

Abstract
Fisetin is a natural flavonol present in edible vegetables, fruits and wine at 2–160 μg/g concentrations and an ingredient in nutritional supplements with much higher concentrations. The compound has been reported to exert anticarcinogenic effects as well as antioxidant and anti-inflammatory activity via its ability to act as an inhibitor of cell proliferation and free radical scavenger, respectively. Our cell-based high-throughput screen for small molecules that override chemically induced mitotic arrest identified fisetin as an antimitotic compound. Fisetin rapidly compromised microtubule drug-induced mitotic block in a proteasome-dependent manner in several human cell lines. Moreover, in unperturbed human cancer cells fisetin caused premature initiation of chromosome segregation and exit from mitosis without normal cytokinesis. To understand the molecular mechanism behind these mitotic errors, we analyzed the consequences of fisetin treatment on the localization and phoshorylation of several mitotic proteins. Aurora B, Bub1, BubR1 and Cenp-F rapidly lost their kinetochore/centromere localization and others became dephosphorylated upon addition of fisetin to the culture medium. Finally, we identified Aurora B kinase as a novel direct target of fisetin. The activity of Aurora B was significantly reduced by fisetin in vitro and in cells, an effect that can explain the observed forced mitotic exit, failure of cytokinesis and decreased cell viability. In conclusion, our data propose that fisetin perturbs spindle checkpoint signaling, which may contribute to the antiproliferative effects of the compound.
Introduction
To maintain their genomic balance, cells have evolved specific signaling networks that monitor fidelity of the cell cycle and integrity of DNA structure. The spindle checkpoint is one of these evolutionary conserved cell cycle pathways and it functions to delay segregation of chromosomes until all chromosomes have achieved proper attachment to spindle microtubules (1). Spindle checkpoint signaling involves specific gene products including members of the Bub, Mad and Cdc20 protein families (1). Altered expression of spindle checkpoint proteins and, to a lesser extend, mutations in the spindle checkpoint genes have been found in various human cancers (1,2). Therefore, it has been hypothesized that errors in spindle checkpoint signaling may contribute to loss or gain of chromosomes (aneuploidy) or induction of polyploidy and thereby drive tumorigenesis. Interestingly, recent animal experiments have demonstrated that while moderate levels of aneuploidy indeed increased oncogenicity (3,4), massive aneuploidy due to malfunction of spindle checkpoint proteins acted as a tumor suppressor (3). Moreover, human cancer cells lacking BubR1 and Mad2 proteins undergo cell death after severe chromosome missegregation caused by premature spindle checkpoint inactivation (2). These findings support the notion that mitotic catastrophe induced by spindle checkpoint inactivation has therapeutic anticancer potential.
Natural products including plant-derived agents have been suggested to possess cancer chemopreventive potency that culminates to induction of apoptosis in various cell lines and animal models (5,6). In this study, we have performed a cell-based high-throughput screen (HTS) for compounds that overcome a chemically hyperactivated spindle checkpoint and cause a precocious mitotic exit. We utilized a novel phenotype-based HTS described in (7). Within the Spectrum Microsource compound library containing 2000 known drugs, experimental bioactives and pure natural products, we identified the flavonoid fisetin (3,3′,4′,7-tetrahydroxyflavone) as a strong inhibitor of the spindle checkpoint. Fisetin is found in fruits, vegetables, nuts and wine at concentrations of 2–160 μg/g with an average daily intake estimate of 0.4 mg (8,9). Fisetin is also added to nutritional supplements at very high concentrations. Fisetin has a variety of established biological effects including antioxidant and anti-inflammatory activity as a free radical scavenger (10,11) and anticarcinogenic potency via its ability to prevent cellular proliferation and in vitro angiogenesis (12,13). At the molecular level, fisetin has been shown to bind and inhibit the activity of cyclin-dependent kinases (Cdks)1, Cdk2, Cdk4 and Cdk6 (14–16) and to act as an antagonist of DNA topoisomerase (Topo) I and II (17,18) and androgen action (19).
The unexpected finding that fisetin caused a proteasome-dependent forced mitotic exit in several human cancer cell lines prompted us to investigate the consequences of fisetin treatment on mitotic signaling in more detail. We found that fisetin reduces the kinetochore affinity of many key spindle checkpoint proteins and causes dephosphorylation of several mitotic proteins. Furthermore, Aurora B kinase was found to be a novel molecular target of fisetin both in vitro and in cultured cells. Based on these findings, we suggest that the forced mitotic exit by fisetin involves inhibition of Aurora B activities that are required for the maintenance of normal spindle checkpoint signaling.
Materials and methods
Cell culture
HeLa cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin/streptomycin, glutamine, non-essential amino acids, N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid and 10% fetal bovine serum (FBS). For the HeLa cells stably expressing histone H2B-GFP fusion protein (20), blasticidin was added to the growth medium (2 μg/ml). MCF-10A was maintained in DMEM:HAM F-12 (1:1) supplemented with insulin (10 μg/ml), hydrocortisone (5 μg/ml), epidermal growth factor (20 ng/ml), cholera toxin (100 ng/ml), glutamine and 5% horse serum. MCF-7 cells were grown in DMEM supplemented with penicillin/streptomycin, glutamine and 10% FBS. PC3 cells were grown in DMEM with glutamine and 10% FBS. For DU145 and A549 cells, RPMI supplemented with penicillin/streptomycin, glutamine and 10% FBS was used. All the cell lines were cultured at 37°C and with 5% CO2.
Chemicals
Fisetin (3,3′,4′,7-tetrahydroxyflavone, C15H10O6, Mw = 286.24) and all other chemicals were from Sigma (St Louis, MO) unless otherwise stated. The Spectrum Collection library (MicroSource Discovery Systems, Gaylordsville, CT) was stored as 10 mM stock plates in dimethyl sulfoxide (DMSO) at −20°C. Fisetin was used in cell-based assays at 6–60 μM, MG132 at 20 μM, nocodazole at 70 nM, 350 nM and 3 μM, taxol (Molecular Probes, Sunnyvale, CA) at 600 nM, monastrol at 100 μM and ZM447439 (a gift from AstraZeneca, Wilmington, DE) at 20 μM concentrations.
