

Abstract
Recently, drug transporters have emerged as significant modifiers of a patient’s pharmacokinetics. In cases where the functioning of drug transporters is altered, such as by drug-drug interactions, by genetic polymorphisms, or as evidenced in knockout animals, the resulting change in volume of distribution can lead to a significant change in drug effect or likelihood of toxicity, as well as a change in half life independent of a change in clearance. Here, we review pharmacokinetic interactions at the transporter level that have been investigated in animals and humans and reported in literature, with a focus on the changes in distribution volume. We pay particular attention to the differing effects of changes in transporter function on the three measures of volume. Further, trends are discussed as they may be used to predict volume changes given the function of a transporter and the primary location of the interaction. Because the liver and kidneys express the greatest level and variety of transporters, we denote these organs as the primary location of transporter-based interactions. We conclude that the liver is a larger contributor to distribution volume than the kidneys, in consideration of both uptake and efflux transporters. Further, while altered distribution due to secondary interactions at tissues other than the liver and kidneys may have a pharmacodynamic effect, these interactions, at least at the blood-brain barrier, do not appear to significantly influence overall distribution volume. The analysis provides a framework for understanding potential pharmacokinetic interactions rooted in drug transporters as they modify drug distribution.
Key words: distribution volume, drug-drug interaction, drug transporters, pharmacokinetics, pharmacogenomics
INTRODUCTION
The pharmacokinetic parameter volume of distribution describes the relationship between the measured systemic concentrations and amount of drug in the body. It is a measure of the extent of tissue distribution, and it usually does not represent any physiological volume. Instead, it is considered a theoretical parameter that is dependent on a variety of drug properties: traditionally, lipophilicity (a measure of tissue affinity), plasma protein binding (1), and tissue components, including proteins. However, with the recent advances in the understanding of the importance of active drug transporters in pharmacokinetics, we sought to understand how transporter activity affects volume of distribution.
Drug transporters are found at numerous tissues in the body, implicating them as players in drug distribution. While a variety of transporters, including P-glycoprotein (P-gp), BCRP, and some members of the OATP family, are heavily expressed at the intestinal epithelium, they should not affect volume of distribution, as volume terms are related to the behavior of the drug once it has entered the systemic circulation. Within the body, the liver and kidneys express the greatest variety and level of drug transporters. At these two organs, transporters modulate access to metabolizing enzymes and excretion processes, both biliary and renal. Consequently, they are likely to also have an effect on other pharmacokinetic parameters, particularly clearance and half life. The majority of published reports, therefore, focus on primary transporter interactions at either the liver or the kidneys.
In those cases where the functioning of drug transporters is altered, such as by a drug-drug interaction or by a genetic polymorphism in the transporter gene or relevant genetic control elements, the resulting change in volume of distribution can lead to a significant change in drug effect or likelihood of toxicity, as well as a change in half life independent of a change in clearance. Generally, because many transporters have a wide range of substrates, drug-drug interactions in this consideration are rooted in the inhibition of a transporter, leading to decreased functionality. Similarly, most polymorphisms will result in a reduced function transporter. Thus, here we primarily examine decreased transporter function and its effects on the distribution volume. It is possible, however, for an increase in transporter function to occur either by up-regulation of the transporter gene under multiple dosing conditions, or by a theoretical polymorphism that creates a transport protein with increased effectiveness or an increased amount of protein. Such transporter induction interactions have been reported to affect bioavailability in the gut; however, these should not affect distribution volume. While induction interactions have been reported at other tissues, including the liver and kidney, in vitro (2-4), clinical, pharmacokinetic interactions with increased functioning transporters have not yet been reported outside the gut.
