

Abstract
The aim of this study was to elucidate the intestinal epithelial cell efflux transport processes that are involved in the intestinal transport of the H2 receptor antagonist nizatidine. The intestinal epithelial efflux transport mechanisms of nizatidine were investigated and characterized across Caco-2 cell monolayers, in the concentration range 0.05–10 mM in both apical–basolateral (AP–BL) and BL–AP directions, and the transport constants of P-glycoprotein (P-gp) efflux activity were calculated. The concentration-dependent effects of various P-gp (verapamil, quinidine, erythromycin, ketoconazole, and cyclosporine A), multidrug resistant-associated protein 2 (MRP2; MK-571, probenecid, indomethacin, and p-aminohipuric acid), and breast cancer resistance protein (BCRP; Fumitremorgin C) inhibitors on nizatidine bidirectional transport were examined. Nizatidine exhibited 7.7-fold higher BL–AP than AP–BL Caco-2 permeability, indicative of net mucosal secretion. All P-gp inhibitors investigated displayed concentration-dependent inhibition on nizatidine secretion in both directions. The IC50 of verapamil on nizatidine P-gp secretion was 1.2 × 10−2 mM. In the absence of inhibitors, nizatidine displayed concentration-dependent secretion, with one saturable (Jmax = 5.7 × 10−3 nmol∙cm−2∙s−1 and Km = 2.2 mM) and one nonsaturable component (Kd = 7 × 10−4 μL∙cm−2∙s−1). Under complete P-gp inhibition, nizatidine exhibited linear secretory flux, with a slope similar to the nonsaturable component. Vmax and Km estimated for nizatidine P-gp-mediated secretion were 4 × 10−3 nmol∙cm−2∙s−1 and 1.2 mM, respectively. No effect was obtained with the MRP2 or the BCRP inhibitors. Being a drug commonly used in pediatrics, adults, and elderly, nizatidine susceptibility to efflux transport by P-gp revealed in this paper may be of significance in its absorption, distribution, and clearance, as well as possible drug–drug interactions.
Key words: BCS class III drugs, caco-2 permeability, efflux transporters, intestinal absorption, nizatidine, P-glycoprotein
INTRODUCTION
The H2-receptor inhibitors (H2RI) have been used in the management of gastroesophageal reflux, prophylaxis, and treatment of peptic and duodenal ulcer and prevention of stress-induced gastric mucosal injury. Nizatidine is the newest member of this class of compounds. Nizatidine is a competitive, reversible, H2-receptor antagonist commonly used in the treatment of peptic ulcer and gastroesophageal reflux disease in adults, pediatrics (1), and elderly (2). Nizatidine is relatively as potent as ranitidine, more potent than cimetidine, and less potent than famotidine in inhibiting gastric acid secretion. However, all four H2RIs are equally effective when equipotent doses are used (3–5).
P-glycoprotein (P-gp) is a 170-kDa glycosylated transmembrane efflux pump, which was first characterized as the ATP-dependent transporter responsible for efflux of chemotherapeutic agents from multidrug resistant cancer cells. P-gp is widely expressed in many tissues, such as the membrane of endothelial cells in the intestine (6,7), liver (6), kidney (8), placenta (9), blood-brain barrier (10), and blood-testis barrier (11). P-gp is present on the villus tip of the apical brush border membrane of gut enterocytes and actively causes efflux of drugs from gut epithelial cells back into the intestinal lumen (7,12,13). Hence, P-gp may play a significant role in drug absorption, disposition, and excretion, as well as in drug–drug and drug–food interactions (12,14,15). Additional ATP-binding cassette efflux membrane transporters known to play a role in drug pharmacokinetics are the 190-kDa multidrug resistant-associated protein 2 (MRP2) and the breast cancer resistance protein (BCRP) (16).
Previous investigations into the intestinal transport of other H2RIs, i.e., ranitidine, famotidine, and cimetidine, have indicated the role of P-gp efflux in the intestinal absorption of these drugs (17–20). However, no reports are available as to whether or not nizatidine is subjected to efflux transport.
The purpose of this study was to investigate and characterize the possible role of efflux transporters in the intestinal absorption of the H2RI nizatidine. The bidirectional transport of nizatidine was evaluated across Caco-2 cell monolayers, in a range of concentrations, and in the presence versus absence of several different P-gp, MRP2, and BCRP inhibitors. This setup allowed us to characterize the inclusive role of intestinal epithelial efflux transport of nizatidine. The susceptibility of nizatidine to P-gp-mediated efflux transport revealed in this paper may be of significance in regard to the absorption, distribution, and clearance of the drug, as well as possible drug–drug and drug–food interactions.
MATERIALS AND METHODS
Materials
Nizatidine, verapamil, quinidine, erythromycin, ketoconazole, cyclosporine A, indomethacin, probenecid, fumitremorgin C (FTC), 2-(N-morpholino)ethanesulfonic acid (MES) buffer, glucose, CaCl2, MgCl2, and trifluoroacetic acid were purchased from Sigma Chemical Co. (St. Louis, MO, USA). MK-571 was purchased from Alexis Biochemicals (Lausen, Switzerland). Potassium chloride, p-aminohippuric acid, and NaCl were obtained from Fisher Scientific Inc. (Pittsburgh, PA, USA). Acetonitrile and water (Acros Organics, Geel, Belgium) were high-performance liquid chromatography (HPLC) grade. All other chemicals were of analytical reagent grade.
Cell Culture
Caco-2 cells (passages 22–27) from American Type Culture Collection (Rockville, MD, USA) were routinely maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 1% nonessential amino acids, 1 mM sodium pyruvate, and 1% l-glutamine. Cells were grown in an atmosphere of 5% CO2 and 90% relative humidity at 37°C.
