WK) and 11.7% (KW) for chromosome 2 QTL and between 2.4% (CW) and 8.9% (BW) for chromosome 7 QTL (Table 4). These values were similar or slightly larger than those obtained by the direct effect of the offspring’s genotype, OORj, in the same genomic regions (between 3.4 and 7.5%).
Maternal QTL for fat pad weight on mouse chromosomes 2 and 7
Chromosome 2 maternal QTL for fat pad weights had similar profiles, although MFP did not reach statistical significance (Fig. 1). A hotspot for genetic maternal contribution to offspring obesity was detected between markers D2Mit477 and D2Mit101 (OR 2.5 and 2.6), although maximum peaks were placed between D2Mit207 and D2Mit101 (P ≤ 2.81 × 10−5; OR 2.6). The effect was favorable to CAST/EiJ alleles promoting obesity in all cases, with increases in fat pad weight ranging between 0.011 ± 0.002 g (RFP) and 0.087 ± 0.019 (TFP; Table 4). These maternal QTL accounted for 2.1% (RFP) to 7.2% (GFP and TFP) of the phenotypic variance (Table 4), similar to the values accounted for by the direct effect of the offspring’s genotype, OORj.
Chromosome 7 P-value profiles for GFP, RFP, FFP, and TFP were very similar, whereas MFP showed slight discrepancies (Fig. 2). Although two maternal QTL were initially detected for MFP, only the one in marker D7Mit90 could be accepted after joint analysis (P = 7.48 × 10−5). The remaining traits showed two significant maternal QTL each, even after the joint analysis of both QTL. Maximum significances for the joint analysis of both QTL were obtained between markers D7Mit90 and D7Mit62 (P = 1.36 × 10−5; OR 7.4) and between markers D7Mit301 and D7Mit373 (P = 3.81 × 10−6; OR 7.8). Nevertheless, all OR between 7.4 and 7.9 showed significant estimates, with the exception of OR 7.7 characterized by marker D7Mit301. All fat pad maternal QTL showed negative estimates, indicating that maternal B6.C-H1 alleles contributed to leaner offspring 4–4.5 months after birth. The maternal QTL accounted for a significant percentage of the phenotypic variance, between 2.5% (MFP and TFP) and 9.7% (GFP).
Discussion
Maternal QTL are scarce in the scientific literature (Jarvis et al. 2005; Wolf et al. 2002). While numerous gene-mapping studies have dissected the mouse genome for direct effects on phenotypic traits, maternal genetic effects have often been ignored or roughly accounted for in QTL models. Maternal genetic effects cannot be addressed in standard F2 designs because all F1 mothers are genetically identical and the maternal contribution is reduced to environmental genetic effects. The unique experimental design in this study examining the F2 progeny originating from overlapping subcongenics provides a very powerful tool to dissect maternal QTL. The indirect effect of a female’s genotype on her offspring’s phenotype (Mousseau and Fox 1998a) can account for much of the phenotypic variance (Hager et al. 2008) and occasionally contribute significant variation in traits expressed later in life (Mousseau and Fox 1998b). Analyses of maternal QTL require appropriate experimental designs and statistical methods should take into account that the genetic background of each female could contribute three times to her offspring, first by the direct inheritance of DNA, second through the maternal in utero environment, and third by the maternal postpartum environment during nursing. Under some experimental designs (Hager et al. 2008), maternal genetic effects and imprinting effects could be difficult to differentiate. Our study distinguishes direct genetic effects from the combined effects of both maternally provided in utero and postpartum environments. No recombinant F2 mice were included in the analysis in order to properly account for the direct genetic contribution of each individual through the effects of the subcongenic strain and the offspring genotype in the chromosomal overlapping region of interest. The two types of maternally provided environments could be distinguished only by using cross-fostering studies. Genomic imprinting cannot be addressed under the F2 experimental design because the F1 heterozygous females (and males) can give rise to both of the reciprocal heterozygotes (background/donor and donor/background, given as paternal/maternal), instead of the experimental designs involving homozygous dams (Bartolomei and Tilghman 1997; Hayward et al. 1998; Reik and Walter 2001). The origin of each allele cannot be identified; therefore, maternal (and paternal) imprinting effects cannot be completely discarded in our study.
