

Abstract
Background:
Nonsteroidal anti-inflammatory drugs (NSAIDs) may prevent Alzheimer dementia (AD).
Methods:
We analyzed the association of prior NSAID exposure with incident dementia and AD in the Adult Changes in Thought population-based cohort aged ≥65 years (median 74.8) at enrollment. Participants were members of Group Health, which provided computerized pharmacy dispensing records from 1977 onward. We studied 2,736 dementia-free enrollees with extensive prior pharmacy data, following them biennially for up to 12 years to identify dementia and AD. Cox proportional hazards regression assessed association of dementia or AD with NSAID use graded in standard daily doses (SDD) dispensed over 2 years (e.g., heavy use = 500+ SDD), with some analyses also adding consecutive biennial self-reports of NSAID use.
Results:
Pharmacy records identified 351 participants (12.8%) with history of heavy NSAID use at enrollment. Another 107 became heavy users during follow-up. Some 476 individuals developed incident dementia, 356 with AD (median onset ages 83.5 and 83.8 years). Contrary to the hypothesis that NSAIDs protect against AD, pharmacy-defined heavy NSAID users showed increased incidence of dementia and AD, with adjusted hazard ratios of 1.66 (95% confidence interval, 1.24–2.24) and 1.57 (95% confidence interval, 1.10–2.23). Addition of self-reported exposure data did not alter these results.
Conclusions:
These findings differ from those of other studies with younger cohorts. The results observed elsewhere may reflect delayed onset of Alzheimer dementia (AD) in nonsteroidal anti-inflammatory drug (NSAID) users. Conceivably, such delay could result in increased AD incidence in late old age. The relation of NSAID use and AD pathogenesis needs further investigation.
GLOSSARY
- ACT
= Adult Changes in Thought;
- AD
= Alzheimer dementia;
- ADL
= activities of daily living;
- aHR
= adjusted hazard ratio;
- CI
= confidence interval;
- DSM-IV
= Diagnostic and Statistical Manual of Mental Disorders, 4th edition;
- GH
= Group Health;
- HR
= hazard ratio;
- NSAID
= nonsteroidal anti-inflammatory drug;
- OR
= odds ratio;
- SDD
= standard daily dose.
Although the pathogenesis of Alzheimer dementia (AD) is poorly understood, inflammatory mechanisms are probably involved. Following up suggestive early observations that incidence of AD is lower among users of nonsteroidal anti-inflammatory drugs (NSAIDs), the NSAIDs–AD relationship has been evaluated in more than 30 observational studies.1 Most have confirmed the original observations, although some well-designed studies have found different results.2,3 NSAIDs are not helpful for people with established AD dementia,4 and the selective cyclooxygenase-2 inhibitor rofecoxib does not allay progression of milder cognitive symptoms to AD.5 NSAIDs also appear to offer no benefit to people whose preclinical AD pathology is sufficiently advanced that they develop dementia symptoms within very few years,6,7 a conclusion that was also supported by early results from the AD Anti-inflammatory Prevention Trial (ADAPT).8 The relationship of NSAID use and AD thus appears to be more complex than was earlier believed.
To investigate these conflicting results, we undertook a prospective analysis of NSAID use and incident dementia or AD in the Adult Changes in Thought (ACT) study, a population-based cohort investigation with several advantages for such work. These included an electronic pharmacy dispensing database with prescription information available since 1977, a prospective design with screening and examination for dementia starting in 1994 and biennial follow-up thereafter for up to 12 years, and a large sample that enabled unusually detailed analyses. We hypothesized that NSAID use would delay the onset of dementia and AD in the ACT cohort, resulting in decreased incidence.
METHODS
ACT study population.
ACT is an ongoing prospective, community-based study of incident dementia and AD. Methods have been described elsewhere.9,10 Briefly, between 1994 and 2003, ACT enrolled 3,392 cognitively intact community-dwelling participants aged 65 years and older from a population base of 23,000 members of Group Health (GH), a large integrated health care delivery system in King County, WA. The ACT cohort was assembled in two phases: between 1994 and 1996, a random sample was drawn from West King County clinics to form an initial cohort of 2,581 individuals; then, using similar methods, an expansion cohort of 811 was enrolled between 2001 and 2003. Participants gave informed consent for all procedures as approved by the Institutional Review Boards of each relevant institution.