Caco-2 cells were seeded on semipermeable filter inserts (12-well Transwell plate, Corning Costar Co., Cambridge, MA, USA). The cells on the insert were cultured for 21 days at 37°C in a humidified incubator containing 5% CO2 in air. The differentiation status of the formed monolayer was evaluated by measuring the transepithelial electrical resistance (TEER; Millicell-ERS epithelial voltohmmeter, Millipore Co., Bedford, MA, USA). Following the 21 days in cell culture, the monolayers developed TEER values above 300 Ωcm2.
Caco-2 Permeability Studies
Transcellular transport studies were performed in a method described previously with minor modifications (21). The uptake buffer contained 1 mM CaCl2, 0.5 mM MgCl2 6H2O, 145 mM NaCl, 3 mM KCl, 1 mM NaH2PO4, 5 mM d-glucose, and 5 mM MES. Similar pH was used in both apical (AP) and basolateral (BL) sides (pH 6.5) in order to maintain constant degree of ionization in both AP–BL and BL–AP direction experiments. This prevents possible influence of this factor on the permeability across the cells and ensures that any asymmetric transport obtained is due to efflux process (22). Briefly, cells were seeded onto collagen-coated membranes (0.4-μm pore size, 12 mm diameter, Costar, Cambridge, MA, USA) and were allowed to grow for 21 days. Mannitol permeability was assayed for each batch of Caco-2 monolayers (n = 3) and TEER measurements were performed on all monolayers. Monolayers with apparent [14C]mannitol permeability <3 × 10−7 cm/s and TEER values >300 Ωcm2 were used for the study. Of drug solution in the uptake buffer (pH 6.5), with or without inhibitor, 0.5 ml was added to the apical side of the monolayer in the AP–BL direction studies, and 1.5 ml of similar uptake buffer was added to the receiver compartment on the basolateral side of the monolayer. In the BL–AP direction studies, 1.5 ml of drug solution, with or without inhibitor, was added to the basolateral side of the monolayer, and 0.5 ml of blank buffer was added to the receiver compartment on the apical side of the monolayer. Samples were taken from the receiver side at various time points up to 120 min (100 μl from basolateral side or 50 μl from apical side), and similar volumes of blank buffer were added following each sample withdrawal. At the last time point (120 min), a sample was taken from the donor side as well, in order to confirm mass balance and to rule out metabolism of the drug throughout the experiment. Samples were immediately assayed for drug content. Caco-2 monolayers were checked for confluence by measuring the TEER before and after the transport study.
Determination of the IC50 of Verapamil on Nizatidine Transport
The concentration-dependent effects of a range of verapamil concentration (0.0001–5 mM) on the secretory (BL–AP) direction transport of 0.25 mM nizatidine were evaluated. The IC50 of verapamil on nizatidine transport was then determined by using the percentage inhibited for each verapamil concentration. The percentage inhibited in each concentration was calculated by dividing the Papp by the control apparent permeability value (0.25 mM nizatidine with no verapamil). Michaelis–Menten parameters and IC50 value were then determined using nonlinear regression according to the Hill equation with GraphPad Prism 4.01 (GraphPad Software Inc., San Diego, CA, USA).
Determination of the Apparent Maximum Velocity (Vmax) and the Michaelis–Menten Constant (Km) of Nizatidine P-gp Secretion
The concentration-dependent membrane transport rate of nizatidine in the secretory (BL–AP) direction was studied in the concentration range 0.1–10 mM, in the absence of inhibitors. This set of experiments was conducted under complete inhibition of the P-gp efflux as well. The conditions for complete P-gp inhibition were identified from the concentration-dependent inhibitory effect of verapamil on nizatidine BL–AP transport. The active P-gp BL–AP secretion was then plotted as described under “Data Analysis” section, and Michaelis–Menten parameters were determined using nonlinear regression with GraphPad Prism 4.01 (GraphPad Software Inc., San Diego, CA, USA).
Inhibition Experiments
The concentration-dependent effects of various P-gp, MRP2, and BCRP inhibitors on the bidirectional transport of nizatidine (0.25 mM) were examined. The P-gp inhibitors used were quinidine (0.2, 0.1, and 0.05 mM), erythromycin (0.2, 0.1, and 0.05 mM), ketoconazole (0.1, 0.05, and 0.01 mM), and cyclosporine A (10, 5, and 1 μM). The MRP2 inhibitors used were MK-571 (0.1, 0.05, and 0.01 mM), probenecid (0.2, 0.1, and 0.05 mM), indomethacin (0.2, 0.1, and 0.05 mM), and p-aminohippuric acid (10, 5, and 1 mM). The BCRP inhibitor used was fumitremorgin C (20, 10, and 5 μM). The results of these experiments were evaluated in comparison to the bidirectional transport of 0.25 mM nizatidine in the absence of inhibitors.
Validation of Efflux Transporters Expression by the Caco-2 Cells
The same batch of Caco-2 cells used in the studies presented in this paper was analyzed for P-gp, MRP2, and BCRP expression using Western blot analysis. Twenty-two days postseeding, cells were washed three times with ice-cold phosphate buffered saline, harvested, and Down’s homogenized in 50 mM Tris buffer containing 150 mM NaCl, pH 7.4, 1% sodium dodecyl sulfate, 1% Triton X-100, and protease inhibitor cocktail (ThermoFisher Scientific, Barrington, IL, USA). The homogenates were ultrasonicated on ice (3 × 10 s), centrifuged (10,000×g; 5 min), and the supernatant was carefully removed and used for protein content (BioRad DC protein assay, Bio-Rad Laboratories Inc., Hercules, CA, USA) and immunoblotting.
