

Abstract
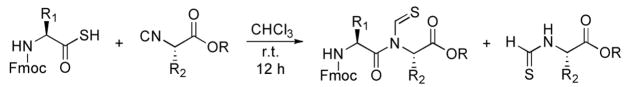
The preparation of N-thioformyl peptides from amino thioacids and isonitriles at room temperature is described.
Since their discovery in the mid 1800s,1,2 isonitriles have been employed in many synthetically useful transformations. Included among these are the Ugi multicomponent couplings3 and the Passerini reaction.4, 5 Recently, we had occasion to explore some new possibilities for amide bond construction. Toward this end, we examined the feasibility of a very simple idea which, remarkably, had thus far been overlooked. 6 It was anticipated that another venerable functional type in organic chemistry, i.e. a carboxyl group, would react with an isonitrile to produce a formimidate carboxy mixed anhydride (cf. systems 3a and 3s which we abbreviate as FCMAs) While, in practice, we have not isolated or characterized either FCMA from the reaction of 1 and 2, we do obtain the N-formylamide 4 when the reaction is conducted at ca 130 °C under microwave thermolysis.7 Clearly, the nitrogen in the context of its two carbonyl attachments corresponds to a mixed imide ensemble. Not surprisingly, nucleophile induced deacylation occurs specifically at the N-formyl bond to afford 5. Other productive options for product 4 were studied and reported.8
While we do not detect any “in hand product” upon mixing of 1 and 2, strong inferential evidence was brought to bear that, in fact, FCMA system 3 is being produced. Thus, when 1 and 2 are heated in the presence of amines (see 6), amides 7 (the apparent products of interdiction of 3) are produced in modest yield, though the acyl transfer product 5 is not detected.7 These findings invite the interpretation that the FCMA structures are, in fact, slowly generated at room temperature but that the presumed 1,3 O→N-acyl transfer requires significant thermal activation.6,9
We feel that this line of chemistry is quite promising because the versatile mixed imide products 4 are generated from acids without the need for either separate external activation of the carboxyl group to a higher acyl donor level, or for coupling agents which function by in situ acyl donor activation. In our chemistry, which we term “2 component coupling strategy” (2CC), the carboxyl is activated in the natural context of its progression to amides.
Of course, we were not unmindful of the potential limitations which ensue from the requirement of thermal activation, particularly as we approach challenging fragment mergers. In this Letter, we report on the use of thioacids, in place of normal carboxylic acids, hoping to realize N-thioformyl imide formation under mild conditions. To the best of our knowledge, there had been only one literature precedent for the formation of N-thioformyl imide, prior to the work documented here, from the reaction of thiocarboxylic acids and isonitriles. It was reported by Chupp and co-workers but only in the context of very simple thioacids (acetic, benzoic thioacids and their derivatives).10 In the study reported below, we sought to evaluate how the substitution of a thioacid in place of 1 would alter the chemistry described above in more complex settings.
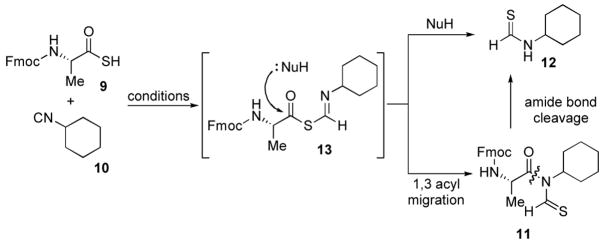
Our initial studies focused on the coupling between Fmoc-Ala-SH 9 11 and commercially available cyclohexyl isonitrile 10. Under mild thermolysis, N-thioformyl imide 11 was indeed obtained in 21% yield, along with a significant amount of cyclohexyl thioformamide 12 (35%). When the reactions were conducted in diethyl ether or chloroform, transformation to product 11 occurred in improved yield with correspondingly diminished formation of small amounts of side product, to the level of a few percent. At this stage, we could not rigorously establish whether 12 arises via adventitious cleavage of 11 or, more likely, by nucleophile induced cleavage of the presumed thio FCMA intermediate 13. It is conceivable that in entries 1 and 2, DMF may serve in the role of NuH.
The generality of the reaction was examined by recourse to different amino acid derivatives reacting with amino acid isonitriles. As demonstrated in Table 2, both sterically hindered thioacids (valine derivative, entries 1–3) and less hindered thioacids (glutamic acid derivative, entries 5 and 6) reacted with isonitriles to give dipeptides in reasonable yields. Reactions of thioacids with aromatic functional groups (tyrosine derivative, entries 7 and 8) proceeded equally well to afford the desired N-thioformyl imides in good yields. In the reaction of valine thioacid, we observed that the yields of the N-thioformyl imides were increased with ascending isonitrile steric hindrance. We surmise that perhaps the rate of cleavage of the thio FCMA is suppressed in hindered cases. Alternatively, and in the same direction, the rate of syn→anti isomerization of intermediate FCMA 23 is increased in the most hindered substrates (Scheme 2). 12 Indeed, in the least hindered glycine isonitrile case (entry 4), the desired imide was isolated in 14% yield along with 68% of thioformyl amide and 67% of the symmetric thioanhydride. Efforts to minimize this unproductive but competitive pathway have thus far proven to be unsuccessful.
Table 2.
 | |||
|---|---|---|---|
| Entry | Producta | Yieldb | [α]D25 |
| 1 |
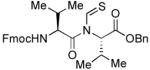
|
60% (82%) | −12.0 |
| 2 |
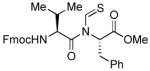
|
50% | −11.6 |
| 3 |
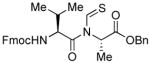
|
28% | −17.0 |
| 4 |
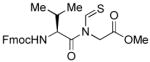
|
14% | +34.4 |
| 5 |
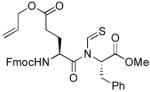
|
49% (61%) | +14.8 |
| 6 |
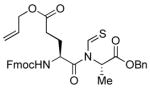
|
59% | −29.7 |
| 7 |
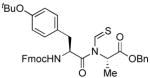
|
60% | −7.2 |
| 8 |
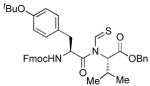
|
63% | −71.1 |
1.0 equivalent of thioacid and 1.0 equivalent of isonitrile were used for all reactions.
Yields refer to isolated yields. The yields in parentheses are based on recovery of the starting isonitriles.
Scheme 2.