The relationship between systemic concentrations and amount of drug in the body can differ depending on the dosing regimen and time at which the necessary parameters are measured. In particular, V1, the initial dilution volume, defined as the dose divided by the initial plasma concentration following an intravenous bolus dose, is likely to be relatively small, because equilibration to the other tissue spaces has not yet occurred. The volume at the terminal phase of elimination, Varea (also known as Vz), will be greater. Most commonly, this represents the phase when distribution is complete and elimination from the plasma is predominant, so drug is re-entering the circulation from the tissue spaces. Varea is defined as the clearance divided by the terminal rate of elimination, and is therefore heavily dependent on the terminal rate of elimination. This rate is often a more difficult parameter to estimate experimentally because it requires concentration data for a long time period following the dose, when concentrations may begin to fall below the limits of sensitivity for some analytical methodology. Finally, the volume at steady state, Vss, is the sum of the distribution volumes of all the compartments in a pharmacokinetic model. It can also be calculated from a single dose as the product of clearance and the mean residence time in the body following non-compartmental analysis. Its value will be between V1 and Varea, and Vss is considered to be a more “accurate” measure of whole body distribution volume, as it is less directly dependent on changes in the elimination processes, a characteristic of Varea.
It is also important to note that calculations of volume are highly model-dependent. The three volume parameters as defined above assume elimination from the central compartment of a pharmacokinetic model (5). The typical compartmental model assumes the liver and kidneys are part of the central compartment, as they are highly perfused organs and assumed to be in rapid equilibrium with the plasma, thus providing for central compartment elimination. However, when elimination does not occur from the central compartment, these three measures will significantly under-predict distribution volume (6, 7). Therefore, it may be important to consider a possible case where transporter dysfunction means the liver or kidneys are not in rapid equilibrium with the plasma, such that elimination occurs from a peripheral compartment. We will return to this topic in the discussion.
Drug transporters can be loosely characterized as either uptake or efflux, denoting whether they facilitate drug entry into a cell or efflux out of a cell. Thus, an uptake transporter with reduced function prevents drug accumulation in the tissue expressing the transporter, while an efflux transporter with decreased function increases accumulation in the tissue expressing the transporter. The effect on total distribution volume depends on the tissue expressing the transporter, whether it is an uptake or efflux transporter, and where the transporter is expressed in this tissue.
Therefore, the objective of this work was to collect and analyze published reports evidencing changes to distribution volume in animals and humans due to drug-drug interactions, genetic polymorphisms, or knockout animals, to determine what conclusions could be drawn.
MATERIALS AND METHODS
Literature searches revealed a number of interactions at the transporter level that have been investigated and reported. A summary of thirty-seven such interactions, including the effects on the primary pharmacokinetic parameters, is presented in Table I. These thirty-seven interactions are all those available where a volume parameter was presented, or where a volume parameter could be calculated from the reported parameters: Where provided, the values in Table I reflect those in the reference article. Otherwise, the missing values were calculated: most commonly, assuming the provided half life is the terminal half life, Varea was calculated from Equation 1. When volume but not half life was provided, a half life was calculated also using Equation 1 under the assumption of a one compartment model. Finally, when clearance and mean residence time were provided, Vss was calculated by the definition of Vss as the product of clearance and mean residence time. Calculated values are indicated by an asterisk throughout this report.
Table I.
