

Abstract
Modeling and simulation of oral drug absorption have been widely used in drug discovery, development, and regulation. Predictive absorption models are used to determine the rate and extent of oral drug absorption, facilitate lead drug candidate selection, establish formulation development strategy, and support the development of regulatory policies. This review highlights the development of recent drug absorption models including dispersion and compartmental models. The compartmental models include the compartmental absorption and transit model; Grass model; gastrointestinal transit absorption model; advanced compartmental absorption and transit model; and advanced dissolution, absorption, and metabolism model. Compared to the early absorption models, the above models developed or extended since the mid-1990s have demonstrated greatly improved predictive performance by accounting for multiple factors such as drug degradation, gastric emptying, intestinal transit, first-pass metabolism, and intestinal transport. For future model development, more heterogeneous features of the gastrointestinal tract (villous blood flow, metabolizing enzymes, and transporters), food effects, and drug–drug interactions should be fully characterized and taken into consideration. Moreover, predicting population inter- and intravariability in oral drug absorption can be useful and important for the evaluation of clinical safety and efficacy of drugs. Establishing databases and libraries that contain accurate pharmaceutical and pharmacokinetic information for commercialized and uncommercialized drugs may also be helpful for model development and validation.
Key words: advanced compartmental absorption and transit (ACAT) model; advanced dissolution, absorption, and metabolism (ADAM) model; compartmental model; dispersion model; oral drug absorption
INTRODUCTION
Oral drug absorption is a complex process. It consists of multiple steps that may include drug disintegration and dissolution, degradation, gastric emptying, intestinal transit, intestinal permeation and transport, intestinal metabolism, and hepatic metabolism. The factors that may have impact on the rate and extent of drug absorption are dosage form, physicochemical and biopharmaceutical properties of the active drug ingredient, and physiology of the gastrointestinal (GI) tract (1,2), as shown in Fig. 1. Knowledge of how these steps and factors influence absorption has fostered the development of predictive models for oral drug absorption. Currently, modeling and simulation of oral drug absorption have been widely used in drug discovery, development, and regulation. The predictive absorption models are used to determine the rate and extent of oral drug absorption, facilitate lead drug candidate selection, establish formulation development strategy, and support the development of regulatory policies.
Fig. 1.

Steps and related factors in oral drug absorption
Based on a previous review by Yu et al. (1), mechanistic approaches are classified into three categories: quasiequilibrium models, steady-state models, and dynamic models. The classification of these models is based upon their dependence on spatial and temporal variables. The quasiequilibrium models, which are independent of spatial and temporal variables, include the pH-partition hypothesis (3) and absorption potential concept (4,5). The steady-state models, which were independent of temporal variables, but dependent on spatial variables, include the film model (6), macroscopic mass balance approach (7,8), and microscopic balance approach (9,10). The steady-state models are limited to prediction of the extent but not the rate of oral drug absorption. The dynamic models consider spatial and temporal variables. As an improvement over the steady-state models, the dynamic models can predict both the rate and extent of oral drug absorption. The dynamic models include dispersion models (11) and compartmental models (12–14). Dispersion models portray the small intestine as a uniform tube with axial velocity, dispersion behavior, and concentration profile across the tube (11). Instead of treating the small intestine as one long cylindrical tube in a dispersion model, compartmental models assume the GI tract as one compartment or a series of compartments with linear transfer kinetics, and each compartment is well mixed with a uniform concentration (12–15). Both dispersion and compartmental models can be linked to pharmacokinetic models to predict plasma concentration-time profiles of drugs.
The article reviews and compares various mechanistic dynamic models developed and extended from the mid-1990s to the present, including the compartmental absorption and transit (CAT) model; Grass model; GI transit absorption (GITA) model; advanced compartmental absorption and transit (ACAT) model; and advanced dissolution, absorption, and metabolism (ADAM) model. In addition, this review explores future developments of oral drug absorption models with better predictability.
DISPERSION MODEL
The dispersion model defines the GI tract as a single tube with spatially varying properties (pH, surface area, etc.) along the tube. The dynamic drug absorption in the small intestine is based on the convection–dispersion equation, as shown below (1,11):
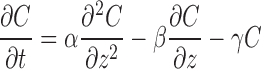 |
1 |
where C is the concentration of a drug in the GI tract, z is the axial distance from the stomach, α is the dispersion coefficient that accounts for mixing by both molecular diffusion and physiologic effect, β is the velocity in the axial direction, and γ is the drug absorption rate constant.
Willmann et al. (16) employed an intestinal transit function TSI (defined as the fraction of the drug dose in a certain GI segment at time t) in the dispersion model to describe the movement of the drugs though the intestine. By accounting for the effects of solubility and permeability on drug absorption, the dispersion model has successfully simulated fractions of dose absorbed for a variety of drugs that cover a wide range of absorption characteristics in rat (16), human (17), and monkey (18). For example, the model predicted the oral absorption of BCS Class I drugs (metoprolol, propranolol, levofloxacin, etc.), BCS Class II drugs (ciprofloxacin, diclofenac, carbamazepine, etc.), BCS Class III drugs (atenolol, nadolol, etc.), and BCS IV drugs (furosemide, etc.) (16–19). It was found that, for most passively absorbed drugs, the dispersion model predicted the extent of absorption very well, and the predicted values agreed with the literature (16–18).