Immunoblot analysis of P-gp, MRP2, and BCRP was performed by a method described before with some modifications (19). From each sample, 100 μg protein were resolved in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Invitrogen Corporation, Carlsbad, CA, USA) followed by electrophoretic transfer onto Hybond ECL nitrocellulose membrane (Amersham, UK). Membranes were blocked overnight in Tris-saline buffer with Tween-20 (TSB-T) solution containing 3% bovine serum albumin at 4°C, followed by 1 h incubation with monoclonal anti-P-gp antibody (C219, 1:200 dilution in TSB-T, Zymed Laboratories Inc., San Francisco, CA, USA), monoclonal anti-MRP2 antibody (M2 III-6, 1:100 dilution in TSB-T, Alexis Biochemicals, Lausen, Switzerland), or monoclonal anti-BCRP antibody (BXP-21, 1:200 dilution in TSB-T, Alexis Biochemicals, Lausen, Switzerland). The secondary antibody was alkaline phosphatase conjugated goat antimouse IgG (1:3,000 dilution, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Blots were developed using ECF substrate (GE Healthcare, NJ) on Typhoon 9200 Variable Mode Imager (Molecular Dynamics, Amersham Pharmacia Biotech, Sweden).
Data Analysis
Permeability coefficient (Papp) across Caco-2 cell monolayers was calculated from the linear plot of drug accumulated in the receiver side versus time, using the following equation:
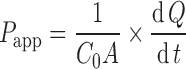 |
1 |
where dQ/dt is the steady-state appearance rate of the drug on the receiver (serosal in the case of AP–BL studies, or mucosal in the case of BL–AP studies) side, C0 is the initial concentration of the drug in the donor side, and A is the monolayer growth surface area (1.12 cm2). Linear regression was carried out to obtain the steady-state appearance rate of the drug on the receiver side (R2 > 0.99 in all experimental groups).
The efflux ratio, ER (i.e., the net efflux of nizatidine), was determined by calculating the ratio of Papp in the secretory (BL–AP) direction divided by the absorptive (AP–BL) Papp direction, according to the following equation:
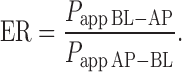 |
2 |
Kinetic constants (Jmax, Km, Kd) for nizatidine concentration-dependent secretory transport data were obtained by fitting a model incorporating saturable and nonsaturable components. The following model was utilized:
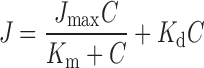 |
3 |
where Jmax is the maximal transport rate, Km is the kinetic constant for the saturable transport component, Kd is the kinetic constant for the nonsaturable transport component, and C is nizatidine concentration.
Delineation of the active P-gp-mediated component of nizatidine transport was determined by subtraction of the passive component from the total transport, in accordance with Gao et al. (21):
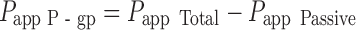 |
4 |
while the passive component was determined by complete inhibition of the active transport process. The conditions for complete inhibition of the transporter in turn were determined by assessing the concentration dependent inhibition of nizatidine secretion by the inhibitor (verapamil).
Analytical Methods
The amount of nizatidine in the Caco-2 medium was assayed using a HPLC system (Waters 2695 Separation Module) with a photodiode array UV detector (Waters 2996). Samples were filtered (Unifilter® 96 wells microplate 0.45 μm filters, Whatman Inc., Florham Park, NJ, USA), and medium aliquots of 40 μl were injected into the HPLC system. The HPLC conditions were as follows: XTerra, RP18, 3.5 μm, 4.6 × 100 mm column (Waters Co., Milford, MA, USA); a gradient mobile phase, going from 70:30% to 90:10% v/v aqueous/organic phase, respectively, over 10 min; the aqueous phase was 0.1% trifluoroacetic acid in water, and the organic phase was 0.1% trifluoroacetic acid in acetonitrile; and flow at a rate of 1 ml/min in room temperature. The detection wavelength was 230 nm, and the retention time was 5.5 min. Separate standard curves were used for each experiment (R2 > 0.999). The inter- and intraday coefficients of variation were <1.0% and 0.5%, respectively.
Statistical Analysis
All experiments were performed across three different unrelated transwells, i.e., n = 3, and the data presented as mean ± standard deviation (SD). To determine statistically significant differences among the experimental groups, the nonparametric Kruskal–Wallis test was used for multiple comparisons and the two-tailed nonparametric Mann–Whitney U test for two-group comparison when appropriate. For comparison of several groups against one control group, one-way analysis of variance followed by Dunnett’s test was performed. A p value of less than 0.05 was termed significant.
RESULTS
Nizatidine Transport Across Caco-2 Monolayers
The flux of nizatidine (0.1 mM) across Caco-2 cell monolayers in the absorptive (AP–BL) and in the secretory (BL–AP) directions and the corresponding Papp values, in the absence or presence of 0.1 mM verapamil, is shown in Fig. 1. It can be seen that nizatidine displayed a polarized transport, i.e., significantly higher Papp value in the BL–AP in comparison to the AP–BL direction, with an efflux ratio (ER; Papp BL–AP/Papp AP–BL) of 7.7. The mucosal secretion of nizatidine was significantly reduced in the presence of the P-gp inhibitor verapamil, in both AP–BL and BL–AP directions, decreasing the ER to 2 under these conditions. The Papp values for nizatidine transport in the absorptive (AP–BL) and in the secretory (BL–AP) directions over the concentration range 0.1–10 mM, as well as the corresponding ER, are summarized in Table I. Increased AP–BL and decreased BL–AP transport, accompanied by significant decrease in ER values with increasing donor drug concentrations, were obtained. Mass-balance analysis at the endpoint of the transport studies indicated no metabolism of nizatidine during the studies.
Fig. 1.

The flux of nizatidine (0.1 mM) across Caco-2 cell monolayers in the absorptive (AP–BL) and secretory (BL–AP) directions and the corresponding P app values, in the presence or absence of the P-gp inhibitor verapamil (0.1 mM). Data presented as mean ± SD; each experimental group represents studies across three transwells (n = 3); **p < 0.01
Table I.
