

Abstract
Objective:
A recent study reported that mutations in a gene on chromosome 2q36-37, GIGYF2, result in Parkinson disease (PD). We have previously reported linkage to this chromosomal region in a sample of multiplex PD families, with the strongest evidence of linkage obtained using the subset of the sample having the strongest family history of disease and meeting the strictest diagnostic criteria. We have tested whether mutations in GIGYF2 may account for the previously observed linkage finding.
Methods:
We sequenced the GIGYF2 coding region in 96 unrelated patients with PD used in our original study that contributed to the chromosome 2q36-37 linkage signal. Subsequently, we genotyped the entire sample of 566 multiplex PD kindreds as well as 1,447 controls to test whether variants in GIGYF2 are causative or increase susceptibility for PD.
Results:
We detected three novel variants as well as one of the previously reported seven variants in a total of five multiple PD families; however, there was no consistent evidence that these variants segregated with PD in these families. We also did not find a significant increase in risk for PD among those inheriting variants in GIGYF2 (p = 0.28).
Conclusions:
We believe that variation in a gene other than GIGYF2 accounts for the previously reported linkage finding on chromosome 2q36-37.
GLOSSARY
- GDS
= Geriatric Depression Scale;
- MMSE
= Mini-Mental State Examination;
- NCRAD
= National Cell Repository for Alzheimer’s Disease;
- PD
= Parkinson disease;
- PSG
= Parkinson Study Group;
- UPDRS
= Unified Parkinson’s Disease Rating Scale.
Parkinson disease (PD) is the second most common neurodegenerative disorder, affecting 3% of the population above age 75.1 Mutations in five genes can result in autosomal dominant or autosomal recessive forms of PD.2 Previously, we reported linkage to an 18 cM region on chromosome 2q36-37 in a sample of 194 multiplex PD kindreds.3 Subsequently, we demonstrated that the evidence of linkage in this region was even greater when the dataset was limited to the subset of pedigrees having a stronger family history of PD, typically consistent with autosomal dominant inheritance.3,4
Within the 18-cM region identified in our linkage study, the gene for Grb10-Interacting GYF protein 2 (GIGYF2) was identified by Giovannone and colleagues5 using yeast two-hybrid screening in a study of novel proteins linked to insulin-like growth factor receptors by the Grb10 adapter. GIGYF2 is hypothesized to modulate IGF-I signaling.5 As the IGFs and insulin have important effects in the CNS and are potentially associated with PD,6-11 the potential involvement of GIGYF2 (also known as TNRC15) in PD was recently investigated.12 The identification of seven missense mutations in GIGYF2 in 12 of 249 unrelated patients with PD was reported. These mutations were not observed in 227 controls.
Owing to the small size of many of the pedigrees and the sampling of only some of the affected individuals, there were very limited data to suggest that these mutations segregate within these families. The goals of this study were to characterize sequence variation within GIGYF2 in a select subset of our large sample of patients with familial PD and to test in our entire sample whether any identified sequence variants increased the risk for PD.
METHODS
Subjects.
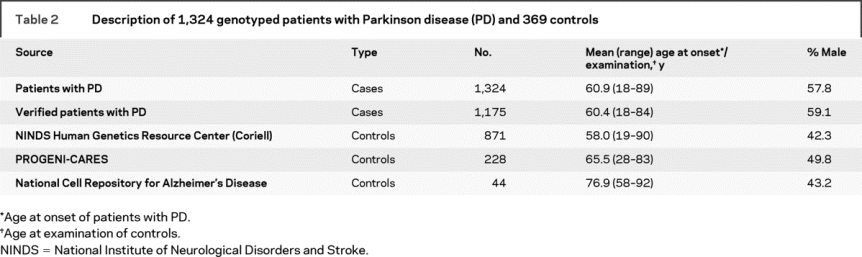
As part of an ongoing study designed to identify genes contributing to PD susceptibility (PROGENI Study), subjects with PD were recruited through the Parkinson Study Group (PSG), a network of 65 participating clinical centers located throughout North America. The inclusion criterion was a sibling pair, both of whom were reported to have a diagnosis of PD or were showing signs of PD. Subjects were seen in person by a movement disorder specialist who completed the Unified Parkinson’s Disease Rating Scale (UPDRS) Parts II and III,13,14 the Mini-Mental State Examination (MMSE),15 the Geriatric Depression Scale (GDS),16 and the Blessed Functional Activity Scale.17 In addition, a Diagnostic Checklist was completed, which consists of inclusion criteria associated with autopsy confirmed PD as well as exclusion criteria corresponding to features associated with other non-PD pathologic diagnoses.18 Responses on the Diagnostic Checklist were used to classify each subject with PD as either verified PD (VPD, n = 871), with all findings consistent with PD, or nonverified PD (NVPD, n = 453), with the subject failing to meet at least one inclusion criterion or meeting one exclusion criterion. Peripheral blood was obtained after completion of appropriate written informed consent approved by each individual institution’s institutional review board.