However, due to the assumption of negligible first-pass, the original dispersion model may overestimate the absorptive fraction values for compounds such as benazepril, bromocriptine, and lovastatin, which undergo enzyme-mediated presystemic metabolism (18,20–22). In addition, the dispersion models only taking into account passive intestinal permeability may result in an overprediction of the fraction absorbed for P-gp substrates such as doxorubicin and ranitidine (23), and conversely, an underprediction for PEPT1 potential substrates such as valacyclovir (24,25), amoxicillin (26), cefalexin (27), and cefadroxil (27).
Therefore, additional inputs of Michaelis–Menten functions for metabolic enzymes and transporters are needed to overcome these limitations. Later, the dispersion model was used as a part of the PK-Sim® (http://www.pk-sim.com/) whole-body physiologically based pharmacokinetic model (28), which encompasses body and organ weight, blood flow, tissue composition, GI physiology, metabolism, active transport, and controlled release to simulate the GI transit and absorption process. Using in vitro dissolution data, PK-Sim® predicted the plasma concentration-time profiles of three cimetidine tablets with different formulation and dissolution profiles (29). In addition, PK-Sim® also predicted the plasma concentration-time profile of nifedipine, a CYP3A substrate with significant first-pass metabolism (30).
COMPARTMENTAL MODELS
CAT Model
The basic equation for the CAT model is described as follows (Eq. 2):
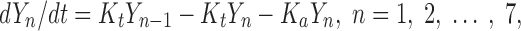 |
2 |
where Yn is the percent of dose at the nth compartment, n is the number of total compartments, Kt is the transit rate constant, and Ka is the absorption rate constant.
The original assumptions for this model include passive absorption, instantaneous dissolution, linear transfer kinetics for each segment, and minor absorption from the stomach and colon (1). This model was originally developed to predict oral drug absorption for nondegradable and highly soluble drugs. Nevertheless, this model was shown to capture the dependence of the fraction of dose absorbed on the effective permeability for various drugs with different absorption characteristics (31). The CAT model could also be linked directly to pharmacokinetic models to predict plasma concentration-time profiles.
By incorporating Michaelis–Menten kinetics for carrier/transporter-mediated absorption, gastric emptying rate constant, and compartment-dependent degradation rate constant into the model, the CAT model was extended for predicting dose-dependent drug absorption with degradation in the small intestine, such as for cefatrizine (32). Moreover, the CAT model was extended to simulate the fraction of dose absorbed in controlled release dosage forms by including a compartment that represents the controlled-release dosage form (1). By taking gastric emptying and dissolution into consideration, the CAT model was also used to predict the fraction of dose absorbed for poorly absorptive drugs such as digoxin, griseofulvin, and panadiplon, and to determine the cause of poor oral absorption (dissolution-, solubility-, or permeability-limited absorption) (33).
Based on the CAT model, a very similar approach was developed by Kortejarvi et al. (34) by considering the process of gastric emptying, drug dissolution, and drug intestinal transit within the GI tract. This model was constructed using Stella software for investigating the effects of different factors including formulation types, physiology of the GI tract, dissolution, absorption, and elimination on biowaiver criteria evaluation (34). The investigators concluded that, based on the simulation, about half of BCS I drugs have a higher risk to fail a bioequivalence study than BCS III drugs do. The above statement can be valid for some BCS I drugs with rapid absorption and elimination vs. BCS III drugs when excipients have no impact on GI transit time and permeability.
ACAT Model
The ACAT model (35) was developed based on the CAT model to include first-pass metabolism and colon absorption (Fig. 2). This model includes linear transfer kinetics and nonlinear metabolism/transport kinetics, six states of drug component (unreleased, undissolved, dissolved, degraded, metabolized, and absorbed), nine compartments (stomach, seven segments of small intestine, and colon), and three states of excreted material (unreleased, undissolved, and dissolved). It takes into consideration physicochemical factors (pKa, solubility, particle size, particle density, and permeability), physiological factors (gastric emptying, intestinal transit rate, first-pass metabolism, and luminal transport), and dosage factors (dosage form and dose) in predicting oral drug absorption.
Fig. 2.

The schematic diagram of the ACAT model developed by Agoram et al. (35). This recent version was provided by Dr. Michael Bolger
It should be noted that there are a variety of drug metabolizing enzymes and transporters localized in the intestinal epithelial cells (Fig. 3). The interaction of the metabolizing enzymes and transporters may have a complex impact on the oral absorption of their cosubstrates. The ACAT model was able to simulate nonlinear saturable Michaelis–Menten kinetics of metabolism and transport in oral drug absorption by using in vitro activity data (Vmax and Km) of enzymes and transporters (35).
Fig. 3.

