

Abstract
We describe a novel array for accurate and reliable genotyping of human papillomavirus (HPV) using peptide nucleic acid (PNA) probes. In order to exploit the superior hybridization properties of PNA with target HPV DNAs, we developed a novel PNA array (PANArray HPV). PANArray HPV enables the detection and genotyping of HPVs using 32 type-specific PNA capture probes for medically important HPVs. All tested HPV types showed highly unique hybridization patterns with type-specific PNA probes. PNA array results showed stable specificities and sensitivities after up to 13 months of storage at room temperature. Also, we demonstrated the superior specificity, sensitivity, and stability of PNA arrays for HPV genotyping. We compared the genotyping results of the PNA array to sequencing with MY09/11 PCR products derived from 72 clinical samples. The results showed excellent agreement between the PNA array and sequencing, except for samples reflecting multiple infections. The results from the PNA array were compared with those of type-specific PCR when discrepant results occurred owing to multiple infections. The results for the PNA array matched those of type-specific PCR in all cases. Newly developed PNA arrays show excellent specificity and sensitivity and long shelf life. Our results suggest that the PNA array represents a reliable alternative to conventional DNA arrays for HPV genotyping, as well as for diagnostics.
Human papillomaviruses (HPVs) comprise more than 100 genotypes, of which over 30 types are associated with sexually transmitted infections. The mucosal types can be divided into high-risk and low-risk types depending on the associated disease risk. HPV infection is a necessary prerequisite for cervical cancer development. Accurate HPV genotyping is increasingly important for investigating the clinical outcomes and epidemiologies of particular types, for the characterization of study populations in HPV vaccination trials, and for monitoring the efficacies of HPV vaccines (17, 20).
Members of the HPV family do not lend themselves to culture in vitro. Thus, detection of HPV is restricted to molecular analyses of HPV DNA sequences (10). Technical improvements in PCR, sequencing, and hybridization assays (ThirdWave Technology hybrid capture, linear array, and oligonucleotide arrays) have increased the sensitivity, specificity, and speed of assays. Although this technology has many advantages, and several working systems have been established, their sensitivity, reproducibility, and reliability are still major issues (6, 8, 11, 21, 26). Most platforms that have been described make use of natural DNA or RNA as capture probes. However, several nucleic acid analogs have demonstrated more favorable hybridization characteristics than standard DNA-based probes. Nucleic acid analogs have been made in order to overcome the limitations of natural nucleic acids for specificity, sensitivity, hybridization kinetics, thermodynamic properties, and stability (2, 4, 12).
Peptide nucleic acids (PNAs) are notable for their exceptional biological and chemical stabilities. In PNA, a negatively charged ribose-phosphate nucleic acid backbone is replaced by an uncharged N-(2-aminoethyl)-glycine scaffold, to which the nucleotide bases are attached via a methylene carbonyl linker. Because all intramolecular distances and configurations of the nucleobases are similar to those of natural DNA molecules, specific hybridization can occur between PNA and DNA. The particular biochemical properties of PNA make it suitable for use in many biological applications. An increasing number of applications for PNA technology have been described, confirming the high potential of PNAs as probes for antisense, PCR clamping, fluorescence in situ hybridization, and microarrays (2, 12, 18). The purpose of this study was to develop a PNA array for the genotyping of 32 medically important HPVs and to evaluate the PANArray HPV (for research use only, not for use in diagnostic procedures) using clinical samples.
MATERIALS AND METHODS
PNA probe synthesis.
PNA probes were synthesized by Panagene's technology using benzothiazole-2-sulfonyl as an amine-protecting group (15). High-performance liquid chromatography (Agilent 1100; Agilent Technologies, Wilmington, DE) was used to purify the PNA probes. The PNA probe quality was assessed by matrix-assisted laser desorption ionization-time of flight (AX1MA-CFR; Shimadzu Co., Kyoto, Japan). The concentrations of PNA probes were determined with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). To select the optimal PNA probes, several candidate probes for each target were designed. PNA probes have a terminal amine group that is required for covalent attachment to an epoxy surface and have spacers that enable the target to approach immobilized probes.
Plasmid clones and sequence analysis.