Our use of subcongenic strains allows for a novel approach to QTL analysis, first applied by Clee et al. (2006) for mapping direct QTL in congenic mice. QTL effects within each overlapping region between strains can be directly assessed as the difference between donor and background subcongenic strains at a given chromosomal location. In this study, the approach described by Clee et al. (2006) has been adapted for the identification of maternal QTL, after accounting for the remaining sources of variation.
We report the identification of several significant maternal QTL on mouse chromosomes 2 and 7, all of them related to growth and obesity traits at later ages. A hotspot was detected in mouse chromosome 2 between 104.5 and 114.8 Mb (OR 2.5–2.6). In this region, the donor alleles of the maternal QTL were responsible for both lighter (9WK, BW, and LW) and fatter (GFP, RFP, FFP, and TFP) offspring (Fig. 1, Table 4). Additional maternal QTL influencing the weight of the spleen, kidneys, and testis were also reported in Fig. 1. On mouse chromosome 7, growth maternal QTL peaked between 98.7 and 103.2 Mb (OR 7.7–7.9), whereas obesity maternal QTL showed significant estimates between 87.5 and 103.2 Mb, with a nonsignificant gap on OR 7.7 (~98.7 Mb). Note that the length and boundaries of this nonsignificant genomic region linked with marker D7Mit301 cannot be properly established. This is due to the presence of unknown donor alleles to the left and right of D7Mit301 in B6.C-A, B6.C-B, and B6.C-L subcongenic strains that can extend up to D7Mit62 (left side) and D7Mit373 (right side). Nevertheless, this does not invalidate the two significant maternal QTL at the left and right of D7Mit301. All the maternal effect QTL accounted for a substantial percentage of the phenotypic variance (1.9-11.7%), agreeing with values reported by Wolf et al. (2002). This result highlights the importance of the maternal genome to the offspring’s phenotype at latter ages. Although this source of variation is often ignored in QTL analyses, our results suggest that maternal effect QTL could be a common and significant source of genetic variation affecting growth and obesity. Maternal QTL donor alleles caused lighter (BW; Table 4) and leaner (GFP, RFP, FFP, and TFP) F2 mice (Table 4).
Our model included the random environmental effect of each litter in order to account for all the remaining litter-specific nongenetic sources of variation (e.g., feeding, temperature). Within this context, only maternal effects with a genetic basis were addressed by the reported QTL. All phenotypic traits involved in significant maternal QTL on chromosome 2 were recorded in 2-month-old mice 6 weeks after weaning and without additional contacts with their mother after the weaning date. The phenotypic traits involved in significant maternal QTL on chromosome 7 were recorded in 4–4.5-month-old mice. Within this context, these delayed indirect maternal genetic contributions must be related to biological mechanisms initiated during fetal or postnatal life and preserved for enough time to substantially modulate the adult phenotype.
According to our survey of the literature, maternal QTL for post-weaning mouse growth are limited to a study by Hager et al. (2008), and maternal QTL for obesity traits in adult mice were first suggested by Jarvis et al. (2005). However, the analysis of maternal effects at later ages has been a field of intense research in animals and humans. This phenomenon is known as fetal programming (Drake and Walker 2004), with the primary hypothesis being that a stimulus or insult acting during critical stages of growth and development may permanently alter tissue structure and function (Barker and Clarck 1997). Gestation and early neonatal life must be considered as potential sources of indirect maternal genetic influences on offspring. During the gestational period, levels of maternal gene products involved in placenta development may influence gene expression in the fetus. In addition, there is a flow of glucose, hormones, and amino acids from mother to offspring that could affect fetus growth and development (Myatt 2006). It has also been shown that during gestation, maternal methyl supplements affect epigenetic variation and DNA methylation of offspring (Cooney et al. 2002). On the other hand, the early neonatal period plays a key role in the development of the immune system (Wheeler et al. 2007) and formation of the hypothalamus-pituitary-adrenal axis. Maternal care and milk quality may influence the formation of both of these systems. In species that give birth to immature young (e.g., mice, rats, and rabbits), much neuroendocrine development occurs in the postnatal period (Kapoor et al. 2006). However, in humans and nonhuman primates, these developmental stages occur in utero. During neuroendocrine development, hormones circulating in the mother’s body can directly regulate the development and activity of the fetus’s central nervous system (Simerly 2002, 2005; Bouret and Simerly 2006). In fact, the formation of the hypothalamus-pituitary-adrenal axis could be one of the most reliable ways to maternally modify growth and adult fat pads of the offspring, given the close relation of the neuroendocrine system to growth and fat metabolism (Björntorp 1997; Müller et al. 1999). Obviously, all these biological mechanisms present a complex scenario in which the reported maternal QTL could be due to multiple genes involved in very different physiologic pathways.