The control sample consisted of 1,447 neurologically normal non-Hispanic Caucasians who provided appropriate written informed consent. The control samples were obtained from three different sources: the National Cell Repository for Alzheimer’s Disease (NCRAD), the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center at the Coriell Cell Repositories (Camden, NJ; DNA), and controls recruited as part of an ongoing PD study at Indiana University (PROGENI-CARES).19
Molecular methods.
PCR and sequencing primers were designed using the chromosome 2 genomic contig sequence NC_000002.10 enabling PCR/sequencing of all 27 coding exons and intron/exon boundaries of GIGYF2 (table e-1 on the Neurology® Web site at www.neurology.org). PCR products were purified and sequenced as previously described.19
TaqMan allelic-discrimination assays (Applied Biosystems, Foster City, CA) were developed to screen for the novel missense variants identified in the 96 sequenced samples as well as the seven point mutations (N56S, T112A, I278V, S335T, N457T, D606E, V1242I) previously reported.12 These 10 assays were used to genotype our complete sample of 566 PD families (with 1,497 family members, 1,324 reported to have PD), as previously described,19,20 as well as the 1447 neurologically normal non-Hispanic Caucasian control subjects (tables 1 and 2).
Table 1 Description of 96 sequenced patients with Parkinson disease (PD)

Table 2 Description of 1,324 genotyped patients with Parkinson disease (PD) and 369 controls
PCR primers were designed flanking the polyglutamine repeat region in exon 25. The forward primer 5′:-GGAGTTTGCCAAGCAGTCC-3′ and the reverse primer 5′:-TACCGCATACACCACACTAC-3′ were used to amplify DNA from ∼200 PD and ∼100 control subjects. The PCR products were analyzed by electrophoresis through 4% composite agarose and visualized by ethidium bromide staining. Based on the results of the gel electrophoresis, exon 25 PCR products corresponding to at least six different patterns were cloned using the TOPO Cloning Kit for Sequencing (Invitrogen, Carlsbad, CA). DNA sequence analysis was performed on miniprep DNA from 15 different clones from each of the cloned patterns. To determine the frequency of each of these different alleles in all 1,497 family members (1,324 PD subjects) and 1,447 controls, the forward primer used to PCR amplify the polyglutamine region was labeled with 6-FAM and fluorescent genotyping was performed on a 3730xl DNA Analyzer (Applied Biosystems) and analyzed using GeneMapper 4.0 (Applied Biosystems). Allele counts were permuted using only one individual per family, and χ2 and odds ratios were calculated separately for each allele using these counts. Genetic association analyses were only performed using non-Hispanic Caucasian samples (cases and controls).
RESULTS
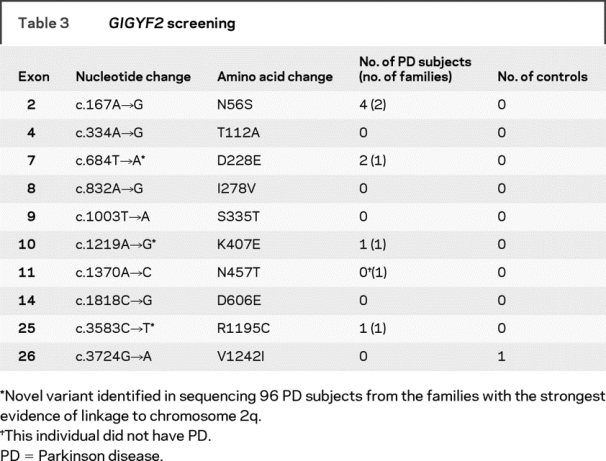
To investigate the frequency of GIGYF2 variants in familial PD, all 27 exons of GIGYF2 were sequenced in one PD case from each of 96 different multiplex PD families (table 1). These PD cases were specifically selected because their families provide evidence of linkage to chromosome 2q.3,4 Sequencing in the 96 index PD cases identified one subject heterozygous for the previously reported N56S variant in exon 2.12 We also identified one subject with each of the following novel variants: D228E in exon 7, K407E in exon 10, and R1195C in exon 25 (table 3). In addition, seven substitutions previously reported as polymorphisms12 were identified in 78 of the 96 index cases: P460T (rs2289912): 1 patient (reported incorrectly as P469T12); E518E (rs2305138): 12 patients; S945S: one patient; Q980Q (rs3816334): 65 patients; delQ1210 (rs10555297): 77 patients; P1217P (rs12328151): 41 patients; S1285S: 5 patients (table 3).