Participants' demographic, medical history, and functional status were collected via in-person interviews at baseline and at biennial follow-up visits for ascertainment of incident dementia. To ensure a thorough accounting of prior NSAID exposure, our analyses considered only ACT enrollees with at least 10 years of GH membership. Primary analyses used pharmacy data for participants with at least one follow-up visit. Secondary analyses considered both pharmacy and self-reported exposures in participants with at least two follow-up visits (see below).
Participant characteristics.
Health and lifestyle characteristics at baseline included measurement of body mass index and self-rating of health, physical activity, alcohol use, and smoking history. APOE genotype was determined as described.9 Participants with one or two APOE ɛ4 alleles were considered APOE ɛ4-positive.11,12 Participants were also asked if a doctor had ever told them they had angina, congestive heart failure, heart attack, stroke, small strokes or transient ischemic attacks, hypertension, diabetes mellitus, osteoarthritis, or cancer, and whether they had ever had coronary bypass surgery or balloon angioplasty. Presence of coronary heart disease was defined by self-reported history of heart attack, angina, angioplasty, or coronary artery bypass surgery.
Ascertainment of dementia and AD.
At baseline and follow-up visits, cognitive function was evaluated using the Cognitive Abilities Screening Instrument.13 Persons scoring ≤85 were referred for a dementia evaluation including neuropsychological testing, medical history, physical examination, laboratory testing, and appropriate neuroimaging. Diagnoses were assigned by a multidisciplinary study team at consensus conferences.9 Criteria for dementia were from DSM-IV14 and diagnoses of probable and possible AD followed National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria.15 Onset date for incident cases was conventionally assigned as midway between the preceding ACT visit and the visit that triggered a positive dementia evaluation.
Time-varying NSAID exposure classification.
We developed two time-varying measures of NSAID exposure, one based on solely pharmacy dispensing data and another integrating pharmacy data with self-report. The GH pharmacy database includes all prescriptions dispensed from March 1977 to the present, including drug name, strength, and amount dispensed. For the primary analyses, we defined standard daily doses for each NSAID,16 much as was done by the Rotterdam study.6 We calculated the number of standard daily doses (SDDs) associated with each prescription in the database, multiplying the amount dispensed by the strength and dividing by the SDD. Then, starting at enrollment in GH or in 1977, whichever occurred later, we considered all prescription fills within a rolling 2-year window of time. Ignoring the last year before dementia onset to avoid consideration of NSAID use influenced by the presence of dementia,6 we counted the number of SDDs over the window. We classified exposure as 1) heavy use, with ≥500 SDD prescribed within 2 years; 2) moderate use, with 60–499 SDD; and 3) light/no use, with <60 SDD. At each time point, a participant's degree of exposure was classified as the highest category attained to date.
Secondary analyses included not only pharmacy-based exposures but also self-reports of NSAID use. For the self-report analyses only, heavy use of NSAIDs required a report of use on two or more successive visits. Again, exposure status over time reflected the highest category to date, whether from pharmacy or self-report data. Because these analyses required two successive interviews for exposure ascertainment and one further visit for detection of incident dementia, the eligible population here was smaller than for the primary, pharmacy-only analyses.
Statistical analyses.
We considered two outcomes: all-cause dementia and AD. In Cox proportional hazards models with time-dependent covariates,17 we used light/nonusers of NSAIDs as the reference group, and participant age as the time axis.18 Individuals without an event were censored at their last ACT visit. All analyses and model diagnostics were performed using PHREG, SAS version 9.1.3 (SAS Institute, Cary, NC).
We fitted three models for each outcome/exposure combination. Model 1 adjusted for potential age cohort effects by stratifying ages at ACT baseline into 5-year age categories, and for study cohort effects by including a variable indicating initial or expansion cohort membership. Model 2 additionally adjusted for characteristics commonly associated with dementia risk: gender, education, and APOE status. To control for other potential confounding, model 3 also included time-dependent covariates for comorbidities plausibly associated with NSAID use or risk of dementia: hypertension, diabetes, obesity, osteoarthritis, and physical activity. We report our main results from the most fully adjusted model 3.