The analysis of maternal QTL on the basis of overlapping subcongenic strains examines each genomic region separately; thus, any one gene in the significant genomic region may have a causative mutation. Although our approach allowed us to fine-map maternal QTL to relatively small genomic regions of less than 5 Mb, we can only suggest some plausible candidate genes. Several genes are valid candidates in the hotspot of mouse chromosome 2 (104.5–114.8 Mb). The acyltransferase-like 3 (Agpat7) gene and the gamma-butyrobetaine dioxygenase (Bbox1) gene are involved in phospholipid and carnitine biosynthetic processes, respectively. Both biosynthetic pathways have important effects during fetal life and the suckling period (Oey et al. 2006; Shennan and Peaker 2000). The brain-derived neurotrophic factor isoform 1 (Bdnf1) gene influences morphology and function of the brain (Liu et al. 2006). Bdnf1 is also expressed in the mammary gland and potentially plays a role in fat deposition. In addition, secretogranin 5 (Scg5) is one of the most promising candidate genes for growth and fat pad weight maternal QTL in chromosome 2, given the previously reported overexpression in our population of subcongenic mice (Farber et al. 2008). For the second maternal QTL region reported in chromosome 7 (OR 7.4–7.6), only one candidate gene can be reported. Prolyl carboxypeptidase (Prcp) is located at 100.0 Mb and is expressed in several tissues including hypothalamic neurons (García-Horsman et al. 2007). It has several known substrates including kallikrein and angiotensin II. Thus, Prcp could be a potential candidate maternal effects gene.
Our findings provide further evidence of the potentially important role maternal genetic effects may play in the determination of growth and body weight phenotypes. It is generally accepted that direct genetic effects modulate obesity, but the evidence presented here shows that maternal genetic effects can play a key role in fat deposition in adult mice. This is an important starting point to study the genetic basis of fetal programming. In addition, our results support the generation and maintenance of subcongenic strains as an important research tool, given that they can be used to characterize and fine-map genomic regions contributing to phenotypic traits of interest and maternal effects, as described here. Although our experimental design did not allow for the identification of specific genetic mechanisms involved in the maternal QTL, the genomic regions of interest were fine-mapped to small sections of chromosomes 2 and 7 spanning less than 10 Mb. Further investigation using our set of subcongenic strains, as well as generation of additional subcongenic strains, will contribute to the characterization of mutations underlying the reported maternal QTL.
Acknowledgements
We appreciate the excellent efforts of Vince De Vera, Noreene Shibata, and Glenda Espinal in mouse husbandry and with phenotypic data collection. This research was supported by the National Research Initiative grant 2005-35205-15453 from the USDA Cooperative State Research, Education, and Extension Service and by NIH grants DK69978 and DK52581. The research contract of J. Casellas was partially financed by Spain’s Ministerio de Ciencia e Innovación (programs Juan de la Cierva and José Castillejo).
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Joaquim Casellas, Email: joaquim.casellas@irta.es.