Table 3GIGYF2 screening
Our entire sample of 566 multiplex PD families, consisting of 1,497 members, including 1,324 reported to have PD, were genotyped for the seven GIGYF2 point mutations previously reported12 as well as the three novel missense variants we identified through sequencing. We identified several additional subjects carrying GIGYF2 variants (figure 1). Among the seven previously reported GIGYF2 variants, the N56S variant identified in one of the 96 sequenced patients with PD was identified in three additional subjects: one from the same family as the subject identified by sequencing and two in an additional family. In family A, both siblings with verified PD carried the N56S variant. In family B, four siblings were initially reported to have symptoms of PD. Following evaluation, three of the siblings met criteria for verified PD, while the fourth sibling did not complete a study visit, but did provide a blood sample. Among these four siblings, two carried the N56S variant while two did not. The mother of these four siblings provided a blood sample but was not evaluated in person. She did not report any symptoms of PD before death at age 90 and did not carry the N56S variant. The father of these siblings died at the age of 39 and was not reported to have any symptoms of PD. In this larger family, the N56S does not segregate completely with disease. Combining our results with those of the previous 249 subjects with PD studied,12 the N56S variant has been identified in 0.9% (3 of 345) of unrelated PD subjects and 0.4% (6 of 1573) of all PD subjects studied.

Figure 1 Segregation of GIGYF2 variants in pedigrees
The GIGYF2 variant identified in each family is indicated above the pedigree. To maintain the anonymity of the pedigree, the gender of all subjects is denoted as female. PD = Parkinson disease.
One individual was shown to be heterozygous for the previously reported N457T variant (family C). This individual was evaluated at age 81 and found to have no evidence of PD. This individual has two siblings with verified PD, neither of whom carries the variant. In addition, the individual carrying the variant also had a daughter with PD; however, she did not carry the variant. Thus, there is no evidence that the N457T variant is segregating with disease in this family.
We did not identify any members of our PD families who carried the T112A, I278V, S335T, D606E, or V1242I variants previously identified.12 While positive controls were not available for these variants for use in the TaqMan allelic discrimination assays, there was 100% correlation between the sequencing results of the 96 subjects with PD and the TaqMan assay results for these same subjects for these five variants.
We also genotyped our full sample for the three novel variants identified in the 96 sequenced patients with PD (D228E, K407E, and R1195C). The D228E variant was found in one additional patient with PD, the sibling of the individual in whom the variant was first identified (family D). The affected sibling of the individual identified by sequence analysis carrying the novel K407E variant does not carry this variant and thus this variant does not segregate with disease (family E). Similarly, the affected sibling of the individual carrying the novel R1195C variant does not carry this variant so this variant does not seem to be segregating with PD either (family F). While previous molecular screening has identified causative mutations in PRKN and LRRK2 in 128 patients with PD in our patient cohort, none of the patients with GIGYF2 variants in this report were shown to carry either a PRKN or LRRK2 mutation.19,21,22
The TaqMan allelic-discrimination assays for the 10 variants previously reported12 or identified in the sequencing of patients with PD as part of this report were also genotyped in 1,447 neurologically normal non-Hispanic Caucasian controls.19 Only one of the 10 variants genotyped (V1242I) was identified in one control sample (table 3).