The intestinal metabolizing enzymes and efflux/influx transporters. The metabolizing enzymes include phase I enzymes such as cytochrome P450s (CYPs) including CYP3A4/3A5, CYP2C9, CYP2C19, CYP2J2, CYP2D6, etc. (67), and phase II enzymes such as UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), and glutathione S-transferases (GSTs). The intestinal transporters are expressed in the intestinal basolateral membrane (MRP1, MRP3, MRP4, MRP5, MCT1, ENT1/2, OATP3A1/4A1, and OCT1/2. MRP, multidrug resistance-associated protein; MCT, monocarboxylic acid transporter; ENT, equilibrative nucleoside transporter; OATP, organic anion-transporting polypeptide transporter; OCT, organic cation transporter) and luminal membrane (MDR1, MRP2, BCRP, MRP4, PEPT1/2, MCT1, OATP1A2, OATP2B1, OCT3, ASBT, CNT1/2 and OCTN1/N2. MDR, multidrug resistance protein; BCRP, breast cancer resistance protein; PEPT, peptide transporter; ASBT, apical sodium-dependent bile acid transporter; CNT, concentrative nucleoside transporter; OCTN, carnitine/organic cation transporter) (19)
For example, the ACAT model successfully predicted oral absorption for drugs undergoing first-pass hepatic metabolism (propranolol), first-pass intestinal and hepatic metabolism (midazolam), efflux transport (digoxin), and first-pass metabolism plus efflux transport (saquinavir, Fig. 4) (35). Furthermore, this model demonstrated the potential to predict food–drug interactions (e.g., grapefruit juice with CYP3A substrates) and drug–drug interactions (e.g., rifampin with P-gp substrate digoxin) during oral drug absorption. However, some features that have an impact on drug absorption, such as local structure of gut enterocytes, cytoplasmic protein binding, segregation of blood flow to the intestine, and the heterogeneous expression and activities of drug metabolizing enzymes and transporters along the GI tract were not included in the original ACAT model (35).
Fig. 4.

Oral bioavailability of saquinavir with and without grape fruit juice simulated by using the ACAT model. Reprinted from Agoram et al. (35) with permission of Elsevier
The commercially available software, GastroPlus™, was developed based on the ACAT mode. This software has undergone several improvements with respect to the capability in predicting oral absorption of a variety of drugs in comparison to the original ACAT model. Gastroplus™ has been used to simulate the in vivo absorption profile of drugs by using in vitro dissolution data, for establishing the in vitro–in vivo correlation (36,37). With an integration of drug physicochemical properties and physiological parameters, Gastroplus™ has been used to aid in justifying biowaivers for selected BCS II compounds (38). Moreover, the impact of different formulation factors such as solubility, particle size, and size distribution on oral drug absorption were also predicted by GastroPlus™ (39–41). Combined with biorelevant solubility, the magnitude of food effects and the oral pharmacokinetics of different drugs under fasted and fed conditions were also predicted by Gastroplus™ (42). In addition to its use for predicting oral drug absorption in the GI tract, whole-body physiologically based pharmacokinetic (43) and combined pharmacokinetic and pharmacodynamic (44) models have been constructed within Gastroplus™ for predicting whole-body pharmacokinetic and pharmacodynamic characteristics in humans.
For drugs that undergo intestinal efflux, such as talinolol (a P-gp substrate), an additional consideration of a heterogeneous expression of P-gp across the intestine was included to simulate the nonlinear pharmacokinetics (dose-dependent absorption) after administration of a series of doses of talinolol immediate-release tablets (44), based on the fact that the P-gp expression level increases from duodenum to jejunum, to ileum, to cecum, and to colon (45).
Grass Model
A physiologically based multiple-compartmental model was developed by Grass (46) for predicting oral drug absorption. This model describes fluid movement (emptying and transit) in the GI tract and calculates the drug absorption (flux) in each compartment (stomach, duodenum, jejunum, ileum, and colon) over time based primarily on three parameters—solubility, permeability, and tissue surface area.
This model was shown to predict plasma concentration-time profiles including the AUC, Cmax, and Tmax for ketorolac (BCS I) and ganciclovir (BCS III) well. The software IDEA™ (47) was developed based upon compartmental models (IDEA™ is not currently available). By using IDEA™, the fractions of dose absorbed for atenolol (BCS III), naproxen (BCS II), and ganciclovir (BCS III) in animals and humans were successfully predicted (47). Similar to GastroPlus™, factors including dose, solubility, and permeability are considered. However, IDEA™ does not take first-pass metabolism and drug transport into account.
The main difference between early versions of GastroPlus™ and IDEA™ is that, for pure in silico prediction, Gastroplus™ requires the aid of QMPRPlus™ to estimate solubility, permeability, and lipophilicity based on the chemical structure input, but IDEA™ only requires chemical structure as input. Another difference is that IDEA™ directly uses permeability data from Caco-2 cells or rabbit intestinal tissue, whereas GastroPlus™ uses human permeability data transformed from in situ intestinal permeability data of rat or dog or Caco-2 data (48,49). A set of 28 drugs was selected for predicting their absorption classes and fractions absorbed by using both early versions of GastroPlus™ and IDEA™; both approaches showed similar predictive abilities (48). In the early discovery phase, IDEA™ is easy and convenient to use for predicting characteristics of drug oral absorption based on chemical structure. For late discovery and development, as well as for formulation evaluation, however, GastroPlus™ is more powerful for integrating data and takes into consideration factors such as first-pass metabolism, transport process, and dosage form.