Juan F. Medrano, Email: jfmedrano@ucdavis.edu
References
- Abbott DH, Padmanabhan V, Dumesic DA (2006) Contributions of androgen and estrogen to fetal programming of ovarian dysfunction. Reprod Biol Endocrinol 4:17 [DOI] [PMC free article] [PubMed]
- Barker DJP (1998) Mothers, babies and health in later life. Churchill Livingstone, Edinburgh
- Barker DJP, Clarck PM (1997) Fetal undernutrition and disease in later life. Rev Reprod 2:105–112 [DOI] [PubMed]
- Bartolomei MS, Tilghman SM (1997) Genomic imprinting in mammals. Annu Rev Genet 31:493–525 [DOI] [PubMed]
- Björntorp P (1997) Neuroendocrine factors in obesity. J Endocrinol 155:193–195 [DOI] [PubMed]
- Bouchard C (1991) Current understanding of the etiology of obesity: genetic and nongenetic factors. Am J Clin Nutr 53:1561S–1565S [DOI] [PubMed]
- Bouret SG, Simerly RB (2006) Developmental programming of hypothalamic feeding circuits. Clin Genet 70:295–301 [DOI] [PubMed]
- Brindley DN, Rolland Y (1989) Possible connections between stress, diabetes, obesity, hypertension and altered lipoprotein metabolism that may result in atherosclerosis. Clin Sci 77:453–461 [DOI] [PubMed]
- Brockmann GA, Kratzsch J, Haley C, Renne U, Schwerin M et al (2000) Single QTL effects, epistasis, and pleiotropy account for two-thirds of the phenotypic F2 variance of growth and obesity in DU6i × DBA/2 mice. Genome Res 10:1941–1957 [DOI] [PMC free article] [PubMed]
- Casellas J, Medrano JF (2008) Within-generation mutation variance for litter size in inbred mice. Genetics 179:2147–2155 [DOI] [PMC free article] [PubMed]
- Clee SM, Yandell BS, Schueler KM, Rabaglia ME, Richards OC et al (2006) Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat Genet 38:688–693 [DOI] [PubMed]
- Cooney CA, Dave AA, Wolff GL (2002) Maternal methyl supplements in mice affect epigenetic variation and DNA methylation of offspring. J Nutr 132:2400S–3293S [DOI] [PubMed]
- Corva PM, Medrano JF (2000) Diet effects on weight gain and body composition in high growth (hg/hg) mice. Physiol Genomics 3:17–23 [DOI] [PubMed]
- Corva PM, Medrano JF (2001) Quantitative trait loci (QTL) mapping for growth traits in the mouse: a review. Genet Sel Evol 33:105–132 [DOI] [PMC free article] [PubMed]
- de Magalhães JP, Cabral JA, de Magalhães D (2005) The influence of genes on the aging process of mice: a statistical assessment of the genetics of aging. Genetics 169:265–274 [DOI] [PMC free article] [PubMed]
- Diament AL, Warden CH (2004) Multiple linked mouse chromosome 7 loci influence body fat mass. Int J Obes 28:199–210 [DOI] [PubMed]
- Drake AJ, Walker BR (2004) The intergenerational effects of fetal programming: non-genomic mechanisms for the inheritance of low birth weight and cardiovascular risk. J Endocrinol 180:1–16 [DOI] [PubMed]
- Farber CR, Medrano JF (2007) Dissection of a genetically complex cluster of growth and obesity QTLs on mouse chromosome 2 using subcongenic intercrosses. Mamm Genome 18:635–645 [DOI] [PubMed]
- Farber CR, Corva PM, Medrano JF (2006) Genome-wide isolation of growth and obesity QTL using mouse speed congenic strains. BMC Genomics 7:102 [DOI] [PMC free article] [PubMed]
- Farber CR, Chitwood J, Lee S-N, Verdugo RA, Islas-Tejero A et al (2008) Overexpression of Scg5 increases enzymatic activity of PCSK2 and is inversely correlated with body weight in congenic mice. BMC Genomics 9:34 [DOI] [PMC free article] [PubMed]
- García-Horsman JA, Männistö PT, Venäläinen JI (2007) On the role of prolyl oligopeptidase in health and disease. Neuropeptides 41:1–24 [DOI] [PubMed]
- Godfrey KM, Barker DJP (2001) Fetal programming and adult health. Public Health Nutr 4:611–624 [DOI] [PubMed]
- Goharkhay N, Sbrana E, Gamble PK, Tamayo EH, Betancourt A et al (2007) Characterization of a murine model of fetal programming of atherosclerosis. Am J Obstet Gynecol 197:e1–e5 [DOI] [PubMed]
- Hager R, Cheverud JM, Wolf JB (2008) Maternal effects as the cause of parent-of-origin effects that mimic genomic imprinting. Genetics 178:1755–1762 [DOI] [PMC free article] [PubMed]
- Hayward BE, Moran V, Strain L, Bonthron DT (1998) Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally and biallelic derived proteins. Proc Natl Acad Sci USA 95:15474–15480 [DOI] [PMC free article] [PubMed]