The Concentration-Dependent Apparent Permeability (P app) of Nizatidine (0.1–10 mM) in the Absorptive (AP–BL) and Secretory (BL–AP) Directions and the Subsequent Efflux Ratio (P app BL–AP/P app AP–BL)
| Group | Donor concentration (mM) | AP–BL | BL–AP | ER |
|---|---|---|---|---|
| P app (×10−6 cm s−1) | ||||
| a | 0.1 | 0.27 ± 0.02 | 2.1 ± 0.2 | 7.7 ± 1.2 (f,g,h,i) |
| b | 0.25 | 0.35 ± 0.07 | 2.9 ± 0.2 | 8.2 ± 2.2 (f,g,h,i) |
| c | 0.5 | 0.36 ± 0.02 | 2.6 ± 0.2 | 7.2 ± 1.1 (f,g,h,i) |
| d | 0.75 | 0.43 ± 0.05 | 2.5 ± 0.3 | 5.8 ± 1.3 (g,h,i) |
| e | 1 | 0.43 ± 0.02 | 2.6 ± 0.2 | 6.0 ± 0.7 (g,h,i) |
| f | 2.5 | 0.52 ± 0.07 | 2.0 ± 0.3 | 3.9 ± 1.0 (a,b,c) |
| g | 5 | 0.66 ± 0.06 | 1.4 ± 0.1 | 2.1 ± 0.3 (a,b,c,d,e) |
| h | 7.5 | 0.82 ± 0.08 | 1.3 ± 0.2 | 1.6 ± 0.3 (a,b,c,d,e) |
| i | 10 | 1.11 ± 0.09 | 1.1 ± 0.1 | 1.0 ± 0.1 (a,b,c,d,e) |
Data presented as mean ± standard deviation; n = 3; letters represent significantly different groups
ER efflux ratio, BL–AP basolateral to apical, AP–BL apical to basolateral
Concentration-Dependent Inhibition of Nizatidine BL–AP Transport by Verapamil
The inhibition of nizatidine (0.25 mM) transport in the secretory (BL–AP) direction by various verapamil concentrations (0.0001–5 mM) across Caco-2 monolayers is shown in Fig. 2. Verapamil displayed a concentration-dependent inhibition on nizatidine secretory transport. The IC50 was determined to be 0.012 ± 0.004 mM. It can be seen that nizatidine BL–AP transport in the presence of 1 mM of verapamil was in the lower plateau region, indicating maximal inhibition of the P-gp secretion. Under these conditions, nizatidine secretory transport was 40% of the control (BL–AP transport of 0.25 mM nizatidine in the absence of verapamil), indicative of passively transported component.
Fig. 2.

Inhibition of nizatidine (0.25 mM) transport in the secretory (BL–AP) direction by various verapamil concentrations (0.0001–5 mM). Data presented as a percentage of the uninhibited control. Each data point represents the mean ± SD of studies across three transwells (n = 3). The IC50 value for verapamil on nizatidine transport was estimated by a nonlinear regression using GraphPad Prism 4.01
Concentration Dependence of Nizatidine BL–AP Transport Under No P-gp Inhibition and Under Complete P-gp Inhibition
The secretory (BL–AP) transport of various nizatidine concentrations (0.05–10 mM) under no P-gp inhibition and under complete P-gp inhibition is shown in Fig. 3. The conditions for complete P-gp inhibition (1 mM of verapamil) were identified from the lower plateau region of the concentration-dependent inhibitory effect of verapamil on nizatidine BL–AP transport (Fig. 2). Under no inhibition of P-gp, nizatidine displayed a concentration-dependent secretory transport, and the data were well described by a model consisting of one saturable and one nonsaturable component. The Jmax and Km estimated for nizatidine secretory transport were 0.0057 ± 0.001 nmol cm−2 s−1 and 2.2 ± 0.7 mM, respectively. The estimated coefficient of the nonsaturable secretory transport, Kd, was 0.0007 ± 0.0001 μL cm−2 s−1. Under complete P-gp inhibition, nizatidine displayed a nonsaturable linear secretory transport, characterized by a slope similar to the nonsaturable component obtained under no P-gp inhibition conditions. The active P-gp secretion in the BL–AP direction could then be determined by subtracting the flux rates obtained under complete P-gp inhibition from the flux rates obtained under no inhibition of P-gp (Fig. 4). Apparent Michaelis–Menten parameters estimated for nizatidine active secretion mediated by P-gp, Vmax and Km, were 0.004 ± 0.0002 nmol cm−2 s−1 and 1.2 ± 0.2 mM, respectively.
Fig. 3.

The concentration dependence of nizatidine (0.05–10 mM) secretory (BL–AP) transport across Caco-2 cell monolayers in the absence of inhibitors (closed circle) and under complete P-gp inhibition (open circle). The data in the absence of inhibitors were well described by a model consisting of one saturable and one nonsaturable component, as described under the “Data Analysis” section. The conditions for complete P-gp inhibition (1 mM of verapamil) were identified from the lower plateau region of the concentration-dependent inhibitory effect of verapamil on nizatidine BL–AP transport. Each data point represents the mean ± SD of studies across three transwells (n = 3)
Fig. 4.

The concentration dependency of nizatidine active transport mediated by P-gp in the secretory (BL–AP) direction across Caco-2 cell monolayers. The P-gp-mediated active transport rates were determined by subtracting the flux rate under complete P-gp inhibition from the flux rate under no inhibition (21). Apparent Michaelis–Menten parameters were estimated using GraphPad Prism 4.01. Data point are means ± SD of studies across three transwells (n = 3)
Nizatidine Transport Across Caco-2 Monolayers in the Presence of Various P-gp, MRP2, and BCRP Inhibitors
Nizatidine (0.25 mM) permeability across Caco-2 cell monolayers in the presence of different concentrations of various P-gp, MRP2, and BCRP inhibitors, in both AP–BL and BL–AP directions, is presented in Fig. 5. All P-gp inhibitors investigated, i.e., quinidine, erythromycin, ketoconazole, and cyclosporine A, displayed a concentration dependent inhibitory effect on nizatidine secretion, in both AP–BL and BL–AP directions in comparison to the control (nizatidine permeability without inhibitors). It can be seen that all of the investigated MRP2 inhibitors, i.e., MK-571, probenecid, indomethacin, and p-aminohippuric acid, as well as the BCRP inhibitor FTC, had no effect on nizatidine transport, neither in the absorptive nor in the secretory directions. The Caco-2 expression of P-gp, MRP2, and BCRP was validated using Western blot analysis (Fig. 6), confirming that these efflux transporters were indeed present in the cell culture experiments in the protein level.