Previously, deletions and insertions in exon 25 of GIGYF2 were reported.12 To determine how many different alleles might be represented in our study samples, we used a combination of agarose gel electrophoresis, cloning and sequencing of individual alleles, and fluorescent genotyping. In our analysis of 1,497 family members from 566 PD families and 1,447 neurologically normal controls, a total of eight different alleles was observed, with six of these corresponding to the six different patterns initially observed by gel electrophoresis (table 4). These six alleles ranged from a deletion of eight amino acids (Del LPQQQQQQ 1209-1216) to an insertion of two amino acids (Ins QQ 1217). Each of these alleles was also previously reported.12 Exon 25 PCR products from two individuals each heterozygous for one of the two new alleles identified by the fluorescent genotyping (142 and 148 bp) were subcloned and sequenced to determine the corresponding change at the DNA/amino acid level. Sequence analysis identified one novel allele (Del QQQQLP 1205-1210) and one allele previously reported (Del PPQQ 1221-1224).12 Figure 2 shows the DNA sequence of the eight alleles detected in our study. The Del QQQQLP 1205-1210 (142 bp) allele was identified in two siblings from a single family but not in any controls while the Del PPQQ 1221-1224 (148 bp) allele was detected in three controls but in none of the PD families. Each of the other six alleles was identified at similar frequencies in both patients with PD and controls and there was no evidence that any of these GIGYF2 insertion/deletions increased the risk for PD (table 4).
Table 4GIGYF2 exon 25 screening
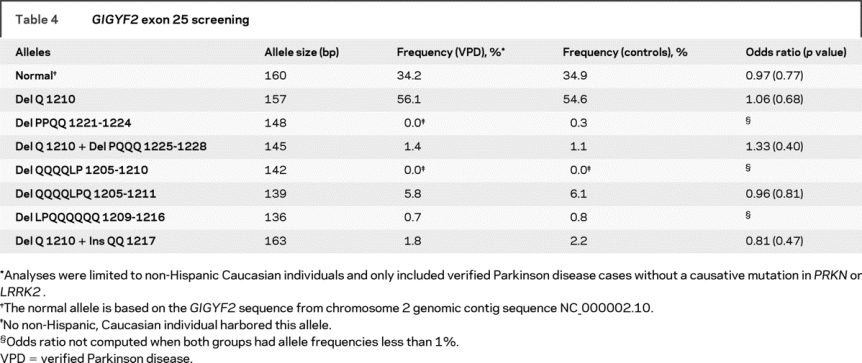

Figure 2 Schematic of GIGYF2 gene (A) and amino acid/DNA sequence of GIGYF2 polymorphic exon 25 alleles (B)
(A) Schematic of GIGYF2 gene showing location of identified variants. The GIGYF2 gene structure is depicted approximately to scale with the exons numbered above the gene. Below the gene are shown variants identified in patients with PD. Those in boxes represent the three novel variants of this report while the remaining seven were previously published. (B) Amino acid/DNA sequence of GIGYF2 polymorphic exon 25 alleles. At top is shown the amino acid sequence corresponding to the 3′ portion of exon 25 encoding residues 1200 to 1228 (numbers above amino acid sequence). Shown directly below the amino acid sequence are the eight different alleles identified in our study sample. Numbers at left designate allele sizes (bp) by fluorescent genotyping. The 160 bp allele is the normal reference allele based on the GenBank sequence. All other alleles are shown relative to the normal 160 bp allele. Missing residues are depicted as underlined gaps in the sequence. The QQ insertion at residue 1217 in allele 163 is indicated by the underlined CAG codons occurring between codons 1216 and 1217 in the normal sequence.
DISCUSSION
The goal of this study was to test whether variants in GIGYF2 could account for the previous evidence of linkage to chromosome 2q36-37 reported in our collection of multiplex PD families.4,23 Our previous study found that a subset of our families, in particular those with the strongest family history of disease, provided the greatest evidence of linkage to this region. Direct sequence analysis of the GIGYF2 coding region in one subject with PD from each of 96 unrelated PD families identified four variants (4.2%) not identified in controls (figure 2). This is similar to the 4.8% frequency previously reported.12 However, our screening of 10 variants (7 previously reported and 3 novel from this study) in 566 families yielded only 6 families with known variants (1.1%). We did not find a significant increase in risk for PD among those inheriting variants in GIGYF2 as compared to controls (p = 0.28). With the identification of so few potential mutations in GIGYF2 in these families, it is very unlikely that these few variants, observed in only six families (and segregating with disease in only two families) (see figure 1), could have accounted for the substantial linkage evidence (lod = 5.1) reported in our sample.4 Therefore, we do not believe that variation in GIGYF2 accounts for the previously reported linkage finding on chromosome 2q36-37.