Compound library screening
The screen was based on the published HTS described in (7). The Spectrum Collection library, comprised of 2000 biologically active and structurally diverse compounds, was screened for inhibitors of the spindle checkpoint. The 10 mM stock library plates were further diluted using Hamilton Microlab Star liquid handling robotics (Hamilton, Reno, NV) to 60, 6, 0.6 and 0.06 μM in growth medium and used in the screen. Cells were accumulated in M phase using a 12 h nocodazole treatment (350 nM). Mitotic cells were harvested using mitotic shake-off and replated on 384-well compound plates in culture medium containing nocodazole (final concentration 70 nM). Five micromolars of Ro 31-8220 (Bisindolylmaleimide IX, LC laboratories, Woburn, MA) was used as a positive control and normal culture medium with 70 nM nocodazole as a negative control. After a 4 h incubation, round mitotic cells were washed out with a Tecan PW384 plate washer (Tecan, Groedig, Austria) and the remaining cells were fixed with 2% paraformaldehyde in PHEM (60 mM PIPES, 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 10 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid, 4 mM MgSO4) containing 0.5% Triton-100 and SYBR Gold nucleic acid stain (1:15 000, Molecular Probes). Fluorescence intensity of DNA was measured with Acumen Cell Cytometer using Acumen Explorer software (TTP LabTech Ltd, Melbourn, UK). Results of the compound screen were normalized for edge effects and for plate/screen variations using the R/bioconductor package cellHTS (21). In the secondary screen, the library drugs that yielded high DNA fluorescence intensity were retested in four different concentrations as duplicates using the primary screening protocol and were further analyzed microscopically to validate the compound effects.
Live cell microscopy
HeLa H2B-GFP cells growing on 35 mm live cell chambers (MatTek Corp., Ashland, MA) were imaged using a Zeiss Axiovert 200M microscope (Carl Zeiss MicroImaging GmbH, Göttingen, Germany) equipped with ×63 (NA 1.4) objective, Orca-ER camera (Hamamatsu Photonics Norden, Solna, Sweden) and MetaMorph imaging software (Molecular Devices, Sunnyvale, CA). Images were captured at 5–10 min intervals with exposure times ∼150 ms for transmitted light and ∼20 ms for fluorescence light.
Immunofluorescence, image acquisition and analysis
Immunofluorescence was performed as described earlier (22). We used primary antibodies against Aurora B (1:1000, Abcam, Cambridge, UK), Bub1 (1:200, Millipore, Billerica, MA), BubR1 (1:400, Abcam), CenpA phosphorylated at Ser7 (pCenpA; 1:500, Millipore), Cenp-F (1:200, BD Biosciences, San Jose, CA), CREST autoimmune serum (1:200, Antibodies, Davies, CA), Hec1 (1:200, Abcam) and Histone H3 phosphorylated at Ser10 (1:1500, Millipore). Secondary fluorescein isothiocyanate, Cy3 or Cy5-conjugated antibodies were used at 1:600–1:800 (Jackson ImmunoResearch, West Grove, PA). Images of the fixed cells were acquired using a Zeiss Axiovert 200M platform (see above) and MetaMorph software as Z-stacks with 0.3 μm step size. Quantification of kinetochore protein signals was done using MetaMorph as described elsewhere (22). For each experiment, a minimum of 50 kinetochores was analyzed in five cells per condition. The p-Histone H3 signal intensities were measured at whole cell level from at least five cells per condition. Statistical testing was performed with student's t-test and P values <0.001 were considered significant.
In vitro kinase assay
Aurora B was expressed in Sf9 insect cells using the Baculogold expression system (BD Biosciences) according to the manufacturer's protocol. For this, the Aurora B open reading frame was amplified from complementary DNA using primers introducing BamHI and SmaI sites and cloned in frame with the glutathione-S-transferase tag open reading frame of the pAcG2T expression vector. Glutathione-S-transferase–Aurora B was purified from Sf9 extracts using glutathione-affinity chromatography (23). Five micrograms of Aurora B and Aurora A substrate myelin basic protein (Millipore) or 0.25 μg of Cdk1 substrate Histone H1 (New England Biolabs, Ipswich, MA) were mixed with the kinase buffer (0.5 μM adenosine triphosphate, 12 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid pH 7.4, 8 mM MgCl2, 0.8 mM dithiothreitol, 0.08 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid) and a concentration series of fisetin or ZM447439 in DMSO. To start the reaction, γ-32P-adenosine triphosphate and 4.5 μg of glutathione-S-transferase–Aurora B kinase, 25 ng of Aurora A (Cell Signaling, Danvers, MA) or 25 ng of Cdk1/cyclin B (Cell Signaling) were added. As a positive control, 5 μM Ro-31-8220 was used for Cdk1/CycB and 0.6 and 5 μM ZM447439 for Aurora B. Reactions were incubated at 37°C for 1 h and terminated by addition of Laemmli sodium dodecyl sulfate sample buffer. Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and phosphorylation was visualized by autoradiography.
Western blotting
Preparation of cell extracts, sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting were done as described elsewhere (22). The blots were incubated with antibodies against Aurora A (1:2000, Abcam), Cenp-F (1:1000, BD Biosciences), cleaved poly (ADP-ribose) polymerase (1:1000, Cell Signaling), Cdc27 (1:1000; a kind gift from Dr Philip Hieter), Bub1 (1:2000; Abcam), cyclin B1 (1 μg/ml; BD Biosciences), Aurora A phosphorylated at Thr288 (1:1000; Cell Signaling) and α-tubulin (1:1000, Abcam). IR Dye® Conjugated secondary antibodies (Rockland Immunochemicals, Gilbertsville, PA) were used at 1:5000. The blotting was performed twice per each experiment. Signals were detected using Odyssey Infrared Imaging System (LI-COR Biotechnology, Lincoln, NE).
Fluorescent-activated cell sorting
Cells were harvested, spun down and fixed in 70% ethanol (−20°C) for at least 30 min at −20°C. The cells were washed once with phosphate-buffered saline before resuspension in 200 μl phosphate-buffered saline containing 100 μg/ml RNase and 20 μg/ml propidium iodide. After 30 min incubation at room temperature under constant agitation, the cells were analyzed with LSR II system (BD Biosciences) and FCS Express 3 software (De Novo Software, Los Angeles, CA).
Results
A cell-based HTS for small molecules inducing forced mitotic exit identifies flavonoids
We performed a HTS for small molecules that cause a forced exit from mitotic arrest induced by the microtubule-destabilizing drug nocodazole (Figure 1). The screen is based on different cell-to-substrate attachment properties of round mitotic and flat interphase HeLa cells. Mitotic cells from cultures incubated overnight in 350 nM nocodazole were replated in the continued presence of 70 nM nocodazole into 384-well compound library plates. Besides the library drugs, each assay plate included positive (Ro-31-8220, Bisindolylmaleimide IX at 5 μM) and negative (70 nM nocodazole) controls. After 4 h incubation, the plates were washed to remove all weakly attached cells (round mitotic and apoptotic cells) and the wash-resistant cells were fixed and stained with a nuclei acid dye (Figure 1A). Wells with compounds that override the spindle checkpoint and force cells out of M phase are expected to have a high mean DNA fluorescence signal due to the resistance of the readhered flat post-mitotic cells to washes. To identify potential inhibitors of the spindle checkpoint among known drugs, experimental bioactives and natural products, we screened the Spectrum Microsource library. The primary screen revealed 34 hit compounds (supplementary Figure S1 is available at Carcinogenesis Online), two of that were confirmed in the secondary screen (Figure 1B) and in further cell-based assays to be strong inducers of forced mitotic exit. The most potent compound identified was the dietary flavonoid fisetin (Figure 1B and C) that caused a rapid escape from a hyperactivated spindle checkpoint in all cell lines tested. The other compound with anti-spindle checkpoint effects was also a flavonoid, eupatorin (Figure 1B, A.-L.Salmela, A.Varis, J.Pouwels, M.J.Kallio, unpublished data).