Fig. 5.

The apparent permeability (P app) of nizatidine (0.25 mM) in the absorptive (AP–BL; black bar) and the secretory (BL–AP; gray bar) directions, in the presence of various concentrations of different P-gp, MRP2, and BCRP inhibitors. Data presented as mean ± SD; each experimental group represents studies across three transwells (n = 3); Asterisk significantly different from control
Fig. 6.

Analysis of P-gp, MRP2, and BCRP expression in the Caco-2 cells used in this paper by Western immunoblotting. P-gp was probed with the monoclonal antibody C219, MRP2 was probed with the monoclonal antibody M2 III-6, and BCRP was probed with the monoclonal antibody BXP-21
DISCUSSION
P-gp may play a significant role in drug absorption, disposition, and excretion, as well as in drug–drug and drug–food interactions. While other H2RIs, i.e., ranitidine, famotidine, and cimetidine, were reported to be P-gp substrates, no reports are available as to whether or not nizatidine is subjected to efflux transport. Hence, the aim of the current study was to elucidate the intestinal epithelial cell efflux transport processes that are important in the intestinal transport of nizatidine.
The results presented in this paper clearly indicate that P-gp is highly involved in the intestinal epithelium transport of nizatidine. Nizatidine displayed a polarized transport, i.e., significantly higher Papp value in the BL–AP in comparison to the AP–BL direction. Increased absorptive (AP–BL) and decreased secretory (BL–AP) permeability with increasing donor concentration were evident for nizatidine transport, accompanied by decreased ER from 7.7 to 1 (Table I), indicating saturation of an apical efflux transporter.
Under complete P-gp inhibition, nizatidine secretory transport was 40% of the control (BL–AP transport of 0.25 mM nizatidine in the absence of verapamil), indicative of passively transported component. This was further evident by nizatidine concentration-dependent secretory transport data. Nizatidine displayed a concentration dependent secretory transport, and the data were well described by a model consisting of one saturable and one nonsaturable component.
Several different inhibitors were used to investigate the membrane transport proteins involved in the intestinal efflux transport of nizatidine. All five P-gp inhibitors, i.e., verapamil, quinidine, erythromycin, ketoconazole, and cyclosporine A, displayed a concentration-dependent inhibition of nizatidine secretory transport, in both AP–BL and BL–AP directions. Nizatidine efflux inhibition by verapamil was in the range of its effect on other H2RIs; 100 μM of verapamil decreased nizatidine BL–AP transport by 50% (Fig. 1), similar to its effect on cimetidine and famotidine BL–AP transport measured in our lab (19). On the other hand, all of the investigated MRP2 inhibitors showed no effect on nizatidine transport in both directions, regardless of their concentration. MK-571 is a specific MRP2 inhibitor (23), and the fact that no effect on nizatidine transport was observed in concentrations as high as 0.1 mM suggests the lack of involvement of MRP2 in nizatidine intestinal transport. p-Aminohippuric acid is a well established MRP2 substrate (24) that again showed no effect on nizatidine permeability even in very high concentrations. Probenecid and indomethacin are both nonspecific inhibitors of MRP2; however, the lack of effect obtained by these compounds supports nizatidine insusceptibility to MRP2-mediated transport. It has been reported that probenecid and indomethacin do not affect the P-gp-mediated activity (25–27), which was also supported by the current study. Hence, it can be concluded that nizatidine is not subjected to intestinal efflux transport mediated by MRP2. The specific BCRP inhibitor fumitremorgin C (28) had no effect on nizatidine permeability, indicating that BCRP is not involved in the intestinal membrane transport of nizatidine. With regard to nizatidine uptake transport, it has been previously reported that the type I organic cation uptake system is involved in nizatidine transport into hepatocytes (29). Additionally, nizatidine oral bioavailability was significantly altered from an apple juice preparation (30). Hence, the exact identity and relative contribution of uptake transporters to nizatidine pharmacokinetics remains to be further investigated.
The data presented in this paper indicate that nizatidine is a low permeability compound. Even under complete P-gp saturation (as indicated by ER = 1), nizatidine AP–BL Papp was 1.1 × 10−6 cm/s. Nizatidine AP–BL permeability increased fourfold in comparison to the nonsaturated P-gp region; however, it was still a low permeability compound with approximately 1% of the total amount transported in the absorptive direction, regardless of the P-gp effect. This is in accordance with nizatidine physicochemical properties (i.e., degree of ionization in the physiological pH) and is similar to the other members of the H2RI class of drugs. Nizatidine’s highest single oral dose is 300 mg, and together with its water solubility (10–33 mg/ml (31)), a dose number (D0) of 0.12 is obtained by the following equation (32):
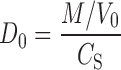 |
5 |
where CS is the solubility, M is the dose, and V0 is the volume of water taken with the dose, which is generally set to be 250 ml. A dose number equals or smaller than 1 indicates that the dissolution of the drug is likely to occur rapidly and that the drug is under high solubility definition. It can be concluded hence that according to the biopharmaceutics classification system (BCS) (32), nizatidine is a class III drug, i.e., high-solubility low-permeability compound. This classification is similar to the other members of the H2RI class of drugs (33,34).