We assessed the robustness of our results with several sensitivity analyses (e-appendix on the Neurology® Web site at www.neurology.org). Additional models included use of histamine H2 blockers or proton pump inhibitors (from self-report), history of coronary heart disease, and impaired activities of daily living. We also tested the proportional hazards assumption by examining possible interactions between age and other covariates. To explore an influence of recency of exposure, we separated heavy users into groups with recent heavy use (within the past 5 years) or with distant heavy use only (at least 5 years before onset.) Other sensitivity analyses considered only NSAID use since the inception of ACT (to simulate the Rotterdam study) or removal or further restriction of the ≥10-year GH enrollment criterion. We also categorized NSAID exposures by the reported capacities of individual agents to reduce production of amyloidogenic Aβ42 in vitro.2 Finally, we considered other outcomes including the more restrictive category of probable AD.15
RESULTS
Baseline characteristics of the analytic sample.
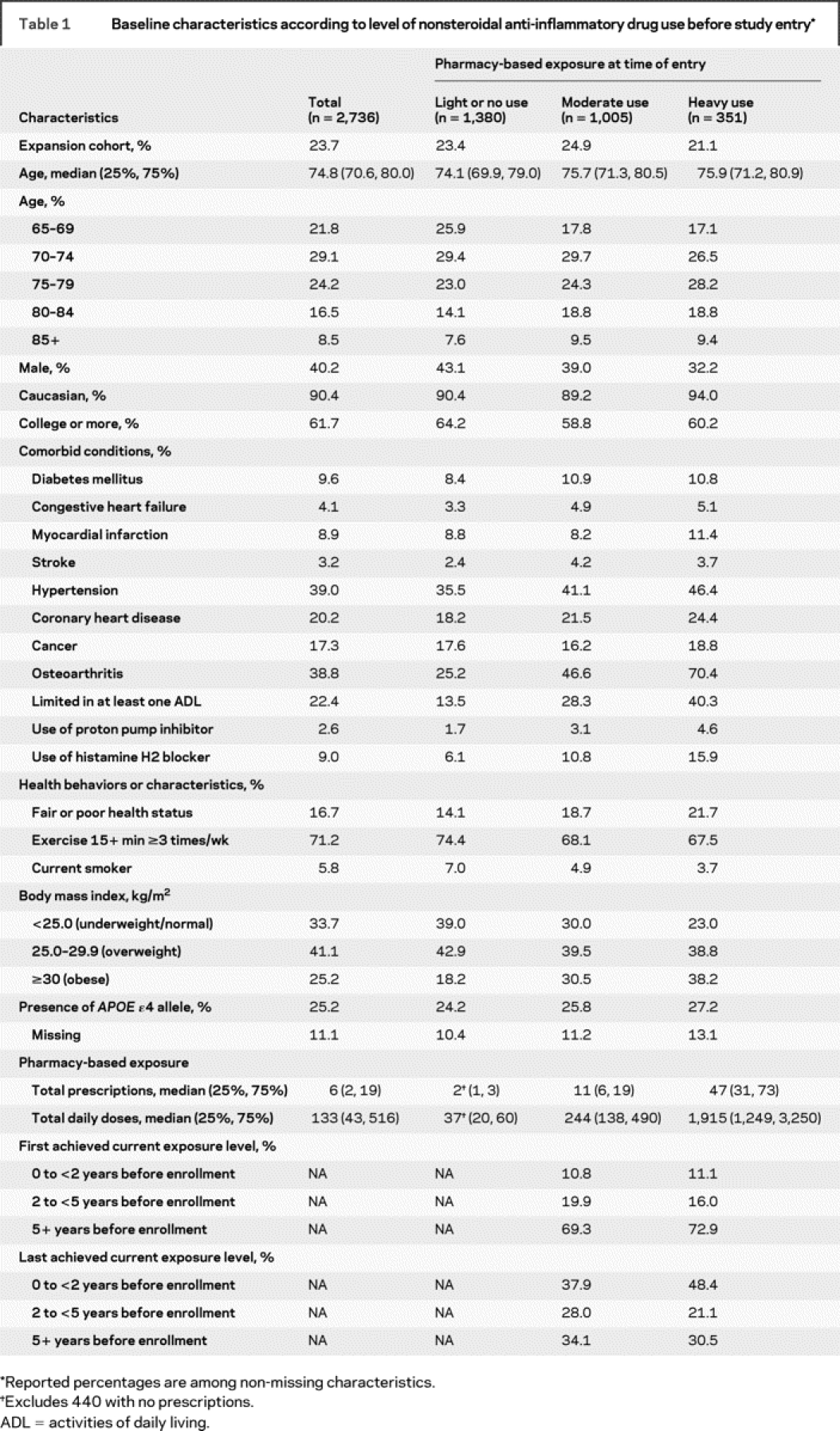
Of the 3,392 ACT participants, 3,026 (89%) had the requisite 10 years of GH enrollment at baseline. Of these, 2,736 (90%) completed at least one follow-up evaluation and were included in our primary analyses, while 2,095 (69%) completed the two or more follow-up visits needed for the secondary analyses. Table 1 provides baseline characteristics stratified by pharmacy-based NSAID exposure level at baseline. The median age at enrollment was 74.8 years (interquartile range, 70.6–80.0). Approximately 40% were male, 90% were Caucasian, and more than 60% had at least some college education. The analytic sample did not differ meaningfully at baseline from the full ACT cohort with regard to the distribution of health and sociodemographic characteristics (not shown). Of the 2,736 participants with at least one follow-up visit, 1,380 (50%) had light or no use at baseline, as defined by pharmacy data, while 1,005 (37%) were moderate users, and 351 (13%) were heavy users. Demographic characteristics and APOE status did not differ substantially across these exposure groups, but moderate or heavy users had more comorbid conditions and adverse health behaviors or characteristics than light/nonusers. Missing data were <2% for all variables with the exception of APOE status.