GITA Model
Intestinal transit time could be variable across different segments of intestine and could impact drug absorption. In addition, the extent of drug absorption in each segment could differ due to heterogeneity in physiology and structures, including heterogeneous expression patterns of metabolizing enzymes and transporters.
Based on the above considerations, the GITA model was proposed (50). This compartmental model divides the GI tract into eight segments (stomach, duodenum, upper jejunum, lower jejunum, upper ileum, lower ileum, cecum, and large intestine), and GI transit and absorption processes in each segment are incorporated. In this model, the absorption rate constant for each segment was determined by a conventional in situ closed-loop method, and the GI transit rate constant for each segment was determined based on in vivo studies using phenol red, a nonabsorbable marker. The following equations (Eqs. 3 and 4) describe drug movement from a segment (i) to the next segment (i + 1) with segmental absorption (first-order absorption):
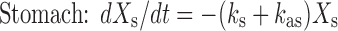 |
3 |
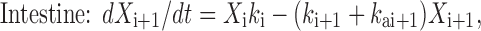 |
4 |
where at t = 0, Xs is the dose of the orally administered drug and X, k, and ka represent the amount, the transit rate constant, and the absorption rate constant, respectively. The subscripts s and i indicate stomach and each intestine site, respectively.
The GITA model was originally proposed to predict oral drug absorption in rats because it is possible and convenient to conduct the in situ closed-loop experiment in animals rather than in humans. Later, Kimura and Higaki (51) demonstrated an application of the GITA model to humans for prediction of oral absorption of theophylline. In this model, the transit rate constant in humans was measured via gamma scintigraphy, and the absorption rate constant in humans was estimated by normalizing the absorption rate constant in rats based on interspecies differences in surface area and luminal volume of the small intestine.
By accounting for the variations of drug absorption and GI transit among these segments, the GITA model was used to predict site-specific oral drug absorption (52). Moreover, this model was used to study the effects of food–drug interactions and drug–drug interactions on oral absorption (51). In the original GITA model, the plasma concentration-time profile was overpredicted for drugs undergoing first-pass metabolism, such as propranolol (50). Incorporating a first-pass effect into the GITA model successfully predicted the oral absorption profile of N-methyltyramine, a compound with first-pass metabolism (53). Furthermore, by incorporating the dissolution process, the GITA model predicted the plasma concentration-time profiles of theophylline (BCS I) and griseofulvin (BCS II) when administered as powders (54,55).
ADAM Model
Similar to the ACAT model, the ADAM model was recently developed based on the CAT mode, and it is a compartmental transit model consisting of seven compartments for the small intestine (56,57). The ADAM model considers the GI physiology including gastric emptying time, small intestinal transit time, and the radius and length of the small intestine, which were defined by the CAT model and literature values.
Similar to the ACAT model, the ADAM model accounts for the processes of dissolution, GI fluid transit, gut wall permeation, drug degradation, intestinal metabolism, and active transport (implicit consideration as stated by Jamei et al.) (56). In addition, the ADAM model also considers the heterogeneity of the GI tract such as heterogeneous distribution of enterocytic blood flow and enzymes in the gut wall. Food effects such as the impact of changes in gastric emptying, splanchnic blood flow, and luminal pH are also taken into consideration and simulated (56). Differing from other approaches using the Noyes–Whitney model (58), the ADAM model uses the generalized diffusion model developed by Wang and Flanagan (59) to describe dissolution under both sink and nonsink conditions.
The model is implemented in the simulation software Simcyp® (http://www.simCYP.com/). Simcyp® has successfully predicted the plasma concentration profiles for three modified release formulations (fast, moderate, and slow) of metoprolol (60). In addition, the midazolam first-pass intestinal extraction ratio and its interindividual variability were also predicted well by Simcyp®. The simulation also predicted the regional fractions of the dose absorbed and metabolized for midazolam, as well as the related interindividual variabilities in different segments of the small intestine (56).
CONCLUSIONS AND FUTURE DIRECTIONS
For the past decade, pharmaceutical scientists have developed and extended mechanistic absorption models to simulate the rate and extent of oral drug absorption. Compared to the early models, the recently developed or improved models have better predictability by accounting for multiple oral absorption factors such as dissolution, degradation, gastric emptying, intestinal transit, first-pass metabolism, and intestinal transport (Table I). In addition, recent models have assessed food effect, explored in vitro–in vivo correlation, and estimated drug–drug interactions.
Table I.