Of the three novel variants identified in this study (table 3), two of them (K407E in exon 10 and R1195C in exon 25) represent nonconservative amino acid substitutions which could potentially alter either structure or function of GIGYF2. The third novel variant (D228E in exon 7) would not be predicted to interfere in protein function. Alignment of the human GIGYF2 protein sequence with 17 other species indicate that the three variants occur within conserved amino acid blocks and involve residues that are highly conserved across species.
While we did not detect sequence variants that appeared consistent with a causative effect on PD, we also explored the possibility that variation in exon 25 in GIGYF2 might increase the susceptibility or risk of PD. The analysis of exon 25 represented a challenge due to the large number of glutamine residues encoded in this exon. Nineteen of the final 27 codons in exon 25 encode glutamine primarily using the CAG codon (18/19) (figure 2). The repeated CAGs result in several different alleles that vary due to insertions/deletions in this region and were also observed in a previous report.12 Many of the 96 samples sequenced were heterozygous for these insertional/deletional events, preventing an accurate determination of the sequence using conventional direct sequence analysis of PCR products as was used for the other exons. Several different sequence patterns were noted on the chromatograms. However, following careful delineation of all insertions and deletions in exon 25 using a combination of cloning/sequencing of the individual alleles and fluorescent genotyping, we did not detect evidence that any of the eight observed alleles were found at higher frequency in subjects with PD as compared with controls (table 4). Our analysis of GIGYF2 was limited to coding sequence alterations and would not identify dosage changes as have been identified in the genes for parkin and α-synuclein in some subjects with PD.2
Despite careful examination of GIGYF2 to identify all sequence variation, we did not detect significant or even suggestive evidence that variation in GIGYF2 can cause or increase the risk of PD even when using a cohort of samples providing evidence of linkage to chromosome 2q36-37. Therefore, we hypothesize that there is another gene within this chromosome 2q region that when mutated results in familial PD. Studies are ongoing to identify this gene or genes. Given the increased interest in genetic testing for PD, it is imperative that the pathogenicity of any newly identified genetic variant be determined before it is included in any panel for diagnostic testing. These data should be available to the clinician to enable proper genetic counseling, especially to those undergoing presymptomatic testing. We urge caution in the implementation of GIGYF2 genetic testing to ensure minimization of the risks of misinterpretation.
AUTHOR CONTRIBUTIONS
Statistical analyses were conducted by Drs. Pankratz and Foroud.
ACKNOWLEDGMENT
The authors thank the subjects for their participation.
Supplementary Material
APPENDIX
Parkinson Study Group–PROGENI Investigators: PROGENI Steering Committee: University of Tennessee Health Science Center: R.F. Pfeiffer; University of Rochester: F. Marshall, D. Oakes, A. Rudolph, A. Shinaman; Columbia University Medical Center: K. Marder; Indiana University School of Medicine: P.M. Conneally, T. Foroud, C. Halter; University of Kansas Medical Center: K. Lyons; Eli Lilly & Company: E. Siemers; Medical College of Ohio: L. Elmers; University of California, Irvine: N. Hermanowicz. PSG-PROGENI Investigators and Coordinators: Albany Medical College: S. Factor, D. Higgins, S. Evans; Barrow Neurological Institute: H. Shill, M. Stacy, J. Danielson, L. Marlor, K. Williamson; Baylor College of Medicine: J. Jankovic, C. Hunter; Beth Israel Deaconess Medical Center: D. Simon, P. Ryan, L. Scollins; Beth Israel Medical Center: R. Saunders-Pullman, K. Boyar, C. Costan-Toth, E. Ohmann; Brigham & Women’s Hospital: L. Sudarsky, C. Joubert; Brown University (Memorial Hospital of RI): J. Friedman, K. Chou, H. Fernandez, M. Lannon; Cleveland