Fig. 1.

HTS for drugs that induce a forced mitotic exit. (A) Schematic representation of the workflow of the primary HTS. (1–2) Subconfluent plate of cycling HeLa cells was treated overnight with nocodazole. (3–4) Mitosis arrested cells were harvested by shake-off, replated on 384-well drug library plates and incubated for 4 h in the continuous presence of nocodazole. (5–6) Wells with treated cells were washed with a plate washer and subsequently stained with SYBR Gold and analyzed for their DNA content. Example micrographs for wells with a low (7, no exit from nocodazole block, most round mitotic cells were lost during the washes) and a high (8, forced mitotic exit from nocodazole block by a library drug, readhered multinucleated cells were resistant to the washes) signal for DNA are shown. Each screen contained a positive control lane (lane 1, cells treated with an experimental drug Ro-31-8220 known to cause premature exit from mitosis) and a negative control lane (lane 2, cells treated with nocodazole only). (B) The secondary screen 384-well plate with 34 library drugs that were selected for further testing on the basis of the primary screen results. The wells in lanes 3–22 contain nocodazole-arrested cells treated with the selected library drugs at four different concentrations (60, 6, 0.6, 0.06 μM). Lanes 23–24 contain only culture medium without cells. Fisetin (positions E9 and E10, 60 and 6 μM concentrations, respectively) and eupatorin (position M9, 60 μM) yielded a high DNA fluorescence intensity from readhered cell population due to induced forced mitotic exit. The lower panel shows quantification of the DNA fluorescence intensity converted into cell numbers for each well of the plate shown above. The arrows point to the nocodazole-arrested cells in wells treated with 60 and 6 μM fisetin. (C) The molecular structure of fisetin.
Fisetin induces a rapid escape from microtubule drug-induced mitotic arrest in a proteasome-dependent manner
To analyze the fisetin-induced forced mitotic exit in detail, we first preincubated HeLa cells stably expressing the DNA marker protein H2B-GFP with 70 nM nocodazole for 16 h prior to addition of 1, 5 or 30 μM fisetin or DMSO and subsequent incubation for 4 h before fixation and determination of mitotic indices. The cells were kept in the continued presence of nocodazole for the duration of all assays. The populations pretreated with 70 nM nocodazole and incubated in the presence of DMSO or 1 μM fisetin remained at M phase arrest, whereas treatment with 5 μM or 30 μM fisetin lowered the mitotic index by 47 or 98%, respectively, compared with cells treated with nocodazole only. The highest fisetin concentration (30 μM) was selected for all further assays due to its clear antimitotic effects. Next, we preincubated the HeLa H2B-GFP cells with 70 nM nocodazole for 16 h prior to addition of vehicle only (DMSO) or 30 μM fisetin and subsequent time-lapse filming for 6 h. Most nocodazole-arrested cells (82%, n = 62) remained in mitosis for at least 6 h after addition of DMSO (Figure 2A and B, supplementary Video 1 is available at Carcinogenesis Online). In sharp contrast, exposure to 30 μM fisetin resulted in a rapid exit from M phase as all nocodazole-blocked mitotic cells (n = 140) decondensed their chromosomes and changed their morphology from a round, loosely attached mitotic cell to a flat adhered cell within 90 min after addition of the drug (Figure 2A and B, supplementary Video 2 is available at Carcinogenesis Online). Increasing the nocodazole concentration to 3 μM, which completely depolymerizes microtubules and maximally activates the spindle checkpoint, did not prevent the forced mitotic exit by fisetin as only 4% of the filmed HeLa cells (n = 100) remained at M phase arrest 6 h after exposure to fisetin (Figure 2B). Similarly, all HeLa H2B-GFP cells preincubated with the Eg5 inhibitor monastrol (n = 30), known to cause M phase arrest with a monopolar spindle (24), or the microtubule-stabilizing drug taxol (n = 160) underwent forced mitotic exit within 90 min after addition of 30 μM fisetin (Figure 2B, supplementary Videos 3 and 4 are available at Carcinogenesis Online). After the expected failure of cytokinesis due to the presence of microtubule drugs in the culture medium, the cells formed polyploid progeny cells that in the subsequent G1 phase displayed a multinuclear phenotype (Figure 2A, supplementary Video 2 is available at Carcinogenesis Online). Importantly, the fisetin-induced forced mitotic exit was independent of the cell type as all other cell types tested, lung adenocarcinoma epithelial cells (A549), breast adenocarcinoma cells (MCF-7), androgen-independent prostate cancer cells (DU145 and PC3) and non-tumorigenic mammary epithelial cells (MCF-10A), behaved in a similar manner as the HeLa cells and exhibited the forced mitotic exit from microtubule drug-induced M phase arrest (data not shown).
Fig. 2.

(A) Fisetin causes a proteasome-dependent escape from chemically induced mitotic arrest. HeLa cells expressing histone H2B-GFP were arrested in mitosis with 70 nM nocodazole and then imaged using time-lapse microscopy without further treatment (upper panel), immediately after addition of 30 μM fisetin (middle panel) or after 1 h treatment with MG132 followed by addition of 30 μM fisetin (lower panel). Fisetin overcomes nocodazole block within 40 min, whereas cells preincubated with MG132 remain arrested for at least 6 h. Scale bars represent 10 μm. Representative time-lapse supplementary videos 1–5 are available at Carcinogenesis Online (B) Quantification of the forced mitotic exit induced by fisetin in cells arrested at M phase with nocodazole, taxol or monastrol. The bars show percentage of cells undergoing forced mitotic exit. (C) Western blot showing the escape from nocodazole block by fisetin. The levels of mitotic markers Aurora A and Cenp-F decrease dramatically within 40 min after the addition of fisetin to the nocodazole-arrested cells compared with the starting time point (0 min) or to the samples incubated in the continued presence of nocodazole to the end of assay (noc only, 180 min). α-Tubulin served as the loading control. The graph shows the average band intensities normalized over tubulin from two separate assays.