The question as to whether or not a certain P-gp substrate is likely to show P-gp efflux-dependent in vivo intestinal absorption is still to be addressed. While several researchers have suggested a very limited influence of P-gp on in vivo intestinal absorption (35–40), other authors have found that P-gp substrates do show P-gp dependent intestinal permeability (41–43). Hence, the in vivo significance of the fact that nizatidine is a substrate for efflux by P-gp remains to be further investigated. However, being a BCS class III drug, i.e., high-solubility low-permeability compound, nizatidine is more susceptible to show P-gp dependent in vivo intestinal absorption. The intrinsic low gut wall permeability of this class of drugs essentially leads to limited amounts of drug inside the enterocyte, with potentially subsaturated P-gp levels (19). On the other hand, nizatidine was reported to have reasonable oral bioavailability (>70%), but it was significantly altered by apple juice (30). The regional differences in GIT P-gp expression levels may also play a role in the in vivo intestinal absorption of nizatidine. Significant differences in P-gp levels along the small intestine were previously reported. In general, P-gp protein expression follows a gradient pattern, increasing from the proximal regions to the distal small intestinal segments (19,44–48). This may lead to different efflux rates along the small intestinal segments, potentially resulting variable intestinal absorption of the drug. Segmental dependent intestinal absorption would also be particularly crucial throughout the development of controlled release products (49–52). In light of recent evaluation of the possibility to develop a controlled release nizatidine product (53), this factor must be taken into account. In addition to intestinal absorption, P-gp might play a role in other pharmacokinetic processes of nizatidine, especially in light of the fact that nizatidine major route of elimination is renal excretion (>90%) of the unchanged drug (>65%) (54).
CONCLUSIONS
In conclusion, it was found that the H2RI nizatidine is a P-gp substrate. P-gp alters nizatidine intestinal epithelial transport, with no involvement of MRP2 or BCRP. Being a drug commonly used in pediatric, adult, and elderly populations, its susceptibility to efflux transport revealed in this paper is of importance and may be of significance in regard to the absorption, distribution, and clearance of the drug, as well as possible drug–drug and drug–food interactions.
References
- 1.Orenstein S. R., Gremse D. A., Pantaleon C. D., Kling D. F., Rotenberg K. S. Nizatidine for the treatment of pediatric gastroesophageal reflux symptoms: An open-label, multiple-dose, randomized, multicenter clinical trial in 210 children. Clin. Ther. 2005;27:472. doi: 10.1016/j.clinthera.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Sasaki M., Sudoh T., Fujimura A. Pharmacokinetics of ranitidine and nizatidine in very elderly patients. Am. J. Ther. 2005;12:223–225. [PubMed] [Google Scholar]
- 3.Feldman M., Burton M. Histamine2-receptor antagonists. Standard therapy for acid-peptic diseases. 2. N. Engl. J. Med. 1990;323:1749–1755. doi: 10.1056/NEJM199012203232507. [DOI] [PubMed] [Google Scholar]
- 4.Feldman M., Burton M. Histamine2-receptor antagonists. Standard therapy for acid-peptic diseases. 1. N. Engl. J. Med. 1990;323:1672–1680. doi: 10.1056/NEJM199012133232405. [DOI] [PubMed] [Google Scholar]
- 5.Knadler M. P., Bergstrom R. F., Callaghan J. T., Rubin A. Nizatidine, an H2-blocker. Its metabolism and disposition in man. Drug Metab. Dispos. 1986;14:175–182. [PubMed] [Google Scholar]
- 6.Benet L. Z., Izumi T., Zhang Y., Silverman J. A., Wacher V. J. Intestinal MDR transport proteins and P-450 enzymes as barriers to oral drug delivery. J. Control Release. 1999;62:25. doi: 10.1016/S0168-3659(99)00034-6. [DOI] [PubMed] [Google Scholar]
- 7.Wacher V. J., Silverman J. A., Zhang Y., Benet L. Z. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J. Pharm. Sci. 1998;87:1322–1330. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 8.Garcia del Moral R., O’Valle F., Andujar M., Aguilar M., Lucena M. A., Lopez-Hidalgo J., Ramirez C., Medina-Cano M. T., Aguilar D., Gomez-Morales M. Relationship between P-glycoprotein expression and cyclosporin A in kidney. An immunohistological and cell culture study. Am. J. Pathol. 1995;146:398–408. [PMC free article] [PubMed] [Google Scholar]
- 9.Ceckova-Novotna M., Pavek P., Staud F. P-glycoprotein in the placenta: Expression, localization, regulation and function. Reprod. Toxicol. 2006;22:400. doi: 10.1016/j.reprotox.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Takano A., Kusuhara H., Suhara T., Ieiri I., Morimoto T., Lee Y. J., Maeda J., Ikoma Y., Ito H., Suzuki K., Sugiyama Y. Evaluation of in vivo P-glycoprotein function at the blood-brain barrier among MDR1 gene polymorphisms by using 11C-verapamil. J. Nucl. Med. 2006;47:1427–1433. [PubMed] [Google Scholar]
- 11.Fromm M. F. Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol. Sci. 2004;25:423. doi: 10.1016/j.tips.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Benet L. Z., Cummins C. L., Wu C. Y. Transporter-enzyme interactions: implications for predicting drug-drug interactions from in vitro data. Curr. Drug Metab. 2003;4:393–398. doi: 10.2174/1389200033489389. [DOI] [PubMed] [Google Scholar]
- 13.Benet L. Z., Cummins C. L., Wu C. Y. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int. J. Pharm. 2004;277:3. doi: 10.1016/j.ijpharm.2002.12.002. [DOI] [PubMed] [Google Scholar]