For evaluation of the sensitivity and specificity of the PNA array, 32 plasmid clones of the following HPV types were used: 6, 11, 16, 18, 26, 31, 32, 33, 34, 35, 39, 40, 42, 43, 44, 45, 51, 52, 53, 54, 55, 56, 58, 59, 62, 66, 68, 69, 70, 73, 81, and 83. PCR-amplified fragments of the HPV L1 region (450 bp) were cloned in the vector pGEM-T (Promega, Madison, WI). Sequence analyses of PCR products used the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Inc., Foster City, CA).
Target DNA preparation. (i) DNA extraction.
Clinical samples were provided by the laboratory service center (Cell Genomics Inc.) for blind evaluation of PNA arrays. Seventy-two cervical scrapes were collected between January and March 2007 and were previously tested by sequencing. The cervical specimens were collected with cervical brush samplers and were transferred to sterile phosphate-buffered saline (PBS). The PBS-cell mixture was centrifuged at 13,000 × g for 30 min, the supernatant was decanted, and the cell pellet was resuspended in 200 μl of PBS.
QuickExtract kits were used to extract DNA according to the manufacturers' protocol (Epicentre, Madison, WI). DNAs were aliquoted and stored at −20°C until they were used.
(ii) PCR amplification.
A single-tube nested PCR was performed to genotype HPV using the PNA array. HPV was amplified by MY09/11 and GP5+/GP6+ primers targeting 450 bp and 130 bp of the HPV L1 open reading frame (9, 23). Only the GP6+ primer was biotinylated at the 5′ terminus to label the target strand of the PCR fragments. Briefly, a 25-μl reaction mixture contained 1 U of Taq DNA polymerase (Solgent, Daejeon, South Korea) and PCR buffer with 2.5 mM MgCl2, 200 μM deoxynucleoside triphosphate mixture, and 10 pM of each primer. Five microliters of target DNA was added to the PCR mixture, and to eliminate carryover contamination, 0.5 U of uracil-DNA glycosylase (Solgent, Daejeon, South Korea) was added. All tubes were incubated for 2 min at 50°C before PCR was started. Ten cycles of 30 s at 94°C, 30 s at 58°C, and 30 s at 72°C were performed, followed by 30 cycles of 30 s at 94°C, 1 min at 47°C, and 30 s at 72°C. PCR was performed with a PCR Thermal Cycler DNAEngine (Bio-Rad Laboratories, Hercules, CA). Type-specific PCR was performed with type-specific PCR primers for each genotype. Two microliters of MY09/11 PCR product was amplified with type-specific PCR primers. Type-specific primers were designed from the polymorphic regions of the L1 sequences of HPV.
PNA probe design.
The L1 region sequences of HPV were obtained from GenBank. Based on multiple-alignment analysis data using CLUSTAL W, 32 type-specific probes were designed from the polymorphic regions of the respective L1 sequences of HPV. The uniqueness of the probe sequences designed from the HPV L1 region was analyzed with a BLAST search. The probes were designed to meet the following parameters. The position of a potential mismatch between similar sequences was close to the center of the probe. The 5′end of each probe was modified by adding a PNA-optimized spacer and linker to enable covalent immobilization on an epoxy-coated glass surface.
Fabrication of the PNA array.
The PNA probes were dissolved in distilled water (DW). A spotting mixture was prepared by making 10 μl spotting buffer containing a 50 μM concentration of PNA probe. The spotting mixture was printed onto epoxy slides using a Qarray mini-Microarrayer equipped with aQu solid pins (Genetix, New Milton, United Kingdom). The spotting layout of PNA probes for genotyping HPV types is shown in Fig. 1. The printed slides were incubated for at least 2 h at room temperature in a humidified microarrayer (the humidity was set to about 70 to 75%). The printed slides were immersed in succinic anhydride and dimethyl formamide, and each slide was sonicated for 2 h at 40°C for blocking. The slides were washed twice with dimethyl formamide, ethanol, and DW. After being washed with DW, the slides were immersed in boiling water and incubated for 5 min. After being dried with compressed air, they were ready for hybridization.
FIG. 1.