Clinic Florida-Weston: N. Galvez-Jimenez, A. Podichetty, K. Thompson; Clinical Neuroscience Center: P. Lewitt, M. DeAngelis; Colorado Neurological Institute: C. O’Brien, L. Seeberger, C. Dingmann, D. Judd; Columbia University Medical Center: K. Marder, J. Fraser, J. Harris; Creighton University: J. Bertoni, C. Peterson; Evanston Northwestern Healthcare: M. Rezak, G. Medalle; Hotel-Dieu Hospital-Chum: S. Chouinard, M. Panisset, J. Hall, H. Poiffaut; Hunter Homes McGuire Veterans Medical Center: V. Calabrese, P. Roberge; Indiana University School of Medicine: J. Wojcieszek, J. Belden; Institute For Neurodegenerative Disorders: D. Jennings, K. Marek, S. Mendick; Johns Hopkins University: S. Reich, B. Dunlop; London Health Sciences Centre: M. Jog, C. Horn; Mayo Clinic Jacksonville: R. Uitti, M. Turk; McFarland Neurosciences: T. Ajax, J. Mannetter; Medical College of Georgia: K. Sethi, J. Carpenter, B. Dill, L. Hatch, K. Ligon, S. Narayan; Medical College of Wisconsin: K. Blindauer, K. Abou-Samra, J. Petit; Medical University of Ohio: L. Elmer, E. Aiken, K. Davis, C. Schell, S. Wilson; Mount Sinai School of Medicine: M. Velickovic, W. Koller (deceased), S. Phipps; North Shore-LIJ Health System: A. Feigin, M. Gordon, J. Hamann, E. Licari, M. Marotta-Kollarus, B. Shannon, R. Winnick; Northwestern University: T. Simuni, A. Videnovic, A. Kaczmarek, K. Williams, M. Wolff; Ochsner Clinic Foundation: J. Rao, M. Cook; Ohio State University: M. Fernandez, S. Kostyk, J. Hubble, A. Campbell, C. Reider, A. Seward; Oregon Health & Science University: R. Camicioli, J. Carter, J. Nutt, P. Andrews, S. Morehouse, C. Stone; Ottawa Hospital Civic Site: T. Mendis, D. Grimes, C. Alcorn-Costa, P. Gray, K. Haas, J. Vendette; Pacific Neuroscience Medical Group: J. Sutton, B. Hutchinson, J. Young; Saskatoon Dist Health Board Royal University Hospital: A. Rajput, A. Rajput, L. Klassen, T. Shirley; Scott & White Hospital/Texas A&M University: B. Manyam, P. Simpson, J. Whetteckey, B. Wulbrecht; The Parkinson’s & Movement Disorder Institute: D. Truong, M. Pathak, K. Frei, N. Luong, T. Tra, A. Tran, J. Vo; Toronto Western Hospital, University Health: A. Lang, G. Kleiner-Fisman, A. Nieves, L. Johnston, J. So; UMDNJ-School of Osteopathic Medicine: G. Podskalny, L. Giffin; University of Alabama at Birmingham: P. Atchison, C. Allen; University of Alberta: W. Martin, M. Wieler; University of Calgary: O. Suchowersky, M. Klimek; University of California Irvine: N. Hermanowicz, S. Niswonger; University of California San Diego: C. Shults (deceased), D. Fontaine; University of California San Francisco: M. Aminoff, C. Christine, M. Diminno, J. Hevezi; University of Chicago: A. Dalvi, U. Kang, J. Richman, S. Uy, J. Young; University of Cincinnati: A. Dalvi, A. Sahay, M. Gartner, D. Schwieterman; University of Colorado Health Sciences Center: D. Hall, M. Leehey, S. Culver, T. Derian; University of Connecticut: T. Demarcaida, S. Thurlow; University of Iowa: R. Rodnitzky, J. Dobson; University of Kansas Medical Center: K. Lyons, R. Pahwa, T. Gales, S. Thomas; University of Maryland School of Medicine: L. Shulman, S. Reich, W. Weiner, K. Dustin; University of Miami: K. Lyons, C. Singer, W. Koller (deceased), W. Weiner, L. Zelaya; University of Minnesota: P. Tuite, V. Hagen, S. Rolandelli, R. Schacherer, J. Kosowicz; University of New Mexico: P. Gordon, J. Werner; University of Puerto Rico School of Medicine: C. Serrano, S. Roque; University of Rochester: R. Kurlan, D. Berry, I. Gardiner; University of South Florida: R. Hauser, J. Sanchez-Ramos, T. Zesiewicz, H. Delgado, K. Price, P. Rodriguez, S. Wolfrath; University of Tennessee Health Science Center: R. Pfeiffer, L. Davis, B. Pfeiffer; University of Texas Southwestern Medical Center: R. Dewey, B. Hayward, A. Johnson, M. Meacham, B. Estes; Wake Forest University School of Medicine: F. Walker, V. Hunt, C. O’Neill; Washington University: B. Racette, L. Good, M. Rundle. Biostatistics and Clinical Trials Coordination Centers Staff: A. Watts, A. Wang, T. Ross, S. Bennett, D. Kamp, E. Julian-Baros, S. Daigneault, R. Doolan.