- 14.Dahan A., Altman H. Food-drug interaction: grapefruit juice augments drug bioavailability-mechanism, extent and relevance. Eur. J. Clin. Nutr. 2004;58:1–9. doi: 10.1038/sj.ejcn.1601736. [DOI] [PubMed] [Google Scholar]
- 15.Lin J. H. Drug-drug interaction mediated by inhibition and induction of P-glycoprotein. Adv. Drug Deliv. Rev. 2003;55:53. doi: 10.1016/S0169-409X(02)00171-0. [DOI] [PubMed] [Google Scholar]
- 16.Takano M., Yumoto R., Murakami T. Expression and function of efflux drug transporters in the intestine. Pharmacol. Ther. 2006;109:137. doi: 10.1016/j.pharmthera.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Bourdet D., Thakker D. Saturable absorptive transport of the hydrophilic organic cation ranitidine in Caco-2 cells: Role of pH-dependent organic cation uptake system and P-glycoprotein. Pharm. Res. 2006;23:1165. doi: 10.1007/s11095-006-0251-4. [DOI] [PubMed] [Google Scholar]
- 18.Collett A., Higgs N. B., Sims E., Rowland M., Warhurst A. G. Modulation of the permeability of H2 receptor antagonists cimetidine and ranitidine by P-glycoprotein in rat intestine and the human colonic cell line Caco-2. J. Pharmacol. Exp. Ther. 1999;288:171–178. [PubMed] [Google Scholar]
- 19.Dahan A., Amidon G. L. Segmental dependent transport of low permeability compounds along the small intestine due to P-glycoprotein: The role of efflux transport in the oral absorption of BCS class III drugs. Mol. Pharmaceutics. 2008;6:19–28. doi: 10.1021/mp800088f. [DOI] [PubMed] [Google Scholar]
- 20.Lee K., Ng C., Brouwer K. L. R., Thakker D. R. Secretory transport of ranitidine and famotidine across Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 2002;303:574–580. doi: 10.1124/jpet.102.038521. [DOI] [PubMed] [Google Scholar]
- 21.Gao J., Murase O., Schowen R. L., Aube J., Borchardt R. T. A functional assay for quantitation of the apparent affinities of ligands of P-glycoprotein in Caco-2 cells. Pharm. Res. 2001;18:171. doi: 10.1023/A:1011076217118. [DOI] [PubMed] [Google Scholar]
- 22.Dahan A., Amidon G. L. Grapefruit juice and its constituents augment colchicine intestinal absorption: Potential hazardous interaction and the role of P-glycoprotein. Pharm. Res. 2009;26:883–892. doi: 10.1007/s11095-008-9789-7. [DOI] [PubMed] [Google Scholar]
- 23.Chang J. H., Benet L. Z. Glucuronidation and the transport of the glucuronide metabolites in LLC-PK1 cells. Mol. Pharmaceutics. 2005;2:428–434. doi: 10.1021/mp050018m. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez-Ibanez M., Nalda-Molina R., Montalar-Montero M., Bermejo M. V., Merino V., Garrigues T. M. Transintestinal secretion of ciprofloxacin, grepafloxacin and sparfloxacin: in vitro and in situ inhibition studies. Eur. J. Pharm. Biopharm. 2003;55:241. doi: 10.1016/S0939-6411(02)00185-6. [DOI] [PubMed] [Google Scholar]
- 25.Draper M. P., Martell R. L., Levy S. B. Indomethacin-mediated reversal of multidrug resistance and drug efflux in human and murine cell lines overexpressing MRP, but not P-glycoprotein. Br. J. Cancer. 1997;75:810–815. doi: 10.1038/bjc.1997.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evers R., Zaman G., van Deemter L., Jansen H., Calafat J., Oomen L., Oude Elferink R., Borst P., Schinkel A. Basolateral localization and export activity of the human multidrug resistance-associated protein in polarized pig kidney cells. J. Clin. Invest. 1996;97:1211–1218. doi: 10.1172/JCI118535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petri N., Tannergren C., Rungstad D., Lennernas H. Transport characteristics of fexofenadine in the Caco-2 cell model. Pharm. Res. 2004;21:1398. doi: 10.1023/B:PHAM.0000036913.90332.b1. [DOI] [PubMed] [Google Scholar]
- 28.Ji Y., Morris M. E. Membrane transport of dietary phenethyl isothiocyanate by ABCG2 (breast cancer resistance protein) Mol. Pharmaceutics. 2005;2:414–419. doi: 10.1021/mp050029f. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura H., Sano H., Yamazaki M., Sugiyama Y. Carrier-mediated active transport of histamine H2 receptor antagonists, cimetidine and nizatidine, into isolated rat hepatocytes: contribution of type I system. J. Pharmacol. Exp. Ther. 1994;269:1220–1227. [PubMed] [Google Scholar]
- 30.Abdel-Rahman S. M., Johnson F. K., Gauthier-Dubois G., Weston I. E., Kearns G. L. The bioequivalence of nizatidine (Axid(R)) in two extemporaneously and one commercially prepared oral liquid formulations compared with capsule. J. Clin. Pharmacol. 2003;43:148–153. doi: 10.1177/0091270002239823. [DOI] [PubMed] [Google Scholar]
- 31.Howard P. H., Meylan W. M. Handbook of physical properties of organic chemicals. New York: CRC; 1996. [Google Scholar]
- 32.Amidon G. L., Lennernas H., Shah V. P., Crison J. R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995;12:413. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]
- 33.Kasim N. A., Whitehouse M., Ramachandran C., Bermejo M., Lennernas H., Hussain A. S., Junginger H. E., Stavchansky S. A., Midha K. K., Shah V. P., Amidon G. L. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharmaceutics. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 34.Takagi T., Ramachandran C., Bermejo M., Yamashita S., Yu L. X., Amidon G. L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharmaceutics. 2006;3:631–643. doi: 10.1021/mp0600182. [DOI] [PubMed] [Google Scholar]