To confirm that the cells have indeed exited M phase rather than underwent a premature chromosome decondensation and a morphological change without leaving mitosis, we analyzed the timely changes in the expression levels of the mitotic markers Cenp-F and Aurora A kinase in nocodazole-blocked mitotic HeLa cells before and after addition of fisetin (Figure 2C). Already 40 min after fisetin addition, the levels of Cenp-F and Aurora A were strongly reduced compared with the starting time point (0 min, Figure 2C) or to the samples incubated in the continued presence of nocodazole to the end of assay (noc only 180 min, Figure 2C). In a similar manner, the expression levels of cyclin B1 were rapidly diminished in fisetin-treated cells compared with nocodazole-arrested controls (data not shown).
One plausible explanation for the observed escape from nocodazole-induced mitotic arrest is that fisetin interferes with the maintenance of normal spindle checkpoint signaling. The anaphase inhibitory complexes formed at the kinetochores of unaligned chromosomes are known to prevent activation of a mitotic ubiquitin ligase termed the anaphase-promoting complex/cyclosome (1). After the spindle checkpoint is satisfied, the anaphase-promoting complex/cyclosome is activated that leads to poly-ubiquitination and subsequent proteasome-mediated degradation of anaphase-promoting complex/cyclosome substrates such as securin and cyclin B1 (1). This is followed by separation of sister chromatids and exit from mitosis. To test if fisetin-induced mitotic exit is dependent on proteasome activity, we treated nocodazole-arrested mitotic H2B-GFP HeLa cells with the proteasome inhibitor MG132, known to induce a metaphase arrest downstream of the spindle checkpoint, for 1 h before addition of fisetin or DMSO and time-lapse imaging (Figure 2A and B and supplementary Video 5 is available at Carcinogenesis Online). All but one of the nocodazole-MG132-fisetin cotreated mitotic cells (n = 81) remained at M phase for at least 6 h. We conclude that fisetin overcomes mitotic arrest induced by nocodazole, taxol and monastrol in various human cell lines and that the fisetin-induced forced mitotic exit is a proteasome-dependent process.
Fisetin causes precocious exit from unperturbed mitosis
In normal cycling mammalian cells, the spindle checkpoint is active from nuclear envelope breakdown until all chromosomes have stable bi-polar attachment to spindle microtubules (1). To test if fisetin causes premature exit from unperturbed mitosis, we treated cycling HeLa H2B-GFP cells with 30 μM fisetin (Figure 3A and B) or DMSO (Figure 3C) followed by time-lapse microscopy. Most prophase and prometaphase cells treated with fisetin (86%, n = 42) were precociously forced out of mitosis despite the presence of unaligned chromosomes (Figure 3A, supplementary Video 6 is available at Carcinogenesis Online). In fisetin-treated prophase cells, the time from nuclear envelope breakdown to premature chromosome decondensation was on average 63 ± 26 min, which corresponded to the average nuclear envelope breakdown-to-anaphase time of DMSO-treated control cells (59 ± 27 min). Upon exit from M phase, sister chromatids of fisetin-treated cells failed to completely separate prior to chromatin decondensation (Figure 3A). Similarly, the vast majority of fisetin-treated metaphase cells (95%, 20 of 21) exhibited defective anaphase without complete separation of the sister chromatids (Figure 3B). Importantly, also cytokinesis was defective in most of these early unperturbed mitotic cells exposed to fisetin. During mitotic exit, each cell exhibited excessive membrane blebbing but the cleavage furrow formation was either soon aborted resulting in multinucleated progeny cell (25 of 63, Figure 3A) or the final abscission of the two separated daughter cells failed leading to cell fusion and formation of binucleate progeny cells with micronuclei (31 of 63, Figure 3B, supplementary Videos 7 and 8 are available at Carcinogenesis Online). In contrast, all cells that were in anaphase at the time of fisetin addition showed normal separation of sister chromatids (n = 11) and most underwent normal cytokinesis (9 of 11, the two abnormal anaphase cells formed binucleate progeny cells after cell fusion). These data demonstrate that fisetin causes precocious escape from early mitotic phases followed by cytokinesis errors. Together with the results from cells with a chemically hyperactivated spindle checkpoint, the data suggest that fisetin interferes with spindle checkpoint signaling components in the mitotic cells.
Fig. 3.

Fisetin causes premature exit without cytokinesis from unperturbed mitosis. (A) Time-lapse microscopy of a prophase H2B-GFP HeLa cell treated with 30 μM fisetin. The chromosomes fail to align at the spindle equator prior to chromatin decondensation. The cell exits mitosis without cytokinesis and forms a polyploidy progeny cell. (B) Time-lapse microscopy of a metaphase H2B-GFP HeLa cell treated with fisetin. The chromosome segregation is initiated but the separation of sister chromatids fails to complete. The cytokinetic furrow (white arrows) cuts through the anaphase chromatid bridges but the abscission of the two progeny cells fails leading to cell fusion and formation of a binucleate progeny cell. In A and B fisetin was added 5 min before the first frame was taken. (C) Time-lapse microscopy of a control H2B-GFP HeLa cell showing normal mitosis with cytokinesis. Scale bars represent 10 μm. Representative time-lapse supplementary videos 6–8 showing the fisetin effects on unperturbed mitotic cells are available at Carcinogenesis Online.