Comparisons of Different Mechanistic Approaches for Predicting Oral Drug Absorption
| Model | Classification | Rate-Limiting Step | Gastric Emptying and Intestinal Transit | Dissolution | Degradation | Passive Diffusion | First-Pass Metabolism | Active Transport | Reference |
|---|---|---|---|---|---|---|---|---|---|
| CAT | Dynamic/compartmental transit | Variable | √ | √ | √ | √ | × | √ | (1,32,33) |
| Grass | Dynamic/compartmental transit | Variable | √ | √a | × | √ | x | x | (46,47) |
| GITA | Dynamic/compartmental transit | Variable | √ | √ | × | √ | √ | × | (50,53–55) |
| ACAT | Dynamic/compartmental transit | Variable | √ | √ | √ | √ | √ | √ | (35) |
| ADAM | Dynamic/compartmental transit | Variable | √ | √ | √ | √ | √ | √b | (56) |
| Dispersion | Dynamic/dispersion | Variable | √ | √a | × | √ | × | × | (16–18) |
All the models listed above are original models as shown in the references.
aOnly solubility and dose are considered.
bImplicit consideration as stated by Jamei et al. (56).
To date, there is no “perfect” model to completely capture the complexity of the oral drug absorption process. In addition, proof of model utility is based on illustration with a small set of drugs or formulations. There are very few cross comparisons of various methods in predicting the same set of drugs and formulations. In general, most compartmental and dispersion models successfully predicted passive oral drug absorption of selected drugs and drug formulations. However, for drugs undergoing first-pass metabolism and transporter-mediated influx/efflux uptake, over- or underpredictions may occur depending on the mechanisms involved. Scientists in the area of drug metabolism and transport have developed several novel approaches, such as the intestinal epithelial cell model (61), which can be used to predict the impact of intestinal CYP3A and P-gp interaction on oral drug absorption, the segregated-flow model (62) and segmental segregated-flow model (63), which can be used to explain “route-dependent” metabolism by assuming only a partial intravenous dose reaches the enterocyte region. These models may complement the oral drug absorption models to improve the predictability of oral drug bioavailability.
Although in vitro and in vivo animal data are often used to inform the models, we need to be cautious since there are significant interspecies differences in expression levels and patterns of metabolizing enzymes between human and rat that may influence the prediction of oral bioavailability in humans (64). In addition, Caco-2 permeability data are suitable to simulate the extent of passive oral dug absorption, but may not be valid for active transport-mediated oral drug absorption since the gene expression of transporters in Caco-2 cells differ qualitatively and quantitatively from those in human intestinal mucosa (65). Furthermore, in the early phase of drug development, different enzyme sources (microsomes, supersomes, and hepatocytes), cell line sources (Caco-2 and MDCK cells), and assay conditions (pH, coenzymes, and ionic strengths) may bring variability into the estimates of Vmax and Km that are used in the model.
For future model development, more heterogeneous features of the GI tract should be characterized and considered, such as heterogeneous distribution of villous blood flow, local structure of gut enterocytes, enterocyte protein binding, and different expression patterns of metabolizing enzymes and drug transporters in the intestine. Other factors, such as food effects and drug–drug interactions, including effects of inducers or inhibitors of enzymes/transporters, may increase inter- and intraindividual variations in oral drug absorption. These factors should also be fully characterized and taken into consideration. Moreover, predicting population inter- and intravariability in oral drug absorption can be useful and important to evaluate clinical safety and efficacy of drugs.
An important aspect in the development of new models will be the acquisition and collection of a large volume of absorption data. By establishing databases and libraries that contain accurate pharmaceutical and pharmacokinetic information for commercialized and uncommercialized drugs, the e-ADME concept that calls for reducing animal and human testing and enhancing computer prediction for drug development and regulation (66) may be implemented.
Acknowledgments
The authors greatly thank Drs. Danny D. Shen and Wallace P. Adams for their reviews and editing of this manuscript.
Footnotes
Opinions expressed in this manuscript are those of Huang, Lee, and Yu and do not necessarily reflect the views or policies of the FDA.