Table 1 Baseline characteristics according to level of nonsteroidal anti-inflammatory drug use before study entry
Exposures.
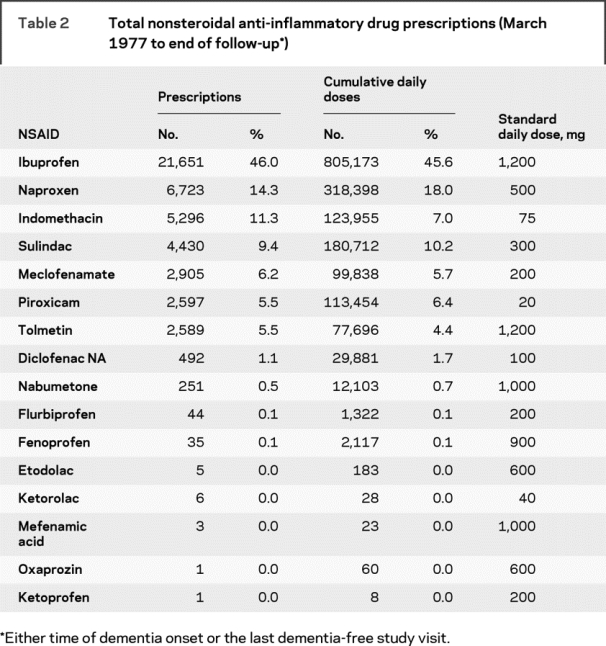
Table 2 shows pharmacy data on NSAIDs for the study cohort. For both prescriptions and SDD, almost half (46%) were for ibuprofen. Together, ibuprofen, naproxen, indomethacin, and sulindac accounted for approximately 80% of all prescriptions and SDD. Among the 1,380 light/nonusers at baseline, subsequent prescriptions led to exposure reclassification such that 231 (17%) became moderate users and 27 (2%) became heavy users. An additional 80 baseline moderate users (8% of 1,005) became heavy users during the follow-up period. For the secondary analyses, of the 1,079 light/nonusers at baseline (52% of 2,095), 195 (18%) became moderate users and 66 (6%) became heavy users on the basis of combined pharmacy and self-report information. Similarly, an additional 121 of 755 (16%) baseline moderate users became heavy users. Table 1 shows timing of moderate and heavy NSAID exposure, including the earliest and most recent time period in which that intensity of exposure was present.
Table 2 Total nonsteroidal anti-inflammatory drug prescriptions (March 1977 to end of follow-up)
Incidence of dementia and AD.
As of December 2006, the 2,736 members of the analytic sample had accrued 16,931 person-years of follow-up: 7,627 person-years among those ultimately characterized as light/nonusers; 6,731 among individuals finally classified as moderate users; and 2,573 among heavy users. There were 476 (17%) cases of incident dementia, with 356 (13% of the sample) attributed to AD. Another 277 (10%) withdrew from ACT and 655 (24%) died before withdrawal or diagnosis.
Regression modeling.