Fisetin modulates kinetochore affinity and phoshorylation status of mitotic proteins
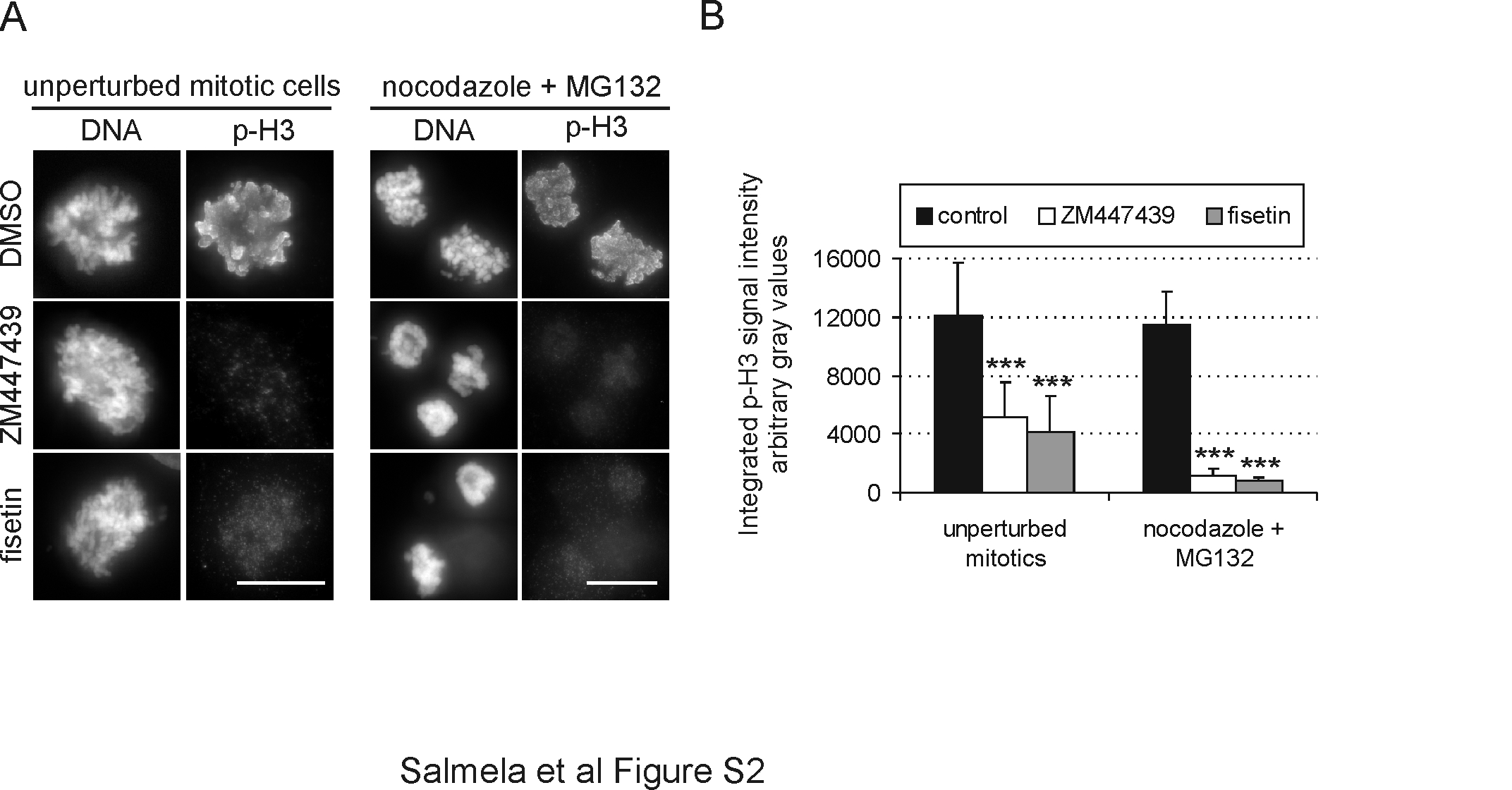
To seek support for the notion that fisetin targets spindle checkpoint proteins, we investigated the drug's ability to modulate the kinetochore affinity of key spindle checkpoint proteins. HeLa cells arrested in mitosis with 70 nM nocodazole were treated with 30 μM fisetin in the continued presence of nocodazole for 3 h before fixation and immunofluorescent staining for Aurora B, Bub1, BubR1, Cenp-F and Hec1 (Figure 4A and B). In all experiments, MG132 was added to the culture medium 1 h before fisetin to prevent premature exit from mitosis. Control cells blocked at mitosis with nocodazole were incubated 4 h with MG132 before fixation and analysis. Quantification of the normalized average fluorescence intensities of spindle checkpoint proteins revealed highly significant (P < 0.001) reductions of Bub1 (down by 94%), BubR1 (down by 94%) and Cenp-F (down by 92%) at the kinetochores of fisetin-treated cells compared with controls (Figure 4A and B). Importantly, localization of the outer kinetochore marker Hec1 was unaffected by fisetin (down by 4% compared with controls) demonstrating that the drug does not cause collapse of the kinetochore structure (Figure 4A and B). Interestingly, Aurora B kinase was mislocalized from inner centromeres to chromosome arms in the fisetin-treated cells (Figure 4A). To assess if fisetin interferes with Aurora B activity in cultured cells, we stained HeLa cells with a phospho-Cenp-A antibody (pCenpA) recognizing phosphorylated serine 7 and with a phospho-Histone H3 antibody recognizing phosphorylated serine 10, both known target residues of Aurora B (25,26). Fisetin treatment led to a significant 98% reduction in the pCenpA signal intensity at kinetochores (P < 0.001, Figure 4A and B). This reduction was similar to that observed in HeLa cells treated with Aurora inhibitor ZM447439 (27) for 3 h, where pCenpA levels were down by 96% compared with controls (P < 0.001). Similarly, fisetin reduced significantly (P < 0.001) the chromosomal phospho-Histone H3 antibody signal intensities in both nocodazole-arrested cells pretreated with MG132 and in unperturbed mitotic cells to levels comparable with ZM447439 treatment (supplementary Figure S2 is available at Carcinogenesis Online). These data suggest that fisetin modulates Aurora B kinase activity in cultured cells.
Fig. 4.

Fisetin perturbs the normal mitotic localization of several spindle checkpoint proteins and inhibits Aurora B activity in cells and in vitro. (A) Nocodazole-arrested HeLa cells were treated 1 h with MG132 followed by 3 h incubation with 30 μM fisetin or DMSO, fixation and staining with antibodies against Bub1, BubR1, Cenp-F, Aurora B, pCenpA and Hec1. ZM447439-treated cells were stained for pCenpA as a positive control for Aurora B inhibition. Scale bars represent 10 μm. The graph (B) shows the quantification of the kinetochore fluorescence intensities in control and drug-treated cells. In fisetin-treated cells Bub1, BubR1, Cenp-F and pCenpA are significantly diminished at the kinetochores compared with controls, and Aurora B is relocalised to the chromosome arms. In contrast, Hec1 staining is not notably different compared with controls. (C) Autoradiograph of in vitro kinase assay for Aurora B, Aurora A and Cdk1/CycB activities determined by γ32-P incorporation to myelin basic protein (Aurora A + B) or Histone H1 (Cdk1/CycB) in the absence or presence of different fisetin concentrations. The graph below shows quantification of the in vitro kinase assay data. At the concentration that cause cells to escape mitosis, fisetin clearly inhibits Aurora B activity (IC50 ∼2.0 μM) but not Aurora A or Cdk1/CycB activity (IC50 > 30 μM). (D) Western blot analysis of extracts prepared from nocodazole-arrested HeLa cells treated with MG132 for 30 min followed by 3 h fisetin or DMSO treatment in the continued presence of MG132 and nocodazole. The autophosphorylation levels of Aurora A (pAurA) and total Aurora A levels (AurA) are not affected by fisetin compared with control. Cyclin B1 levels are also unchanged indicating that the cells have remained at M phase arrest. α-Tubulin was used as loading control.