References
- 1.Yu LX, Lipka E, Crison JR, Amidon GL. Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev. 1996;19:359–376. doi: 10.1016/0169-409X(96)00009-9. [DOI] [PubMed] [Google Scholar]
- 2.Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42:620–643. doi: 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- 3.Schanker LS. On the mechanism of absorption of drugs from the gastrointestinal tract. J Med Pharm Chem. 1960;2:343–359. doi: 10.1021/jm50011a001. [DOI] [PubMed] [Google Scholar]
- 4.Dressman JB, Amidon GL, Fleisher D. Absorption potential: estimating the fraction absorbed for orally administered compounds. J Pharm Sci. 1985;74:588–589. doi: 10.1002/jps.2600740523. [DOI] [PubMed] [Google Scholar]
- 5.Macheras PE, Symillides MY. Toward a quantitative approach for the prediction of the fraction of dose absorbed using the absorption potential concept. Biopharm Drug Dispos. 1989;10:43–53. doi: 10.1002/bdd.2510100106. [DOI] [PubMed] [Google Scholar]
- 6.Amidon GL, Sinko PJ, Fleisher D. Estimating human oral fraction dose absorbed: a correlation using rat intestinal membrane permeability for passive and carrier-mediated compounds. Pharm Res. 1988;5:651–654. doi: 10.1023/A:1015927004752. [DOI] [PubMed] [Google Scholar]
- 7.Sinko PJ, Leesman GD, Amidon GL. Predicting fraction dose absorbed in humans using a macroscopic mass balance approach. Pharm Res. 1991;8:979–988. doi: 10.1023/A:1015892621261. [DOI] [PubMed] [Google Scholar]
- 8.Sinko PJ, Leesman GD, Amidon GL. Mass balance approaches for estimating the intestinal absorption and metabolism of peptides and analogues: theoretical development and applications. Pharm Res. 1993;10:271–275. doi: 10.1023/A:1018999130076. [DOI] [PubMed] [Google Scholar]
- 9.Oh DM, Curl RL, Amidon GL. Estimating the fraction dose absorbed from suspensions of poorly soluble compounds in humans: a mathematical model. Pharm Res. 1993;10:264–270. doi: 10.1023/A:1018947113238. [DOI] [PubMed] [Google Scholar]
- 10.Lennernas H, et al. Regional jejunal perfusion, a new in vivo approach to study oral drug absorption in man. Pharm Res. 1992;9:1243–1251. doi: 10.1023/A:1015888813741. [DOI] [PubMed] [Google Scholar]
- 11.Ni PF, Ho NFH, Fox JF. Theoretical model studies of intestinal drug absorption v: nonsteady-state fluid flow and absorption. Int J Pharm. 1980;5:33–47. doi: 10.1016/0378-5173(80)90048-4. [DOI] [Google Scholar]
- 12.Goodacre BC, Murray PJ. A mathematical model of drug absorption. J Clin Hosp Pharm. 1981;6:117–133. doi: 10.1111/j.1365-2710.1981.tb00983.x. [DOI] [PubMed] [Google Scholar]
- 13.Dressman JB, Fleisher D, Amidon GL. Physicochemical model for dose-dependent drug absorption. J Pharm Sci. 1984;73:4–9. doi: 10.1002/jps.2600730922. [DOI] [PubMed] [Google Scholar]
- 14.Dressman JB, Fleisher D. Mixing-tank model for predicting dissolution rate control or oral absorption. J Pharm Sci. 1986;75:109–116. doi: 10.1002/jps.2600750202. [DOI] [PubMed] [Google Scholar]
- 15.Oberle RL, Amidon GL. The influence of variable gastric emptying and intestinal transit rates on the plasma level curve of cimetidine; an explanation for the double peak phenomenon. J Pharmacokinet Biopharm. 1987;15:529–544. doi: 10.1007/BF01061761. [DOI] [PubMed] [Google Scholar]
- 16.Willmann S, Schmitt W, Keldenich J, Dressman JB. A physiologic model for simulating gastrointestinal flow and drug absorption in rats. Pharm Res. 2003;20:1766–1771. doi: 10.1023/B:PHAM.0000003373.72652.c0. [DOI] [PubMed] [Google Scholar]
- 17.Willmann S, et al. A physiological model for the estimation of the fraction dose absorbed in humans. J Med Chem. 2004;47:4022–4031. doi: 10.1021/jm030999b. [DOI] [PubMed] [Google Scholar]
- 18.Willmann S, Edginton AN, Dressman JB. Development and validation of a physiology-based model for the prediction of oral absorption in monkeys. Pharm Res. 2007;24:1275–1282. doi: 10.1007/s11095-007-9247-y. [DOI] [PubMed] [Google Scholar]
- 19.Custodio JM, Wu CY, Benet LZ. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Adv Drug Deliv Rev. 2008;60:717–733. doi: 10.1016/j.addr.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Webb R, Miller D, Traina V, Gomez H. Benazepril. Cardiovasc Drug Rev. 1990;8:89–104. doi: 10.1111/j.1527-3466.1990.tb00432.x. [DOI] [Google Scholar]
- 21.Schran HF, Tse FL, Bhuta SI. Pharmacokinetics and pharmacodynamics of bromocriptine in the rat. Biopharm Drug Dispos. 1985;6:301–311. doi: 10.1002/bdd.2510060305. [DOI] [PubMed] [Google Scholar]
- 22.Kivisto KT, Kantola T, Neuvonen PJ. Different effects of itraconazole on the pharmacokinetics of fluvastatin and lovastatin. Br J Clin Pharmacol. 1998;46:49–53. doi: 10.1046/j.1365-2125.1998.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm Res. 2003;20:1210–1224. doi: 10.1023/A:1025001131513. [DOI] [PubMed] [Google Scholar]
- 24.Sinko PJ, Balimane PV. Carrier-mediated intestinal absorption of valacyclovir, the L-valyl ester prodrug of acyclovir: 1. Interactions with peptides, organic anions and organic cations in rats. Biopharm Drug Dispos. 1998;19:209–217. doi: 10.1002/(SICI)1099-081X(199805)19:4<209::AID-BDD93>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 25.Han H, et al. 5′-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm Res. 1998;15:1154–1159. doi: 10.1023/A:1011919319810. [DOI] [PubMed] [Google Scholar]
- 26.Bretschneider B, Brandsch M, Neubert R. Intestinal transport of beta-lactam antibiotics: analysis of the affinity at the H+/peptide symporter (PEPT1), the uptake into Caco-2 cell monolayers and the transepithelial flux. Pharm Res. 1999;16:55–61. doi: 10.1023/A:1018814627484. [DOI] [PubMed] [Google Scholar]
- 27.Ganapathy ME, et al. Differential recognition of beta -lactam antibiotics by intestinal and renal peptide transporters, PEPT 1 and PEPT 2. J Biol Chem. 1995;270:25672–25677. doi: 10.1074/jbc.270.43.25672. [DOI] [PubMed] [Google Scholar]
- 28.Willmann S, Lippert J, Sevestre M. PK-Sim®: a physiologically based pharmacokinetic ‘whole-body’ model. Biosilico. 2003;1:121–124. doi: 10.1016/S1478-5382(03)02342-4. [DOI] [Google Scholar]
- 29.Kleine-Besten M, Willmann S, Eddington AN. The prediction of cimetidine absorption profiles with PK-Sim® using in vitro dissolution data. German Chapter Annual Meeting of the Controlled Release Society February: 23–24 (2006).