- Horvat S, Medrano JF (2001) Lack of Socs2 expression causes the high-growth phenotype in mice. Genomics 72:209–212 [DOI] [PubMed]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y et al (2002) The Ensembl genome database project. Nucleic Acids Res 30:38–41 [DOI] [PMC free article] [PubMed]
- Jarvis JP, Kenney-Hunt J, Ehrich TH, Pletscher LS, Semenkovich CF et al (2005) Maternal genotype affects adult offspring lipid, obesity, and diabetes phenotypes in LGXSM recombinant inbred strains. J Lipid Res 46:1692–1702 [DOI] [PubMed]
- Johnson PR, Hirsch J (1972) Cellularity of adipose depots in six strains of genetically obese mice. J Lipid Res 13:2–11 [PubMed]
- Kapoor A, Dunn E, Kostaki A, Andrews MH, Matthews SG (2006) Fetal programming of hypothalamo-pituitary-adrenal function: prenatal stress and glucocorticoids. J Physiol 572:31–44 [DOI] [PMC free article] [PubMed]
- Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A et al (2003) The UCSU genome browser database. Nucleic Acids Res 31:51–54 [DOI] [PMC free article] [PubMed]
- Lew EA, Garfinkel L (1979) Variations in mortality by weight among 750,000 men and women. J Chronic Dis 32:563–576 [DOI] [PubMed]
- Liu Q-R, Lu L, Zhu X-G, Gong J-P, Shaham Y et al (2006) Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res 1067:1–12 [DOI] [PubMed]
- Maes HH, Neale MC, Eaves LJ (1997) Genetic and environmental factors in relative body weight and human adiposity. Behav Genet 27:325–351 [DOI] [PubMed]
- Mignon-Grasteau S, Piles M, Varona L, Rochambeau H, Poivey JP et al (2000) Genetic analysis of growth curve parameters for male and female chickens resulting from selection based on juvenile and adult body weights simultaneously. J Anim Sci 78:2515–2524 [DOI] [PubMed]
- Mousseau TA, Fox C (1998a) Maternal effects as adaptations. Oxford University Press, New York
- Mousseau TA, Fox C (1998b) The adaptative significance of maternal effects: moms do more than make babies. Trends Ecol Evol 13:403–406 [DOI] [PubMed]
- Müller EE, Locatelli V, Cocchi D (1999) Neuroendocrine control of growth hormone secretion. Physiol Rev 79:511–607 [DOI] [PubMed]
- Myatt L (2006) Placental adaptive responses and fetal programming. J Physiol 572:25–30 [DOI] [PMC free article] [PubMed]
- Oey NA, Van Vlies N, Wijburg FA, Wanders RJA, Attie-Bitach T et al (2006) L-carnitine is synthesized in the human fetal-placental unit: Potential roles in placental and fetal metabolism. Placenta 27:841–846 [DOI] [PubMed]
- Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev 2:21–32 [DOI] [PubMed]
- Remacle C, Bieswal F, Reusens B (2004) Programming of obesity and cardiovascular disease. Int J Obes 28:S46–S53 [DOI] [PubMed]
- Schwartz J, Morrison JL (2005) Impact and mechanisms of fetal physiological programming. Am J Physiol Regul Integr Comp Physiol 288:R11–R15 [DOI] [PubMed]
- Shennan DB, Peaker M (2000) Transport of milk constituents by the mammary gland. Physiol Rev 80:925–951 [DOI] [PubMed]
- Simerly RB (2002) Wired for reproduction: organization and development of sexually dimorphic circuits in the mammalian forebrain. Annu Rev Neurosci 25:507–536 [DOI] [PubMed]
- Simerly RB (2005) Wired on hormones: endocrine regulation of hypothalamic development. Curr Opin Neurobiol 15:81–85 [DOI] [PubMed]
- Warden CH, Fisler JS, Shoemaker SM, Wen P-Z, Svenson KL et al (1995) Identification of four chromosomal loci determining obesity in a multifactorial mouse model. J Clin Invest 95:1545–1552 [DOI] [PMC free article] [PubMed]
- Wheeler TT, Hodgkinson AJ, Prosser CG, Davis SR (2007) Immune components of colostrums and milk—a historical perspective. J Mammary Gland Biol Neoplasia 12:237–247 [DOI] [PubMed]
- Wolf JB, Vaughn TT, Pletscher LS, Cheverud JM (2002) Contribution of maternal effect QTL to genetic architecture of early growth in mice. Heredity 89:300–310 [DOI] [PubMed]
- Wu G, Bazer FW, Wallace JM, Spencer TE (2006) Intrauterine growth retardation: implications for the animal sciences. J Anim Sci 84:2316–2337 [DOI] [PubMed]
- Yura S, Itoh H, Sagawa N, Yamamoto H, Masuzaki H et al (2005) Role of premature leptin surge in obesity resulting from intrauterine undernutrition. Cell Metab 1:371–378 [DOI] [PubMed]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L et al (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 [DOI] [PubMed]