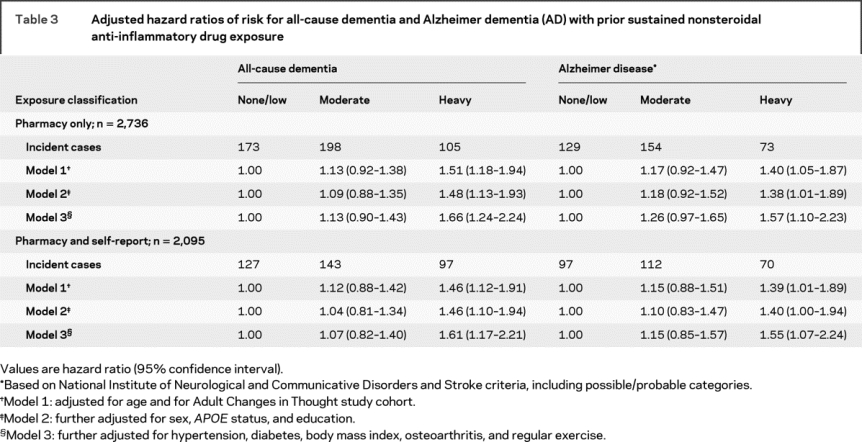
Table 3 shows results from the main regression analyses. In primary analyses, the adjusted hazard ratio (aHR) for all-cause dementia in heavy users was 1.66 (95% confidence interval [CI], 1.24–2.24), for AD 1.57 (95% CI, 1.10–2.23). Omnibus Wald tests jointly assessed effects across the three NSAID exposure groups, yielding p values of 0.003 and 0.036 for the primary analyses. These results were consistent across all models. Results for the secondary analyses combining pharmacy and self-reported exposures yielded similar results. Sensitivity analyses showed no evidence of an interaction with age (e.g., aHR 1.02, p = 0.581, for the analysis of all-cause dementia and heavy NSAID exposure). Splitting heavy users into distant and recent users yielded aHRs for all-cause dementia of 1.51 (95% CI, 1.03–2.20) for distant users and 1.85 (95% CI, 1.28–2.66) for recent users. Results for probable AD, and for all other sensitivity analyses, were similar, and there was no appreciable distinction in the effect of so-called Aβ-lowering NSAIDs vs other agents2 (see e-appendix for details on the sensitivity analyses).
Table 3 Adjusted hazard ratios of risk for all-cause dementia and Alzheimer dementia (AD) with prior sustained nonsteroidal anti-inflammatory drug exposure
DISCUSSION
In a large cohort study of an elderly population-based sample, we observed no reduction in risk of dementia or AD among users of NSAIDs. Instead, we found that prior sustained NSAID exposure was associated with increased incidence of dementia and AD. This result was robust to comprehensive sensitivity analyses investigating features of both the design and analytic approach.
Interest in NSAIDs for AD treatment or prevention began in 1990, when patients with rheumatoid arthritis (for the most part, obligatory users of NSAIDs) were reported to show reduced rates of Alzheimer pathology.19 Several early studies20,21 then spurred speculation that anti-inflammatory treatments including NSAIDs could delay or prevent the onset of AD. Subsequently, numerous observational studies reported data consistent with this notion. Notably, a population-based prospective study from Rotterdam examined individuals aged 55 years and older who had NSAID prescription information from a computerized pharmacy dispensing database.6 A 2004 meta-analysis of three carefully conducted case-control studies and four cohort studies showed convergent odds ratios (ORs) or hazard ratios (HRs) near 0.6, suggesting a 40% lower incidence of AD among those exposed.22 A more recent meta-analysis of six population-based prospective studies suggested an HR of 0.75 (95% CI, 0.64–0.89),2 but no difference in apparent effects of categories of NSAIDs that were distinguished by their ability to reduce production of the amyloidogenic peptide Aβ42 in vitro. A large recent case-control study using the national VA database found an OR of 0.76 (95% CI, 0.68–0.85) with 5 years of NSAID use.23
However, the AD–NSAIDs relationship may be more complicated. NSAIDs are not helpful for people with established AD,4 and rofecoxib does not allay progression of milder cognitive symptoms to AD.5 NSAIDs also appear to offer no benefit to people whose preclinical AD pathology is sufficiently advanced to produce dementia symptoms within very few years.6,7 ADAPT, which followed participants for only 24 months on average after randomization, yielded relative risks of 1.99 (95% CI, 0.80–4.97) and 2.35 (95% CI, 0.95–5.77) in those assigned to celecoxib or naproxen vs placebo.