- 30.Blank K, Becker C, Jantratid E. PBPK modeling to demonstrate the impact of variability in CYP3A-mediated metabolism of nifedipine in different subpopulations. The 6th World Meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology April: 7–10 (2008).
- 31.Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186:119–125. doi: 10.1016/S0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 32.Yu LX, Amidon GL. Saturable small intestinal drug absorption in humans: modeling and interpretation of cefatrizine data. Eur J Pharm Biopharm. 1998;45:199–203. doi: 10.1016/S0939-6411(97)00088-X. [DOI] [PubMed] [Google Scholar]
- 33.Yu LX. An integrated model for determining causes of poor oral drug absorption. Pharm Res. 1999;16:1883–1887. doi: 10.1023/A:1018911728161. [DOI] [PubMed] [Google Scholar]
- 34.Kortejarvi H, Urtti A, Yliperttula M. Pharmacokinetic simulation of biowaiver criteria: the effects of gastric emptying, dissolution, absorption and elimination rates. Eur J Pharm Sci. 2007;30:155–166. doi: 10.1016/j.ejps.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 35.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 36.Lobenberg R, et al. Dissolution testing as a prognostic tool for oral drug absorption: dissolution behavior of glibenclamide. Pharm Res. 2000;17:439–444. doi: 10.1023/A:1007529020774. [DOI] [PubMed] [Google Scholar]
- 37.Okumu A, Dimaso M, Lobenberg R. Dynamic dissolution testing to establish in vitro/in vivo correlations for montelukast sodium, a poorly soluble drug. Pharm Res. 2008;25:2778–2785. doi: 10.1007/s11095-008-9642-z. [DOI] [PubMed] [Google Scholar]
- 38.Tubic-Grozdanis M, Bolger MB, Langguth P. Application of gastrointestinal simulation for extensions for biowaivers of highly permeable compounds. AAPS J. 2008;10:213–226. doi: 10.1208/s12248-008-9023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dannenfelser RM, et al. Development of clinical dosage forms for a poorly water soluble drug I: application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J Pharm Sci. 2004;93:1165–1175. doi: 10.1002/jps.20044. [DOI] [PubMed] [Google Scholar]
- 40.Kuentz M, Nick S, Parrott N, Rothlisberger D. A strategy for preclinical formulation development using Gastroplus as pharmacokinetic simulation tool and a statistical screening design applied to a dog study. Eur J Pharm Sci. 2006;27:91–99. doi: 10.1016/j.ejps.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Wei H, et al. Physicochemical characterization of five glyburide powders: a BCS based approach to predict oral absorption. Eur J Pharm Biopharm. 2008;69:1046–1056. doi: 10.1016/j.ejpb.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 42.Jones HM, Parrott N, Ohlenbusch G, Lave T. Predicting pharmacokinetic food effects using biorelevant solubility media and physiologically based modelling. Clin Pharmacokinet. 2006;45:1213–1226. doi: 10.2165/00003088-200645120-00006. [DOI] [PubMed] [Google Scholar]
- 43.De Buck SS, et al. Prediction of human pharmacokinetics using physiologically based modeling: a retrospective analysis of 26 clinically tested drugs. Drug Metab Dispos. 2007;35:1766–1780. doi: 10.1124/dmd.107.015644. [DOI] [PubMed] [Google Scholar]
- 44.Tubic M, et al. In silico modeling of non-linear drug absorption for the P-gp substrate talinolol and of consequences for the resulting pharmacodynamic effect. Pharm Res. 2006;23:1712–1720. doi: 10.1007/s11095-006-9020-7. [DOI] [PubMed] [Google Scholar]
- 45.Mouly S, Paine MF. P-glycoprotein increases from proximal to distal regions of human small intestine. Pharm Res. 2003;20:1595–1599. doi: 10.1023/A:1026183200740. [DOI] [PubMed] [Google Scholar]
- 46.Grass GM. Simulation models to predict oral drug absorption from in vitro data. Adv Drug Deliv Rev. 1997;23:199–219. doi: 10.1016/S0169-409X(96)00436-X. [DOI] [Google Scholar]
- 47.Norris DA, Leesman GD, Sinko PJ, Grass GM. Development of predictive pharmacokinetic simulation models for drug discovery. J Control Release. 2000;65:55–62. doi: 10.1016/S0168-3659(99)00232-1. [DOI] [PubMed] [Google Scholar]
- 48.Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of Gastroplus and IDEA. Eur J Pharm Sci. 2002;17:51–61. doi: 10.1016/S0928-0987(02)00132-X. [DOI] [PubMed] [Google Scholar]