Layout of the HPV PNA array. The PNA probes printed on the array are indicated by their names in the boxes. Their sequences are given in Table 1. Each PNA probe was spotted in duplicate. PM, position marker.
Hybridization and scanning.
The biotin-labeled PCR fragments were denatured for 5 min at 95°C and then chilled on ice for 2 min. Five microliters of biotin-labeled target DNA was mixed with 95 μl of the PNA hybridization buffer containing Cy5-streptavidin (Amersham Pharmacia Biotech UK Limited, Little Chalfont, Buckinghamshire, England) and then applied to the PNA array. The PNA array was incubated for 1 h at 40°C. After hybridization, the slides were washed twice for 5 min each time with PNA wash buffer. They were ready for scanning after drying. Array images were taken using a nonconfocal fluorescent scanner (GenePix 4000B; Axon Instruments, Union City, CA) with a typical laser power of 100% and a photomultiplier tube gain of 700. The fluorescence signal intensities represented the hybridization signals of the probe-target duplexes.
RESULTS
Development of a PNA array for HPV genotyping. (i) Optimum hybridization buffer conditions of the PNA array.
To optimize the hybridization conditions for PNA arrays, the effects of phosphate (0 to 200 mM) and salt (0 to 1000 mM) concentrations on the signal intensity and signal-to-background ratio (SBR) were examined. The results showed a slight decrease in signal intensity and SBR when a phosphate buffer concentration greater than 150 mM was used. The range of 50 to 200 mM phosphate concentrations provided the maximum signal intensity and SBR for the PNA array. The signal intensity remained nearly constant for salt concentrations from 50 to 200 mM and decreased at concentrations greater than 200 mM. As the salt concentration increased from 50 to 200 mM, the SBR generally increased (Fig. 2A and B). The optimal phosphate and salt concentrations appeared to be 50 mM and 200 mM, respectively. We also tested the effects of several concentrations of formamide on the signal intensity and SBR for the PNA array. The hybridization was carried out at 40°C for 1 h. The use of a 20% formamide concentration resulted in a high signal intensity and SBR. Concentrations lower or higher than 20% formamide tended to show low signal intensities and SBRs. These hybridization buffer conditions were used to genotype HPVs on PNA arrays.
FIG. 2.

Effects of phosphate and salt concentrations on signal intensity and SBR of hybridization of the PNA probes and DNA targets. This was followed by hybridization for 1 h with 50 μM PNA probes and PCR fragments. Shown are the effects of the phosphate concentration (1 to 200 mM) (A), salt concentration (1 to 1,000 mM) (B), and formamide concentration (0 to 40%) (C) on the signal intensities and SBRs of the PNA probes and DNA targets. The error bars indicate standard deviations for signal intensity.
(ii) Design and selection of HPV type-specific PNA probes.
There were polymorphic regions of L1 that could be used to design HPV type-specific probes. The small region of the L1 gene was used as a foundation to generate highly informative phylogenetic comparisons. The L1 gene is at least 10% dissimilar to that of any other HPV types. As with DNA probes, the PNA probes were also designed to maximize their hybridization efficiencies and to minimize any nontarget hybridizations. The type-specific PNA probes were designed to be between 15 and 20 nucleotides long in consideration of melting-temperature (Tm) values (Table 1). Each type of PNA probe designed was immobilized on a glass slide and hybridized with PCR fragments of extracted DNA from the 32 HPV clones. To select optimum type-specific PNA probes, a high hybridization signal intensity and SBR were taken into consideration.
TABLE 1.