Next, we determined if fisetin could directly inhibit Aurora B kinase activity using an in vitro kinase assay. Based on the concentration-dependent reduction of γ-32P incorporation to myelin basic protein, we estimated an IC50 of 2.0 μM for inhibition of Aurora B by fisetin (Figure 4C). Importantly, the activities of Aurora A and Cdk1 were only moderately inhibited by fisetin in vitro (IC50 > 30 μM for both kinases, Figure 4C) in parallel assays. In our in vitro assay setup, the inhibitory potency of ∼2.0 μM fisetin corresponded with the effect of ∼0.3 μM ZM447439, which both yielded ∼50% reduction in the Aurora B kinase activity (supplementary Figure S3 is available at Carcinogenesis Online). In the original study, an IC50 of 0.130 μM for ZM447439 was reported (27) indicating that fisetin is less effective inhibitor of Aurora B in vitro. On the other hand, as shown in the western blot in Figure 4D, the cellular levels of Aurora A auto-phosphorylated on Thr288, a marker for Aurora A activity (28), were comparable between the fisetin-treated and control cells, giving support for the notion that fisetin has higher selectivity toward Aurora B than Aurora A. In conclusion, the flavonoid fisetin prevents the normal kinetochore accumulation of Aurora B, Bub1, BubR1 and Cenp-F without causing any major structural damage to the kinetochores. Furthermore, fisetin inhibits the activity of Aurora B in vitro (IC50 ∼2.0 μM) and in cultured cells.
Fisetin increases apoptosis in cycling cells
To test if fisetin treatment triggers apoptosis, we incubated a panel of cell lines (A549, DU145, HeLa, PC3 and MCF-10A cells) with DMSO or fisetin for 1, 3 or 5 days (single 30 μM fisetin pulse on day 0) before cell harvest and analysis by fluorescent-activated cell sorting (FACS) (Figure 5). Judged by the appearance of a sub-G1 peak in the FACS profiles of fisetin-treated cell populations, indicative of the apoptotic fraction due to fragmented DNA, fisetin induces a massive apoptosis in A549 (75 and 83%) and DU145 (60 and 74%) cells and moderate levels of apoptosis in HeLa (27 and 20%) and PC3 (17 and 21%) cells at 3 and 5 day time points, respectively (Figure 5). Interestingly, the MCF-10A cells appear less sensitive to the fisetin-induced cell killing as only 6–7% of these cells are in the sub-G1 population at 1, 3 and 5 day time points. Together with the time-lapse filming, the FACS profiles indicate that fisetin treatment causes either a rapid apoptosis without significant accumulation to 4N or 8N populations (DU145) or a moderate apoptosis with induction of polyploidy (PC3). We conclude that long-term exposure to fisetin provokes variable amounts of apoptosis in cancer cell lines, whereas the non-tumorigenic MCF-10A cells appear more resistant to the induced cell killing.
Fig. 5.

Fisetin induces apoptosis in A549, DU145, HeLa and PC3 cells, but not in MCF-10A cells. (A) Cycling cell populations were incubated for 1, 3 or 5 days in the presence of 30 μM fisetin or DMSO after the samples were analyzed by FACS. The treated cell types could be categorized into three different FACS profile groups with the following examples: rapid apoptosis without accumulation to ≥4N cell populations (DU145); moderate apoptosis with some accumulation to 4N/8N cell populations (PC3) and low levels of apoptosis with only minor cell cycle effects (MCF-10A). (B) Percentages of fisetin-induced apoptosis at various time points. The bars represent the sub-G1 populations of the FACS profiles, indicative of fragmented DNA by apoptosis.
Discussion
Deregulation of spindle checkpoint signaling is one potential source for chromosomal instability and aneuploidy found in many human primary tumors and tumor cell lines (2). In an effort to find small molecules with anti-spindle checkpoint activity, we executed a newly designed HTS based on different cell-to-substrate attachment properties of round mitotic and flat interphase cells (7). The discovery of the dietary flavonoid fisetin as a drug that overcomes chemically induced mitotic block provides a proof-of-principle for the screening strategy. Our results show that fisetin interferes with kinetochore accumulation of several key mitotic proteins and inhibits Aurora B kinase in cultured cells and in vitro. As Aurora B activity has been shown to be required for the maintenance of spindle checkpoint signaling, we propose that fisetin may exerts its antimitotic activity, at least partially, through inhibition of Aurora B. It also remains possible that Aurora B function is inhibited indirectly in the cells, e.g. by fisetin inhibiting an enzyme required for the activation of the kinase.
Fisetin and other plant-derived flavonoids have been shown to exhibit antiproliferative effects in several normal and tumor cell lines primarily through activation of the programmed cell death pathways (12,29). In case of fisetin, these potential chemopreventive activities can be partially explained by the previously reported inhibition of Cdk1/2/4/6, nuclear factor-kappa B and Topo II activities (14–17). This also indicates that the drug most probably targets multiple proteins and pathways simultaneously. We demonstrate that fisetin treatment rapidly compromised a mitotic block imposed by the microtubule-depolymerizing drug nocodazole, the microtubule-stabilizing drug taxol or the Eg5 inhibitor monastrol, indicating that fisetin's mechanisms of action are independent of microtubule–kinetochore interactions and/or spindle morphology. Moreover, rapid bypass of a chemically induced mitotic block was observed with all cell lines tested this far, proposing perturbation of a conserved pathway. Finally, the fact that proteasome inhibition prevented the mitotic exit induced by fisetin suggests that the drug's molecular targets reside upstream of onset of anaphase where a set of spindle checkpoint kinases, including Aurora B kinase, BubR1, Plk1 and Cdk1, work to maintain spindle checkpoint activity.
The cellular effects of fisetin on untreated mitotic cells we report here phenocopy those caused by Aurora B RNAi (30), dominant-negative Aurora B mutants (23) and experimental Aurora drugs such as ZM-class of inhibitors (31), Hesperadin (32) and VX-680 (33); cells bypassed the spindle checkpoint and exited M phase despite presence of multiple unaligned chromosomes and failed in cytokinesis leading to formation of progeny cells with 4N DNA content. These findings suggest that the inhibitory effect of fisetin in mitotic cells is primarily on Aurora B kinase rather than Aurora A, which when silenced or genetically perturbed does not compromise the spindle checkpoint or induce cytokinesis errors (34,35). In support of this argument, in vitro kinase assays showed that fisetin inhibits Aurora B more strongly than Aurora A (Figure 4C). Furthermore, phosphorylation of CenpA at serine 7, a specific Aurora B phosphorylation site, was significantly reduced by fisetin in cells (Figure 4A and B), whereas no major effects were observed on Aurora A autophosphorylation (Figure 4D). The observed resistance of anaphase cells to fisetin is most probably a consequence of drug kinetics; the full effect of fisetin may require >10 min, which is the average duration of anaphase in unperturbed cells. Alternatively, spatial modulation of Aurora B activity, or that of other possible fisetin targets, in anaphase cells may include protein modifications that prevent fisetin–kinase interaction in late mitotic cells (36).