Similarly, the present findings appear to contradict the straightforward hypothesis of reduced incidence with NSAID exposure, and they contrast with results from many prior studies, including the methodologically similar Rotterdam study.6 Why? Possibly owing to several methodologic strengths, we may have observed a truer representation of the association of NSAIDs and dementia risk than others before us. These strengths include not only a community-based sample but also biennial assessment for dementia and AD, rigorous exposure classification based on pharmacy dispensing records beginning in 1977 (17 or more years before initial enrollment into ACT), large numbers of incident dementia or AD cases affording good statistical power, and consideration to self-reported as well as pharmacy-based exposure data. Some confidence in the robustness of our observations is also suggested by the consistency of the associations observed in a variety of models using several methods of exposure classification and multiple outcomes, including several sensitivity analyses. On balance, these strengths appear to outweigh the weaknesses in our study, which include a sample which, although representative of its population base, might not generalize well to other populations, lack of precise dosing information, and (inherent in all observational studies) the possibility of bias from inadequately measured or unsuspected confounders.
Upon reflection, however, we pursued other explanations for the discrepancy. For example, unlike the Rotterdam investigators, we had detailed information on NSAID exposure for many years before enrollment. This might arguably have reduced misclassification of individuals as nonusers who had heavy NSAID exposure before enrollment. However, sensitivity analyses ignoring earlier exposures did not change our results (e-appendix). Alternatively, NSAID use might have promoted AD onset in a particular group of susceptible individuals with unrecognized advanced preclinical AD pathology, as appeared likely in ADAPT.8 Arguably, these nondemented individuals had advanced AD pathology because they developed dementia within such a short time and, given its advanced age, the ACT cohort may have included many such individuals. Again, however, this explanation was not supported by a sensitivity analysis of distant vs recent exposures, which found no reduction in dementia or AD risk in participants with distant heavy exposures only (e-appendix).
We therefore considered whether the most likely explanation of the discrepant findings related to differences in participant ages across studies. The ACT cohort is older than most previously studied populations, with a minimum enrollment age of 65 years and a median age of 83.5 years (interquartile range, 79.6–86.9) at dementia onset. By contrast, the Rotterdam cohort was much younger, with 45% of participants younger than 65 years at enrollment. The Cache County Study7 cohort was also younger, with a mean age of 74 years at dementia assessment. The latter figure is broadly typical for most prior studies, which have formed the evidence base for reduced incidence of AD in NSAID users.2 The Cache County Study also found an interaction between age and AD incidence in NSAID users, such that an aHR of 0.38 at the mean age of 74 was increased by 7% with each year of age. We regarded this sort of modification of effect by age as potentially important because, similar to present results, two other studies of substantially older cohorts, the Religious Orders Study3 and the MoVIES project (median dementia onset near age 80),24 failed to show a reduced risk of AD, with aHRs of 1.19 (95% CI, 0.87–1.62) and 1.16 (95% CI, 0.82–1.63; data from reference 7). Upon closer inspection, however, this level of interaction appeared too modest to explain our unexpected results, and probably those of the Religious Orders Study or MoVIES as well.
How else might differences in cohort age account for different results? Others have suggested that differences across age groups may be attributable to selection bias.25 We attempted to control for such cohort effects by stratifying our analyses by 5-year age groups at enrollment. However, this technique produced little change in results (data not shown). We also considered one other explanation that, if substantiated, might simultaneously explain our results and also account for opposite findings from younger cohorts. A part of our hypothesis was that NSAID exposure delays the onset of AD. Indeed, it is commonly conjectured that most, if not all, AD risk factors act by accelerating or retarding dementia onset. This phenomenon has been shown repeatedly for APOE genotype,26,27 and it appears also to apply to head injury.28 In fact, we know of no AD risk-modifying factor that demonstrably acts otherwise. If NSAID exposure defers the onset of AD, then exposed members of younger cohorts would logically show a reduced frequency of disease, but NSAID users in older cohorts could be enriched for cases that would otherwise have appeared earlier.
AUTHOR CONTRIBUTIONS
Statistical analyses were performed by S.J.P.A.H. and R.W.
Supplementary Material
Received October 20, 2008. Accepted in final form January 26, 2009.
Address correspondence and reprint requests to Dr. Breitner, GRECC (S-182), VA Puget Sound Health Care System, 1660 South Columbian Way, Seattle, WA 98108 jcsb@u.washington.edu
Supplemental data at www.neurology.org
Editorial, page 1884
e-Pub ahead of print on April 22, 2009, at www.neurology.org.