Sequences of specific PNA probes for HPV genotyping
| No. | HPV type | Sequence (N to C) | GC% | Tm (°C)a |
|---|---|---|---|---|
| 1 | 6 | ATCCGTAACTACATCTTC | 38 | 68 |
| 2 | 11 | CTGTGTCTAAATCTGCTA | 38 | 69 |
| 3 | 16 | TGCCATATCTACTTCAG | 41 | 67 |
| 4 | 18 | CACAGTCTCCTGTACCT | 51 | 71 |
| 5 | 26 | CAGCATCTGCATCCACT | 52 | 74 |
| 6 | 31 | CTGCAATTGCAAACAGTG | 44 | 78 |
| 7 | 32 | CTACTGTAACAACTGAAG | 38 | 73 |
| 8 | 33 | ACACAAGTAACTAGTG | 37 | 71 |
| 9 | 34 | AGGTACACAATCCAC | 46 | 72 |
| 10 | 35 | GCTGTGTCTTCTAGTGA | 47 | 72 |
| 11 | 39 | TAGAGTCTTCCATACCT | 41 | 68 |
| 12 | 40 | CACACCAACCCAT | 53 | 67 |
| 13 | 42 | ACATCTGGTGATACATAT | 33 | 71 |
| 14 | 43 | TATGACAATGCAAAGTT | 29 | 72 |
| 15 | 44 | TCCGTCTACATATACTAGT | 36 | 69 |
| 16 | 45 | CTACACAAAATCCTGTG | 41 | 71 |
| 17 | 51 | CTGCCACTGCTGCG | 71 | 74 |
| 18 | 52 | TACCTTCGTCATGGC | 53 | 69 |
| 19 | 53 | CTACATATAATTCAAAGC | 27 | 67 |
| 20 | 54 | TACAGCATCCACGC | 57 | 71 |
| 21 | 55 | TACAACTCAGTCTCC | 46 | 65 |
| 22 | 56 | TGCTACAGAACAGTTA | 37 | 71 |
| 23 | 58 | TTATGCACTGGAAGTAA | 35 | 74 |
| 24 | 59 | ACTACTTCTTCTATTCCTAATG | 31 | 69 |
| 25 | 62 | TTTGCGACACACGGA | 53 | 78 |
| 26 | 66 | AGCTAAAAGCACATTA | 31 | 71 |
| 27 | 68 | TTGTCTACTACTACTGAA | 33 | 68 |
| 28 | 69 | CTGCCACTTTTAAACCAT | 38 | 70 |
| 29 | 70 | TAGCCCTACAAAGTTT | 37 | 69 |
| 30 | 73 | TAGCTCTACTACAAC | 40 | 63 |
| 31 | 81 | TTTGCACAGCTACATCT | 41 | 70 |
| 32 | 83 | CAGGCTAATGAATAC | 40 | 68 |
The Tm values of PNA probes were calculated using the ABI Web program.
(iii) Specific hybridization patterns of the PNA array.
We selected the optimal type-specific probes by hybridization with the cloned DNAs of 32 HPV types. The results of independent experiments for genotyping HPV types are shown in Fig. 3. The optimal type-specific probes for HPV genotyping were selected (data not shown). As shown in Fig. 3, all HPV-specific probes hybridized specifically to the corresponding targets for each genotype, and there were no cross-hybridizations with other HPVs. For example, HPV-16 was expected to hybridize with probe 16 (Fig. 3), and other HPVs did not hybridize with probe 16. Each type-specific probe was hybridized specifically with HPV. The type-specific probes could genotype HPV under uniform hybridization conditions.
FIG. 3.

Scanning images of the PNA array were obtained through hybridization of 32 HPV types. All HPV-specific probes were highly discriminating and did not show cross-hybridization with any other targets.
(iv) Detection limit of the PNA array.
The detection limit of the PNA array was determined by amplification and hybridization of serial dilutions of HPV DNA (101 to 107 clones/μl of the initial DNA concentration of HPV-11 and -58). The detection limits for the PNA array were as low as 10 copies/μl of the initial DNA concentrations of HPV-11 and -58. We show only the results for hybridizations with 101, 102, 103, 104, 105, 106, and 107 copies/μl of the initial DNA concentrations of HPV-11 and -58. These were several hundreds to 10,000 times the SBR value for 102 copies/μl of the initial DNA concentration of HPV. At 10 copies/μl of the initial DNA concentration, the SBR was too low to detect the target (Fig. 4).
FIG. 4.

Signal intensities and SBRs generated for HPV types 11 and 58. Serial dilutions were made with 107 to 101 copies. Each curve is based on an average of three to five replicates. The error bars indicate standard deviations for signal intensity.
(iv) Shelf life of the PNA array.