- 49.Bohets H, et al. Strategies for absorption screening in drug discovery and development. Curr Top Med Chem. 2001;1:367–383. doi: 10.2174/1568026013394886. [DOI] [PubMed] [Google Scholar]
- 50.Sawamoto T, et al. Prediction of the plasma concentration profiles of orally administered drugs in rats on the basis of gastrointestinal transit kinetics and absorbability. J Pharm Pharmacol. 1997;49:450–457. doi: 10.1111/j.2042-7158.1997.tb06823.x. [DOI] [PubMed] [Google Scholar]
- 51.Kimura T, Higaki K. Gastrointestinal transit and drug absorption. Biol Pharm Bull. 2002;25:149–164. doi: 10.1248/bpb.25.149. [DOI] [PubMed] [Google Scholar]
- 52.Yokoe J, et al. Analysis and prediction of absorption behavior of colon-targeted prodrug in rats by GI-transit-absorption model. J Control Release. 2003;86:305–313. doi: 10.1016/S0168-3659(02)00424-8. [DOI] [PubMed] [Google Scholar]
- 53.Kimura T, et al. Analysis and prediction of absorption profile including hepatic first-pass metabolism of n-methyltyramine, a potent stimulant of gastrin release present in beer, after oral ingestion in rats by gastrointestinal-transit-absorption model. Drug Metab Dispos. 2000;28:577–581. [PubMed] [Google Scholar]
- 54.Kadono K, et al. Analysis and prediction of absorption behavior for theophylline orally administered as powders based on gastrointestinal-transit-absorption (GITA) model. Drug Metab Pharmacokinet. 2002;17:307–315. doi: 10.2133/dmpk.17.307. [DOI] [PubMed] [Google Scholar]
- 55.Fujioka Y, et al. Prediction of oral absorption of griseofulvin, a BCS class II drug, based on GITA model: utilization of a more suitable medium for in-vitro dissolution study. J Control Release. 2007;119:222–228. doi: 10.1016/j.jconrel.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Jamei M, Yang J, T. D. A novel physiologically-based mechanistic model for predicting oral drug absorption: the advanced dissolution, absorption, and metabolism (ADAM) model. 4th World Conference on Drug Absorption, Transport and Delivery (WCDATD) June: 20–22 (2007).
- 57.Dokoumetzidis A, Kalantzi L, Fotaki N. Predictive models for oral drug absorption: from in silico methods to integrated dynamical models. Expert Opin Drug Metab Toxicol. 2007;3:491–505. doi: 10.1517/17425255.3.4.491. [DOI] [PubMed] [Google Scholar]
- 58.Horter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46:75–87. doi: 10.1016/S0169-409X(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 59.Wang J, Flanagan DR. General solution for diffusion-controlled dissolution of spherical particles. 1. Theory. J Pharm Sci. 1999;88:731–738. doi: 10.1021/js980236p. [DOI] [PubMed] [Google Scholar]
- 60.Polak S, Jamei M, Turner D. Prediction of the in vivo behaviour of modified release formulations of metoprolol from in vitro dissolution profiles using the ADAM model (Simcyp®v8). 10th European ISSX Meeting May: 18–21 (2008).
- 61.Ito K, Kusuhara H, Sugiyama Y. Effects of intestinal CYP3A4 and P-glycoprotein on oral drug absorption—theoretical approach. Pharm Res. 1999;16:225–231. doi: 10.1023/A:1018872207437. [DOI] [PubMed] [Google Scholar]
- 62.Cong D, Doherty M, Pang KS. A new physiologically based, segregated-flow model to explain route-dependent intestinal metabolism. Drug Metab Dispos. 2000;28:224–235. [PubMed] [Google Scholar]
- 63.Tam D, Tirona RG, Pang KS. Segmental intestinal transporters and metabolic enzymes on intestinal drug absorption. Drug Metab Dispos. 2003;31:373–383. doi: 10.1124/dmd.31.4.373. [DOI] [PubMed] [Google Scholar]
- 64.Cao X, et al. Why is it challenging to predict intestinal drug absorption and oral bioavailability in human using rat model. Pharm Res. 2006;23:1675–1686. doi: 10.1007/s11095-006-9041-2. [DOI] [PubMed] [Google Scholar]
- 65.Sun D, et al. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm Res. 2002;19:1400–1416. doi: 10.1023/A:1020483911355. [DOI] [PubMed] [Google Scholar]
- 66.Yu L. E-adme: predicting bioavailability and permeability. Gordon Research Conference Drug Metabolism. 2001, July.
- 67.Paine MF, et al. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]