*These authors contributed equally.
Supported by the US Department of Veterans Affairs and NIH Grants U01-AG-06781, R01-AG-24010, U01-AG-15477, and K23-AG-28954 (Paul Beeson award to S.D., supported in part by the American Federation for Aging Research, the Hartford Foundation, the Atlantic Philanthropies, and the Starr Foundation).
Disclosure: The authors report no disclosures.
REFERENCES
- 1.Szekely CA, Town T, Zandi PP. NSAIDs for the chemoprevention of Alzheimer's disease. Subcell Biochem 2007;42:229–248. [DOI] [PubMed] [Google Scholar]
- 2.Szekely CA, Green RC, Breitner JC, et al. No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology 2008;70:2291–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvanitakis Z, Grodstein F, Bienias JL, et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 2008;70:2219–2225. [DOI] [PubMed] [Google Scholar]
- 4.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 2003;289:2819–2826. [DOI] [PubMed] [Google Scholar]
- 5.Thal LJ, Ferris SH, Kirby L, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005;30:1204–1215. [DOI] [PubMed] [Google Scholar]
- 6.in't Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 2001;345:1515–1521. [DOI] [PubMed] [Google Scholar]
- 7.Zandi PP, Anthony J, Hayden K, et al. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology 2002;59:880–886. [DOI] [PubMed] [Google Scholar]
- 8.AD APT Research Group. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007;68:1800–1808. [DOI] [PubMed] [Google Scholar]
- 9.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 2002;59:1737–1746. [DOI] [PubMed] [Google Scholar]
- 10.Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006;144:73–81. [DOI] [PubMed] [Google Scholar]
- 11.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990;31:545–548. [PubMed] [Google Scholar]
- 12.Emi M, Wu LL, Robertson MA, et al. Genotyping and sequence analysis of apolipoprotein E isoforms. Genomics 1988;3:373–379. [DOI] [PubMed] [Google Scholar]
- 13.Teng EL, Hasegawa K, Homma A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr 1994;6:45–58; discussion 62. [DOI] [PubMed]
- 14.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. Washington, DC: American Psychiatric Association; 1994:886. [Google Scholar]
- 15.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 16.Drug Facts and Comparisons 2008, 62nd ed. St. Louis: Lippincott Williams & Wilkins; 2007. [Google Scholar]
- 17.Fleming TR, Harrington DP. Counting Processes and Survival Analysis. New York, NY: John Wiley & Sons; 1991. [Google Scholar]
- 18.Korn EL, Graubard BI, Midthune D. Time-to-event analysis of longitudinal follow-up of a survey: choice of the time-scale. Am J Epidemiol 1997;145:72–80. [DOI] [PubMed] [Google Scholar]
- 19.McGeer PL, McGeer E, Rogers J, Sibley J. Anti-inflammatory drugs and Alzheimer disease. Lancet 1990;335:1037. [DOI] [PubMed] [Google Scholar]
- 20.Rogers J, Kirby LC, Hempelman SR, et al. Clinical trial of indomethacin in Alzheimer's disease. Neurology 1993;43:1609–1611. [DOI] [PubMed] [Google Scholar]
- 21.Breitner JCS, Gau BA, Welsh KA, et al. Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology 1994;44:227–232. [DOI] [PubMed] [Google Scholar]
- 22.Szekely C, Thorne J, Zandi P, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology 2004:159–169. [DOI] [PubMed] [Google Scholar]
- 23.Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 2008;70:1672–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganguli M, Dodge HH, Chen P, et al. Ten-year incidence of dementia in a rural elderly US community population: the MoVIES Project. Neurology 2000;54:1109–1116. [DOI] [PubMed] [Google Scholar]
- 25.Hernan MA, Alonso A, Logroscino G. Cigarette smoking and dementia: potential selection bias in the elderly. Epidemiology 2008;19:448–450. [DOI] [PubMed] [Google Scholar]
- 26.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261:921–923. [DOI] [PubMed] [Google Scholar]
- 27.Khachaturian KS, Corcoran C, Mayer LS, et al. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study. Arch Gen Psychiatry 2004;61:518–524. [DOI] [PubMed] [Google Scholar]
- 28.Gedye A, Beattie BL, Tuokko H, et al. Severe head injury hastens age of onset of Alzheimer's disease. J Am Geriatr Soc 1989;37:970–973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.