For reliable results in practical applications, arrays should maintain their SBR (specificity) and signal intensity (sensitivity) while stored. PNAs are completely resistant to nucleases and other degradation factors in biological systems. Hence, it was expected that the PNA array would be effective after long-term storage. To evaluate the shelf life of the arrays, stability tests were performed on the PNA and DNA arrays after storage. The arrays were stored in plastic slide boxes at room temperature. As shown in Fig. 5, the PNA array showed the same level of intensity as a freshly fabricated (0-month) PNA array after 3 months of storage, whereas that of the DNA arrays decreased rapidly after 3 months of storage. The PNA arrays still showed identical signal intensities and SBRs after 13 months at room temperature.
FIG. 5.

Stabilities of the PNA and DNA arrays when stored at room temperature. The hybridization signals of the PNA and DNA arrays after 0 (fresh fabrication) to 13 months of storage. Shown are the results of a single representative experiment among three independent hybridization experiments. The results were obtained through hybridization with PCR fragments of an HPV clone.
Clinical evaluation with PNA array assays.
We tested 72 clinical samples with the PNA array. We used two approaches to identify the clinical samples. The clinical specimens were analyzed blindly, and the results were compared with sequencing results that were done at a later date. The evaluation showed excellent agreement between the results obtained with the PNA array and sequencing, except for samples reflecting multiple infections. The results with the PNA array were compared with those for type-specific PCR when some discrepant results occurred owing to the multiple infections. The results with the PNA array matched those of type-specific PCR in all cases (Table 2). Multiple infections were detected by the array, but the sequencing assay could not detect multiple infections (Fig. 6). The PNA array allowed the genotyping of a number of HPVs in one reaction. This is advantageous for diagnostic purposes when genotyping HPV in clinical samples. Based on these results, we expect that the PANArray HPV can greatly facilitate HPV genotyping of clinical samples.
TABLE 2.
Comparison of results obtained by PNA array, conventional direct sequencing, and specific PCRa
| PNA array | Identification by:
|
|
|---|---|---|
| Sequencing | Specific PCR | |
| 6 (15) | 6 (15) | -c |
| 11 (5) | 11 (5) | - |
| 16 (6) | 16 (6) | - |
| 18 (5) | 18 (5) | - |
| 31 (1) | 31 (1) | - |
| 35 (1) | 35 (1) | - |
| 53 (5) | 53 (5) | - |
| 61 (0)b | 61 (1) | - |
| 62 (1) | 62 (1) | - |
| 66 (3) | 66 (3) | - |
| 70 (2) | 70 (2) | - |
| 81 (3) | 81 (3) | - |
| 83 (1) | 83 (1) | - |
| 84 (0)b | 84 (1) | - |
| 11,45d | 11 | 11,45 |
| 53,70d | 53 | 53,70 |
| 53,56d | 53 | 53,56 |
| 18,44d | 18 | 18,44 |
| 53,35d | 53 | 53,35 |
| 62,58d | 62 | 62,58 |
| 31,53d | 31 | 31,53 |
| 6,39d | 6 | 6,39 |
| 53,66,70d | 66 | 53,66,70 |
| 16,53 | 16,53 | - |
| 16,53,54d | 16,53 | 16,53,54 |
We used three approaches to identify the clinical samples. The numbers are HPV types. The numbers in parentheses are the numbers of the types detected. Total, 72; negative, 12; positive, 60.
Probes for HPV types 61 and 84 were not included in the PNA array.
Type-specific PCR was not performed in cases where PNA array results and sequencing results were in good agreement.
These 10 case of multiple infections were confirmed to have mixed infection on analysis by type-specific PCR and sequencing.
FIG. 6.