Comparison of the anti-Aurora B potencies of fisetin (IC50 ∼2.0 μM) and ZM447439 [IC50 ∼300 nM in the present study and IC50 130 nM in the original report (27)] indicates that fisetin is less effective in suppression of the Aurora B kinase activity in vitro. Also, the cellular effects of fisetin in the anti-microtubule drug pre-challenged cell populations are slightly different from those of ZM447439, a selective Aurora inhibitor ZM447439 overrides taxol-induced M phase arrest more readily compared with nocodazole block when used at 1–5 μM concentration (27), whereas fisetin causes premature escape from both nocodazole and taxol blocks at 30 μM concentration. This discrepancy is most probably due to differences in the effective fisetin and ZM447439 drug concentrations as increasing ZM447439 concentration over 10 μM causes forced mitotic exit from nocodazole block (27). It is also probably that fisetin-induced forced mitotic exit depends on perturbed function of other proteins besides Aurora B. For instance, inhibition of Cdk1 during mitosis could provide an explanation for the observed forced mitotic exit. However, the inhibition of Cdk1 by fisetin was reported to occur at a higher drug concentration of 60 μM (14) compared with the concentration used here that resulted in complete override of the spindle checkpoint. Moreover, our in vitro kinase assays demonstrate notably lower IC50 value for Aurora B inhibition compared with that of Cdk1 and Aurora A. Topo II is another confirmed fisetin target (17) whose activity is essential for normal mitosis where it is required for decatenation of entangled DNA strands and for proper DNA condensation. However, treatment of cells with anti-Topo II drugs such as VM-26 (37) or VP-16 (our unpublished data) does not override chemically activated spindle checkpoint but instead appears to cause a transient metaphase arrest (38,39). In contrast, Topo II-depleted Drosophila S2 and HeLa cells can successfully satisfy the spindle checkpoint without any significant mitotic delay (40). Interestingly, in the same study, Topo II RNAi caused an indirect inhibition of Aurora B kinase activity through BubR1-mediated action (40). We conclude that the observed inhibition of Aurora B activity by fisetin can provide a sufficient explanation for the forced mitotic exit but we cannot exclude the possibility that fisetin inappropriately turns off the spindle checkpoint by simultaneous inhibition of more than one mitotic target involved in this process such as Mps1, Bub1 and BubR1.
Previous reports with FACS analyses of fisetin-treated (10–60 μM; 24–48 h) human prostate (PC-3) or colon (HT-29) cancer cells have shown a significant accumulation of cells in G2/M phase followed by apoptosis (14,41). In light of our findings that fisetin rapidly overrides mitotic control and abrogates cytokinesis even in unperturbed cells, we propose that the earlier FACS analyses showing increase in the G2/M peaks after fisetin treatment are, at least partially, due to formation of G1 cells with 4N DNA content after forced mitotic exit without cytokinesis. In agreement with previous studies, we show that fisetin treatment triggers cell suicide mechanisms in many cancer cell lines leading to apoptosis starting 24 h after exposure to the drug. It appears that certain cell types are more sensitive to the induction of cell death by fisetin as they undergo massive apoptosis rapidly after addition of the drug without showing significant increase in polyploidy (e.g. A549 and DU145 cells) whereas some cell types show accumulation of 4N and 8N populations followed by moderate levels of apoptosis (e.g. PC3). Interestingly, the non-carcinogenic MCF-10A cells were less sensitive to the fisetin-induced cell killing, which proposes that cell line-specific variations exists either in the mode of action of fisetin, in its pharmacokinetics, or in cellular processes of the drug response pathways. Future testing of broader cell line panels for the antiproliferative effects of fisetin and other flavonoids is, however, essential before any conclusions can be made on cancer cell-specific killing.
Due to its reported cardioprotective, memory-enhancing and anticarcinogenic properties fisetin is often added to nutritional supplements at very high concentrations under the assumption that it will possess enhanced beneficial health effects. The concentration of fisetin in these dietary supplements can far exceed the daily dose attained from a typical vegetarian diet. Importantly, previous studies suggested that maternal ingestion of flavonoids might induce breaks and translocations affecting the mixed lineage leukemia gene in the fetus, and eventually lead to infant leukemia, through possible effects on the catalytic activities of Topo II (42). Also, Aurora kinases are known to have essential roles in embryonic cell divisions (43,44) and their inhibition may disturb normal fetal development. Our finding that fisetin inhibits the activity of Aurora B kinase in cells provides a new mechanism of action whereby fisetin may elicit its previously reported antiproliferative effects and suggest that flavonoids in general may contribute to the preventive effect of a plant-based diet on serious diseases, including solid tumors. However, the biological effects of ingesting flavonoids at high concentrations are not known and precaution should be taken when using nutritional supplements rich with fisetin, especially during pregnancy.
Supplementary material
Supplementary Figures S1–S3 and Videos 1–8 can be found at http://carcin.oxfordjournals.org/
Funding
EUFP6 Marie Curie EXT grant (002697 to M.J.K.), Academy of Finland (120804) and Centre of Excellence for Translational Genome-Scale Biology (M.J.K. and O.K.); Turku Graduate School of Biomedical Sciences to A-L.S.; National Institute of General Medical Sciences, McCasland Foundation (G.J.G.); EUFP6 Marie Curie EXT grant (002728 to P.H.and M.P.).