- 35.Berggren S., Hoogstraate J., Fagerholm U., Lennernas H. Characterization of jejunal absorption and apical efflux of ropivacaine, lidocaine and bupivacaine in the rat using in situ and in vitro absorption models. Eur. J. Pharm. Sci. 2004;21:553. doi: 10.1016/j.ejps.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Chiou W., Chung S., Wu T., Ma C. A comprehensive account on the role of efflux transporters in the gastrointestinal absorption of 13 commonly used substrate drugs in humans. Int. J. Clin. Pharmacol. Ther. 2001;39:93–101. doi: 10.5414/cpp39093. [DOI] [PubMed] [Google Scholar]
- 37.Chiou W. L., Chung S. M., Wu T. C. Apparent lack of effect of P-glycoprotein on the gastrointestinal absorption of a substrate, tacrolimus, in normal mice. Pharm. Res. 2000;17:205. doi: 10.1023/A:1007573531947. [DOI] [PubMed] [Google Scholar]
- 38.Lee Y. J., Chung S. J., Shim C. K. Limited role of P-glycoprotein in the intestinal absorption of cyclosporin A. Biol. Pharm. Bull. 2005;28:760. doi: 10.1248/bpb.28.760. [DOI] [PubMed] [Google Scholar]
- 39.Lin J. How significant is the role of P-glycoprotein in drug absorption and brain uptake? Drugs Today. 2004;40:5–22. doi: 10.1358/dot.2004.40.1.799434. [DOI] [PubMed] [Google Scholar]
- 40.Toyobuku H., Tamai I., Ueno K., Tsuji A. Limited influence of P-glycoprotein on small-intestinal absorption of cilostazol, a high absorptive permeability drug. J. Pharm. Sci. 2003;92:2249–2259. doi: 10.1002/jps.10490. [DOI] [PubMed] [Google Scholar]
- 41.Stephens R. H., Tanianis-Hughes J., Higgs N. B., Humphrey M., Warhurst G. Region-dependent modulation of intestinal permeability by drug efflux transporters: In vitro studies in mdr1a(−/−) mouse intestine. J. Pharmacol. Exp. Ther. 2002;303:1095–1101. doi: 10.1124/jpet.102.041236. [DOI] [PubMed] [Google Scholar]
- 42.Tamura S., Ohike A., Ibuki R., Amidon G. L., Yamashita S. Tacrolimus is a class II low-solubility high-permeability drug: The effect of P-glycoprotein efflux on regional permeability of tacrolimus in rats. J. Pharm. Sci. 2002;91:719–729. doi: 10.1002/jps.10041. [DOI] [PubMed] [Google Scholar]
- 43.Terwogt J. M. M., Beijnen J. H., Huinink W. W. t. B., Rosing H., Schellens J. H. M. Co-administration of cyclosporin enables oral therapy with paclitaxel. Lancet. 1998;352:285. doi: 10.1016/S0140-6736(98)24030-X. [DOI] [PubMed] [Google Scholar]
- 44.Cao X., Yu L. X., Barbaciru C., Landowski C. P., Shin H. C., Gibbs S., Miller H. A., Amidon G. L., Sun D. Permeability dominates in vivo intestinal absorption of P-gp substrate with high solubility and high permeability. Mol. Pharmaceutics. 2005;2:329–340. doi: 10.1021/mp0499104. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez-Alvarez I., Fernandez-Teruel C., Casabo-Alos V. G., Garrigues T. M., Polli J. E., Ruiz-Garcia A., Bermejo M. In situ kinetic modelling of intestinal efflux in rats: functional characterization of segmental differences and correlation with in vitro results. Biopharm. Drug Dispos. 2007;28:229–239. doi: 10.1002/bdd.548. [DOI] [PubMed] [Google Scholar]
- 46.Landowski C. P., Sun D., Foster D. R., Menon S. S., Barnett J. L., Welage L. S., Ramachandran C., Amidon G. L. Gene expression in the human intestine and correlation with oral valacyclovir pharmacokinetic parameters. J. Pharmacol. Exp. Ther. 2003;306:778–786. doi: 10.1124/jpet.103.051011. [DOI] [PubMed] [Google Scholar]
- 47.Tam D., Tirona R. G., Pang K. S. Segmental intestinal transporters and metabolic enzymes on intestinal drug absorption. Drug Metab. Dispos. 2003;31:373–383. doi: 10.1124/dmd.31.4.373. [DOI] [PubMed] [Google Scholar]
- 48.Valenzuela B., Nacher A., Ruiz-Carretero P., Martin-Villodre A., Lopez-Carballo G., Barettino D. Profile of P-glycoprotein distribution in the rat and its possible influence on the salbutamol intestinal absorption process. J. Pharm. Sci. 2004;93:1641–1648. doi: 10.1002/jps.20071. [DOI] [PubMed] [Google Scholar]
- 49.Dahan A., Duvdevani R., Dvir E., Elmann A., Hoffman A. A novel mechanism for oral controlled release of drugs by continuous degradation of a phospholipid prodrug along the intestine: In-vivo and in-vitro evaluation of an indomethacin-lecithin conjugate. J. Control Release. 2007;119:86. doi: 10.1016/j.jconrel.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 50.Hoffman A. Pharmacodynamic aspects of sustained release preparations. Adv. Drug Deliv. Rev. 1998;33:185. doi: 10.1016/S0169-409X(98)00027-1. [DOI] [PubMed] [Google Scholar]
- 51.Lobenberg R., Kim J. S., Amidon G. L. Pharmacokinetics of an immediate release, a controlled release and a two pulse dosage form in dogs. Eur. J. Pharm. Biopharm. 2005;60:17. doi: 10.1016/j.ejpb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 52.Tubic M., Wagner D., Spahn-Langguth H., Weiler C., Wanitschke R., Bocher W. O., Langguth P. Effects of controlled-release on the pharmacokinetics and absorption characteristics of a compound undergoing intestinal efflux in humans. Eur. J. Pharm. Sci. 2006;29:231. doi: 10.1016/j.ejps.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Blum R. A., Braverman A. J., Rice P., Johnson F. K. Pharmacokinetics and pharmacodynamics of a novel nizatidine controlled-release formulation in healthy subjects. J. Clin. Pharmacol. 2003;43:74–83. doi: 10.1177/0091270002239709. [DOI] [PubMed] [Google Scholar]
- 54.Callaghan J. T., Bergstrom R. F., Rubin A., Chernish S., Crabtree R., Knadler M. P., Obermeyer B., Offen W. W., Schneck D. W., Aronoff G., Lasseter K. C. A pharmacokinetic profile of nizatidine in man. Scand. J. Gastroenterol. 1987;22:9–17. doi: 10.3109/00365528709094480. [DOI] [PubMed] [Google Scholar]