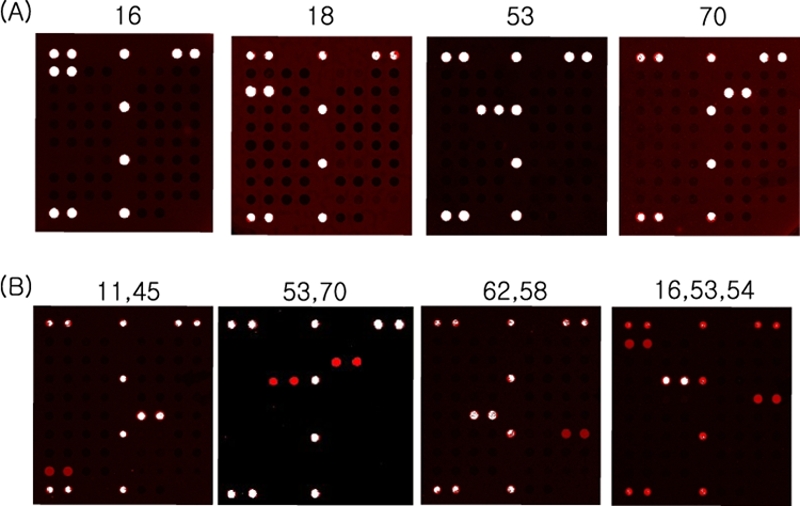
Representative results of the HPV PNA array. The PANArray HPV is available for detection of 32 HPV genotypes, all of which were amplified from HPV-PCR with clinical samples. (A) A single infection. (B) Double and triple infections.
DISCUSSION
The identification of HPV types prompted the development of effective methods for early cancer screening. In order to meet these demands, various technologies have been used and have accelerated HPV diagnosis (20). Oligonucleotide microarrays are highly parallel hybridization platforms that allow rapid, simultaneous identification of many different viruses in a single assay (16). The technology uses hybridization of PCR products to type-specific probes immobilized on different formats, such as membranes (27), liquid beads (20, 24), and glass slides (5, 13, 14).
Most microarray platforms have used DNA oligonucleotides as immobilized probes. However, many nucleic acid analogs have emerged during recent years that demonstrate more favorable hybridization characteristics than standard DNA-based probes. Many forms of promising nucleic acid analogs and mimics are available, such as PNAs, locked nucleic acids, and morpholinos. The synthetic DNA mimic exhibits superior hybridization characteristics and improved chemical and enzymatic stabilities relative to nucleic acids. Several studies have demonstrated that PNA oligonucleotides exhibit unprecedented thermal stability when hybridized with their DNA target molecules (2, 3, 22). Because PNAs increase Tm values by approximately 1.0°C per base pair for a PNA-DNA duplex and ΔTm values of PNA-DNA duplexes are 2.5 to 8.5°C higher than those of the corresponding DNA-DNA duplexes, PNA probes are expected to be more accurate. The following factors probably contribute to the higher sensitivity of these arrays: (i) improved detection of sequence variations by novel PNA probes, (ii) improved detection limits of PNA probes, and (iii) overall higher sensitivity of PNA arrays (3, 18). PNA-DNA hybrids offer greater stability than their DNA-DNA or DNA-RNA counterparts. These properties motivated us to apply PNA probes as capture probes for the microarray-based genotyping of HPV.
We have developed a novel PNA-based array for HPVs genotyping that takes advantage of the high sensitivity and specificity and long shelf life of these arrays. Optimum PNA array fabrication and performance must take into consideration the factors of probe design, spacer, solid substrate, probe immobilization, labeling, hybridization, and washing (1, 7, 25). Fabricating the PNA arrays also requires amine-modified probes and spacers for immobilization and efficient hybridization, as for DNA arrays. Some reviews of DNA mimics indicated that PNAs are difficult to handle in practical applications due to their poor solubility (19). To address this problem, PNA probes that were applied to arrays were first conjugated to a linker to improve solubility.
The uncharged backbones of PNA probes are independent of ionic strength. As expected, PNA-DNA duplex stabilities were found to be reduced depending on salt concentrations. As shown in Fig. 2, as the ionic strength changed, the signal intensity and perfect match signal/mismatch signal ratio of the PNA-DNA duplex were not significantly affected. These results suggest that concentrations of 50 mM phosphate and 200 mM salt are an adequate compromise between specificity (SBR) and signal intensity (sensitivity) in order for PNA arrays to discriminate sequence mismatches. The synthetic backbone of PNAs also means that they are not degraded by nucleases or proteases. Because of this resistance to enzyme degradation, the lifetime of PNAs is extended both in vivo and in vitro (18). The PNA arrays were found to have a shelf life longer than 13 months when stored in slide boxes at room temperature (Fig. 6). Because the shelf life of an array depends on the storage conditions, such as humidity, temperature, and wrapping methods, the shelf life of the arrays was experimentally investigated using the same storage conditions. PNA arrays have a significantly longer shelf life than DNA arrays. The long shelf life of these arrays is also important for diagnostic applications.