Address correspondence and reprint requests to Dr. William C. Nichols, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229 bill.nichols@cchmc.org
Supplemental data at www.neurology.org
Editorial, page 1882
e-Pub ahead of print on March 11, 2009, at www.neurology.org.
*The Parkinson Study Group–PROGENI Investigators are listed in the appendix.
Supported by R01 NS37167, MO1 RR-00750. Control samples and clinical data were provided by the National Cell Repository for Alzheimer’s Disease (U24 AG021886) and the National Institute of Neurological Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds).
Disclosure: The authors report no disclosures.
Medical Devices: 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA).
Received July 8, 2008. Accepted in final form December 30, 2008.
REFERENCES
- 1.de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts: Neurologic Diseases in the Elderly Research Group. Neurology 2000;54:S21–S23. [PubMed] [Google Scholar]
- 2.Pankratz N, Foroud T. Genetics of Parkinson disease. Genet Med 2007;9:801–811. [DOI] [PubMed] [Google Scholar]
- 3.Pankratz N, Nichols WC, Uniacke SK, et al. Genome-wide linkage analysis and evidence of gene-by-gene interactions in a sample of 362 multiplex Parkinson disease families. Hum Mol Genet 2003;12:2599–2608. [DOI] [PubMed] [Google Scholar]
- 4.Pankratz N, Nichols WC, Uniacke SK, et al. Significant linkage of Parkinson disease to chromosome 2q36-37. Am J Hum Genet 2003;72:1053–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannone B, Lee E, Laviola L, Giorgino F, Cleveland KA, Smith RJ. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J Biol Chem 2003;278:31564–31573. [DOI] [PubMed] [Google Scholar]
- 6.Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr Rev 2005;26:916–943. [DOI] [PubMed] [Google Scholar]
- 7.Schulingkamp RJ, Pagano TC, Hung D, Raffa RB. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev 2000;24:855–872. [DOI] [PubMed] [Google Scholar]
- 8.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 2004;3:169–178. [DOI] [PubMed] [Google Scholar]
- 9.Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007;30:842–847. [DOI] [PubMed] [Google Scholar]
- 10.Offen D, Shtaif B, Hadad D, Weizman A, Melamed E, Gil-Ad I. Protective effect of insulin-like-growth-factor-1 against dopamine-induced neurotoxicity in human and rodent neuronal cultures: possible implications for Parkinson’s disease. Neurosci Lett 2001;316:129–132. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi M, Yamada T, Tooyama I, et al. Insulin receptor mRNA in the substantia nigra in Parkinson’s disease. Neurosci Lett 1996;204:201–204. [DOI] [PubMed] [Google Scholar]
- 12.Lautier C, Goldwurm S, Durr A, et al. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet 2008;82:822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahn S, Elton R, Committee UD. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden C, Goldstein M, eds. Recent Developments in Parkinson’s Disease. Florham Park, NY: Macmillan Healthcare Information; 1987:153–163. [Google Scholar]
- 14.Lang AE, Fahn S. Assessment of Parkinson’s disease. In: Munsat T, ed. Quantification of Neurologic Deficit. Boston: Butterworths; 1989:285–309. [Google Scholar]
- 15.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 16.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 1982;17:37–49. [DOI] [PubMed] [Google Scholar]
- 17.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 1968;114:797–811. [DOI] [PubMed] [Google Scholar]
- 18.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nichols WC, Elsaesser VE, Pankratz N, et al. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology 2007;69:1737–1744. [DOI] [PubMed] [Google Scholar]
- 20.Nichols WC, Pankratz N, Kissell DK, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age of onset. Neurology 2009;72:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foroud T, Uniacke SK, Liu L, et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003;60:796–801. [DOI] [PubMed] [Google Scholar]
- 22.Nichols WC, Pankratz N, Uniacke SK, et al. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson’s disease families. J Med Genet 2002;39:489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pankratz N, Nichols WC, Uniacke SK, et al. Genome screen to identify susceptibility genes for Parkinson disease in a sample without parkin mutations. Am J Hum Genet 2002;71:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.