Acknowledgments
We thank P.Kohonen for his assistance in the statistical analysis, N.Sahlberg for help in HTS, P.Terho for assistance in FACS assays and T.Holmström for assistance in kinase assays. We thank Dr P.Hieter for providing the anti-Cdc27 antibody and Astrazeneca for the ZM447439.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- Cdk
cyclin-dependent kinase
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulfoxide
- FACS
fluorescent-activated cell sorting
- FBS
fetal bovine serum
- HTS
high-throughput screen
- Topo
topoisomerase
References
- 1.Musacchio A, et al. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 2.Kops GJ, et al. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 3.Weaver BA, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 4.Sotillo R, et al. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kandaswami C, et al. The antitumor activities of flavonoids. In Vivo. 2005;19:895–909. [PubMed] [Google Scholar]
- 6.Wang LS, et al. Anthocyanins and their role in cancer prevention. Cancer Lett. 2008;269:281–290. doi: 10.1016/j.canlet.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demoe JH, et al. A high throughput, whole cell screen for small molecule inhibitors of the mitotic spindle checkpoint identifies OM137, a novel Aurora kinase inhibitor. Cancer Res. 2009;69:1509–1516. doi: 10.1158/0008-5472.CAN-08-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arai Y, et al. Comparison of isoflavones among dietary intake, plasma concentration and urinary excretion for accurate estimation of phytoestrogen intake. J. Epidemiol. 2000;10:127–135. doi: 10.2188/jea.10.127. [DOI] [PubMed] [Google Scholar]
- 9.Kimira M, et al. Japanese intake of flavonoids and isoflavonoids from foods. J. Epidemiol. 1998;8:168–175. doi: 10.2188/jea.8.168. [DOI] [PubMed] [Google Scholar]
- 10.Woodman OL, et al. Vascular and anti-oxidant actions of flavonols and flavones. Clin. Exp. Pharmacol. Physiol. 2004;31:786–790. doi: 10.1111/j.1440-1681.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 11.Park HS, et al. Antioxidant flavone glycosides from the leaves of Sasa borealis. Arch. Pharm. Res. 2007;30:161–166. doi: 10.1007/BF02977689. [DOI] [PubMed] [Google Scholar]
- 12.Fotsis T, et al. Flavonoids, dietary-derived inhibitors of cell proliferation and in vitro angiogenesis. Cancer Res. 1997;57:2916–2921. [PubMed] [Google Scholar]
- 13.Kuntz S, et al. Comparative analysis of the effects of flavonoids on proliferation, cytotoxicity, and apoptosis in human colon cancer cell lines. Eur. J. Nutr. 1999;38:133–142. doi: 10.1007/s003940050054. [DOI] [PubMed] [Google Scholar]
- 14.Lu X, et al. Fisetin inhibits the activities of cyclin-dependent kinases leading to cell cycle arrest in HT-29 human colon cancer cells. J. Nutr. 2005;135:2884–2890. doi: 10.1093/jn/135.12.2884. [DOI] [PubMed] [Google Scholar]
- 15.Sung B, et al. Fisetin, an inhibitor of cyclin-dependent kinase 6, down-regulates nuclear factor-kappaB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein-regulated IkappaBalpha kinase activation. Mol. Pharmacol. 2007;71:1703–1714. doi: 10.1124/mol.107.034512. [DOI] [PubMed] [Google Scholar]
- 16.Lu H, et al. Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin. J. Med. Chem. 2005;48:737–743. doi: 10.1021/jm049353p. [DOI] [PubMed] [Google Scholar]
- 17.Olaharski AJ, et al. Chromosomal malsegregation and micronucleus induction in vitro by the DNA topoisomerase II inhibitor fisetin. Mutat. Res. 2005;582:79–86. doi: 10.1016/j.mrgentox.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Constantinou A, et al. Flavonoids as DNA topoisomerase antagonists and poisons: structure-activity relationships. J. Nat. Prod. 1995;58:217–225. doi: 10.1021/np50116a009. [DOI] [PubMed] [Google Scholar]
- 19.Khan N, et al. A novel dietary flavonoid fisetin inhibits androgen receptor signaling and tumor growth in athymic nude mice. Cancer Res. 2008;68:8555–8563. doi: 10.1158/0008-5472.CAN-08-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanda T, et al. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 21.Boutros M, et al. Analysis of cell-based RNAi screens. Genome Biol. 2006;7:R66. doi: 10.1186/gb-2006-7-7-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pouwels J, et al. Shugoshin 1 plays a central role in kinetochore assembly and is required for kinetochore targeting of Plk1. Cell Cycle. 2007;6:1579–1585. doi: 10.4161/cc.6.13.4442. [DOI] [PubMed] [Google Scholar]
- 23.Honda R, et al. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol. Biol. Cell. 2003;14:3325–3341. doi: 10.1091/mbc.E02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer TU, et al. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 25.Hsu JY, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 26.Zeitlin SG, et al. CENP-A is phosphorylated by Aurora B kinase and plays an unexpected role in completion of cytokinesis. J. Cell Biol. 2001;155:1147–1157. doi: 10.1083/jcb.200108125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ditchfield C, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter AO, et al. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene. 2000;19:4906–4916. doi: 10.1038/sj.onc.1203847. [DOI] [PubMed] [Google Scholar]
- 29.Syed DN, et al. Dietary agents for chemoprevention of prostate cancer. Cancer Lett. 2008;265:167–176. doi: 10.1016/j.canlet.2008.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giet R, et al. Drosophila aurora B kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 2001;152:669–682. doi: 10.1083/jcb.152.4.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girdler F, et al. Validating Aurora B as an anti-cancer drug target. J. Cell Sci. 2006;119:3664–3675. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 32.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrington EA, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat. Med. 2004;10:262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 34.Glover DM, et al. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 35.Katayama H, et al. Interaction and feedback regulation between STK15/BTAK/Aurora-A kinase and protein phosphatase 1 through mitotic cell division cycle. J. Biol. Chem. 2001;276:46219–46224. doi: 10.1074/jbc.M107540200. [DOI] [PubMed] [Google Scholar]
- 36.Fuller BG, et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature. 2008;453:1132–1136. doi: 10.1038/nature06923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roberge M, et al. The topoisomerase II inhibitor VM-26 induces marked changes in histone H1 kinase activity, histones H1 and H3 phosphorylation, and chromosome condensation in G2 phase and mitotic BHK cells. J. Cell Biol. 1990;111:1753–1762. doi: 10.1083/jcb.111.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mikhailov A, et al. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 2004;166:517–526. doi: 10.1083/jcb.200405167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel C, et al. The mitotic spindle checkpoint is a critical determinant for topoisomerase-based chemotherapy. J. Biol. Chem. 2005;280:4025–4028. doi: 10.1074/jbc.C400545200. [DOI] [PubMed] [Google Scholar]
- 40.Coelho PA, et al. Dual role of topoisomerase II in centromere resolution and aurora B activity. PLoS Biol. 2008;6:e207. doi: 10.1371/journal.pbio.0060207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haddad AQ, et al. Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines. Prostate Cancer Prostatic Dis. 2006;9:68–76. doi: 10.1038/sj.pcan.4500845. [DOI] [PubMed] [Google Scholar]
- 42.Strick R, et al. Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc. Natl Acad. Sci. USA. 2000;97:4790–4795. doi: 10.1073/pnas.070061297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schumacher JM, et al. A highly conserved centrosomal kinase, AIR-1, is required for accurate cell cycle progression and segregation of developmental factors in Caenorhabditis elegans embryos. Development. 1998;125:4391–4402. doi: 10.1242/dev.125.22.4391. [DOI] [PubMed] [Google Scholar]
- 44.Yao LJ, et al. Aurora-A is a critical regulator of microtubule assembly and nuclear activity in mouse oocytes, fertilized eggs, and early embryos. Biol. Reprod. 2004;70:1392–1399. doi: 10.1095/biolreprod.103.025155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}