Our studies have established a PNA array platform technology for the genotyping of 32 HPV types. The turnaround time of PNA arrays (DNA extraction, PCR, hybridization, and scanning) was 4.5 h. The accuracy and reliability of the PNA array in HPV genotyping have been demonstrated, especially with regard to sensitivity, specificity, and stability. We believe that these results, especially the long shelf life of PNA arrays at room temperature, can enable many practical applications of HPV genotyping assays. Major improvements for the sensitivity, accuracy, and shelf life of the PNA chip were achieved by using PNA probes. This study is the first application of a PNA array in HPV genotyping, as well as for diagnostics. We demonstrated that PNA arrays are promising assays for clinical applications.
Footnotes
Published ahead of print on 15 April 2009.
REFERENCES
- 1.Auburn, R. P., D. P. Kreil, L. A. Meadows, B. Fischer, S. S. Matilla, and S. Russell. 2005. Robotic spotting of cDNA and oligonucleotide microarrays. Trends Biotechnol. 23374-379. [DOI] [PubMed] [Google Scholar]
- 2.Beuvink, I., F. A. Kolb, W. Budach, A. Garnier, J. Lange, F. Natt, U. Dengler, J. Hall, W. Filipowicz, and J. Weiler. 2007. A novel microarray approach reveals new tissue-specific signatures of known and predicted mammalian microRNAs. Nucleic Acids Res. 7e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt, O., and J. D. Hoheisel. 2004. Peptide nucleic acids on microarrays and other biosensors. Trends Biotechnol. 22617-622. [DOI] [PubMed] [Google Scholar]
- 4.Castoldi, M., S. Schmidt, V. Benes, M. Noerholm, A. E. Kulozik, M. W. Hentze, and M. U. Muckenthaler. 2006. A sensitive array for microRNA expression profiling (miChip) based on locked nucleic acids (LNA). RNA 12913-920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho, N. H., H. J. An, J. K. Jeong, S. Kang, J. W. Kim, Y. T. Kim, and T. K. Park. 2003. Genotyping of 22 human papillomavirus types by DNA chip in Korean women: comparison with cytologic diagnosis. Am. J. Obstet. Gynecol. 18856-62. [DOI] [PubMed] [Google Scholar]
- 6.Draghici, S., P. Khatri, A. C. Eklund, and Z. Szallasi. 2006. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 22101-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman, W. M., D. J. Robertson, and K. E. Vrana. 2003. Fundamentals of DNA hybridization arrays for gene expression analysis. BioTechniques 291042-1055. [DOI] [PubMed] [Google Scholar]
- 8.Gharizadeh, B., B. Zheng, M. Akhras, M. Ghaderi, O. Jejelowo, B. Strander, P. Nyrén, K. L. Wallin, and N. Pourmand. 2006. Sentinel-based DNA genotyping using multiple sequencing primers for high-risk human papillomaviruses. Mol. Cell. Probes 20230-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haws, A. L. F., Q. Hea, P. L. Rady, L. Zhang, J. Grady, T. K. Hughes, K. Stisser, R. Konig, and S. K. Tyring. 2004. Nested PCR with the PGMY09/11 and GP5+/6+ primer sets improves detection of HPV DNA in cervical samples. J. Virol. Methods 12287-93. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard, R. A. 2003. Human papillomavirus testing methods. Arch. Pathol. Lab. Med. 127940-945. [DOI] [PubMed] [Google Scholar]
- 11.Jeney, C., T. Takács, A. Sebe, and Z. Schaff. 2007. Detection and typing of 46 genital human papillomaviruses by the L1F/L1R primer system based multiplex PCR and hybridization. J. Virol. Methods 14032-42. [DOI] [PubMed] [Google Scholar]