Unfortunately, we have not been able to extend this chemistry effectively to the coupling of a dipeptide with a single amino acid. We attempted to accomplish this in each of the two possible directions. In the case of the reaction of the C-terminal dipeptide thio acid, with the phenylalanine derived isonitrile 25, there was obtained only the phenylalanine derived thioformamide 27. A reasonable interpretation of this breakdown presupposes oxazalone 28 formation from the activated thio FCMA 26. When conducted in the opposite sense, i.e. dipeptide isonitrile 25 and valine derived thioacid 22, there was obtained, albeit in only 18%, the tripeptide 29 and also thioformamide 27. We note that in this case, internal cleavage of the thio FCMA via oxazalone formation could not occur, hence it can not be the sole course of the problem.
The viability of methodology for native peptide bond formation required dethioformylation of 21. An initial attempt at dethioformylation was performed according to the procedure reported by Chupp and coworkers.11 However, treatment of 21 with aniline hydrochloride in toluene for 20 h under reflux afforded thioformamide 30 only. The same regioselectivity was observed under basic conditions as well. This surprising result indicates how a subtle structural change (from formyl to thioformyl) can control fundamental chemical properties.
Fortunately dethioformylation could be accomplished by a 2-step protocol (Scheme 5). Treatment of 21 with mCPBA in dichloromethane at low temperature afforded the corresponding N-formyl imide, which was smoothly converted in 80% yield to the desired amide 31 whose spectroscopic properties are identical to the authentic sample prepared by other standard methods.13 To our knowledge, the work described here is the first recognition and solution of this problem.
Scheme 5.

In summary, we have shown that the reaction of thioacids with isonitriles can be used to generate N-thioformylamides at rt in contrast to the requirement for thermolysis in the carboxy counterpart. The reaction is applicable to the synthesis of dipeptides. It is, however, complicated by the high vulnerability of the thio FCMA intermediate to interdiction, competitive with the key S→N acyl transfer. This problem is, at this writing, unmanageable in attempted 2 + 1 couplings in peptides. Finally effective methodology to accomplish the transformylation of N-thioformyl amides is described.
Applications of these general ideas to some synthetic targets which are otherwise difficult to access will be described in due course.
Supplementary Material
Scheme 1.

Scheme 3.

Scheme 4.

Table 1.
 | ||
|---|---|---|
| Entry | Conditionsa | Yieldb |
| 1 | DMF, 40 ºC, 2 h | 21%c |
| 2 | DMF, microwave, 60 ºC,25 min | 22% |
| 3 | Ether, r.t., 6 h | 50% |
| 4 | CHCl3, r.t., 6 h | 53% d |
1.0 equiv. of thioacid and 2.0 equiv. of isonitrile were used for all reactions.
isolated yields for 11.
35% of the cyclohexyl thioformamide was isolated.
71% based on the recovery of thioacid.
Acknowledgments
This work was supported by Grant CA103823.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gautier A. Liebigs Ann Chem. 1867;142:289. [Google Scholar]
- 2.Hofmann AW. Liebigs Ann Chem. 1867;144:114. [Google Scholar]
- 3.Ugi I, Lohberger S, Karl R. The Passerini and Ugi Reactions, Chapter 4.6. Comprehensive Organic Synthesis. 1991;2:1083. [Google Scholar]
- 4.Passerini M, Simone L. Gazz Chim Ital. 1921;51:126. [Google Scholar]
- 5.Passerini M, Ragni G. Gazz Chim Ital. 1931;61:964. [Google Scholar]
- 6.In this regard, some initiatives, which invoked incorrect structural assignments, have been reported in the literature. For details, see: Li X, Yuan Y, Berkowitz WF, Todaro LJ, Danishefsky SJ. J J Am Chem Soc. 2008;130:13222. doi: 10.1021/ja8047078.
- 7.Li X, Danishefsky SJ. J Am Chem Soc. 2008;130:5446. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corriu RJP, Lanneau GF, Perrot-Petta M, Mehta VD. Tetrahedron Lett. 1990;31:2585. [Google Scholar]
- 9.Li X, Yuan Y, Kan C, Danishefsky SJ. J Am Chem Soc. 2008;130:13225. doi: 10.1021/ja804709s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chupp JP, Leschinsky KL. J Org Chem. 1975;40:66. [Google Scholar]
- 11.Fu X, Jiang S, Li C, Xin J, Yang Y, Ji R. Bioorg Med Chem Lett. 2007;17:465. doi: 10.1016/j.bmcl.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Marcelli T, Himo F. Eur J Org Chem. 2008:4751. [Google Scholar]
- 13.Significant level of epimerization was observed, if the reaction was performed at higher temperature.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.