- 12.Karkare, S., and D. Bhatnagar. 2006. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 71575-586. [DOI] [PubMed] [Google Scholar]
- 13.Kim, C. J., J. K. Jeong, M. Park, T. S. Park, T. C. Park, S. E. Namkoong, and J. S. Park. 2003. HPV oligonucleotide microarray-based detection of HPV genotypes in cervical neoplastic lesions. Gynecol. Oncol. 89210-217. [DOI] [PubMed] [Google Scholar]
- 14.Klaassen, C. H. W., C. F. M. Prinsen, H. A. de Valk, A. M. Horrevorts, M. A. F. Jeunink, and F. B. J. M. Thunnissen. 2004. DNA microarray format for detection and subtyping of human papilomavirus. J. Clin. Microbiol. 422152-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee, H., J. H. Jeon, J. C. Lim, H. Choi, Y. Yoon, and S. K. Kim. 2007. Peptide nucleic acid synthesis by novel amide formation. Org. Lett. 93291-3293. [DOI] [PubMed] [Google Scholar]
- 16.Loy, A., and L. Bodrossy. 2006. Highly parallel microbial diagnostics using oligonucleotide microarrays. Clin. Chim. Acta 363106-119. [DOI] [PubMed] [Google Scholar]
- 17.Molijn, A., B. Kleter, W. Quint, and L. J. van Doorn. 2005. Molecular diagnosis of human papillomavirus (HPV) infections. J. Clin. Virol. 32SS43-S51. [DOI] [PubMed] [Google Scholar]
- 18.Pellestor, F., and P. Paulasova. 2004. The peptide nucleic acids, efficient tools for molecular diagnosis. Int. J. Mol. Med. 13521-525. [PubMed] [Google Scholar]
- 19.Qu, W., G. Jiang, Y. Cruz, C. J. Chang, G. Y. F. Ho, R. S. Klein, and R. D. Burk. 1997. PCR detection of human papillomavirus: comparison between MY09/MY11 and GP51/GP61 primer systems. J. Clin. Microbiol. 351304-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmitt, M., I. G. Bravo, P. J. F. Snijders, L. Gissmann, M. Pawlita, and T. Waterboer. 2006. Bead-based multiplex genotyping of human papillomaviruses. J. Clin. Microbiol. 44504-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song, J. Y., H. G. Park, S. O. Jung, and J. C. Park. 2005. Diagnosis of HNF-1a mutations on a PNA zip-code microarray by single base extension. Nucleic Acids Res. 33e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenvang, J., A. N. Silahtaroglu, M. Lindowb, J. Elmenb, and S. Kauppinen. 2008. The utility of LNA in microRNA-based cancer diagnostics and therapeutics. Semin. Cancer Biol. 1889-102. [DOI] [PubMed] [Google Scholar]
- 23.Strauss, S., J. Z. Jordens, U. Desselberger, and J. J. Gray. 2000. Single-tube real-time nested polymerase chain reaction for detecting human papillomavirus DNA. Diagn. Mol. Pathol. 9151-157. [DOI] [PubMed] [Google Scholar]
- 24.Wallace, J., B. A. Woda, and G. Pihan. 2005. Facile, comprehensive, high-throughput genotyping of human genital papillomaviruses using spectrally addressable liquid bead microarrays. J. Mol. Diagn. 772-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton, S. P., M. N. Mindrinos, and R. W. Davis. 2006. Analysis of hybridization on the molecular barcode GeneChip microarray. Biochem. Biophys. Commun. 348689-696. [DOI] [PubMed] [Google Scholar]
- 26.Wong, C. W., C. L. W. Heng, L. W. Yee, S. W. L. Soh, C. B. Kartasasmita, E. A. F. Simoes, M. L. Hibberd, W. K. Sung, and L. D. Miller. 2007. Optimization and clinical validation of a pathogen detection microarry. Genome Biol. 8R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woo, Y. L., I. Damay, M. Stanley, R. Crawford, and J. Sterling. 2007. The use of HPV linear array assay for multiple HPV typing on archival frozen tissue and DNA specimens J. Virol. Methods 142226-230. [DOI] [PubMed] [Google Scholar]