Summary of transporter-based interactions and their effect on volume, clearance, and half life
| Drug | Interaction | V | CL | t1/2 | Mechanism of clearance in reference organism | Related Transporter Comments | Reference |
|---|---|---|---|---|---|---|---|
| 1. Adriamycin | Verapamil | ↑ 31.2% (Vss) | ↓ 32.6% | ↑ 37.7% | Metabolism > Biliary > Renal | Inhibition of P-gp efflux in humans after iv dosing; in rats, renal transport is saturable but biliary is not, indicating a transporter is involved in renal elimination | (8, 9) |
| 2. Atorvastatin | Rifampin | ↓ 94.3% (Vss/F) | ↓ 87.0% (CL/F) | ↓ 63.1% | Metabolism | Inhibition of OATP1B1 uptake into the liver in humans after oral dosing | (10) |
| 3. Atorvastatin | OATP1B1 reduced function allele | ↓ 57.6%* (Varea/F) | ↓ 59.2%* (CL/F) | ↔ | Metabolism | Reduced OATP1B1 uptake into the liver in humans after oral dosing | (11) |
| 4. Cefazolin | Probenecid | ↔ (V1) | ↓ 32.1% | ↑ 68.8% | Renal | Inhibition of OAT-mediated uptake into the renal tubules in humans after intramuscular or iv dosing, likely OAT2, OAT3 | (12-15) |
| 5. Cerivastatin | Cyclosporine | ↓ 66.7% (V1/F) | ↓ 73.3% (CL/F) | ↔ | Metabolism | Inhibition of OATP1B1 uptake into the liver in humans after oral dosing | (16, 17) |
| 6. Cetirizine | Pilsicainide | ↑ 102%* (Vss/F) | ↓ 28.9% (CL/F); ↓ 37.6% (CLr) | ↑ 188% | Renal > Metabolism | Inhibition of P-gp efflux in humans after oral dosing, possible inhibition of OCT2 as well, but P-gp effect is more significant | (18) |
| 7. Ciprofloxacin | Probenecid | ↔ (Vss) | ↓ 41.2% (CL); ↓ 64.0% (CLr) | ↑ 51.5% | Renal > Metabolism | Inhibition of OAT-mediated uptake into the renal tubules in humans after iv dosing | (19) |
| 8. Colchicine | SDZ PSC 833 | ↔ (Varea) | ↓ 47.0% | ↔ | Biliary > Renal > Metabolism | Inhibition of P-gp efflux in rats after iv dosing | (20, 21) |
| 9. Daunomycin | Verapamil | ↓ 63.7%* (Varea) | ↓ 89.1% | ↑ 232% | Metabolism > Renal ~ Biliary | Inhibition of P-gp efflux in rats after iv dosing | (22, 23) |
| 10. Digoxin | Rifampin | ↓ 70.8% (Vss) | ↓ 54.2% | ↔ | Metabolism | Inhibition of Oatp1a4 uptake into the liver under Cyp3a-induced conditions in rats after iv dosing | (24) |
| 11. Digoxin | Ritonavir | ↑ 76.7% (Vss) | ↓ 41.8% | ↑ 156% | Renal | Inhibition of P-gp efflux in humans after iv dosing | (25) |
| 12. Famotidine | Probenecid | ↓ 39.5% (V1/F) | ↓ 64.0% (CLr) | ↔ | Renal | Inhibition of uptake into the renal tubules, likely OAT3, in humans after oral dosing | (26, 27) |
| 13. Glyburide | Rifampin | ↓ 67.4% (Vss/F) | ↓ 54.6% (CL/F) | ↔ | Metabolism | Reduced OATP2B1 uptake into the liver in humans after oral dosing | (28) |
| 14. Metformin | OCT1 reduced function allele | ↓ 53.9% (Varea/F) | ↓ 37.5% (CL/F) | ↔ | Renal | Reduced OCT1 uptake into the liver in humans after oral dosing; OCT1 does not mediate elimination at the renal tubules | (29) |
| 15. Methotrexate | Pantoprazole | ↓ 21.6%* (Varea) | ↓ 45.7% | ↑ 44.4% | Biliary > Renal | Inhibition of Bcrp mediated efflux in mice after iv dosing | (30) |
| 16. Penicillin G | oat3-/- mice | ↓ 33.5% (Vss) | ↓ 44.8% | ↑ 72.9% | Renal > Metabolism | Decreased renal clearance in mice after iv dosing | (31) |
| 17. Pilsicainide | Cetirizine | ↑ 106%* (Vss/F) | ↓ 26.6% (CL/F); ↓ 41.3% (CLr) | ↑ 206% | Renal >> Metabolism | Inhibition of P-gp efflux after oral dosing in humans, possible inhibition of OCT2 as well, but P-gp effect is more significant | (18) |
| 18. Pilsicainide | Cimetidine | ↔ (V1/F) | ↓ 26.4% (CL/F); ↓ 28.0% (CLr) | ↑ 24.5% | Renal >> Metabolism | Inhibition of uptake into the renal tubules, likely OCT2, in humans after oral dosing | (18, 32) |
| 19. Pitavastatin | OATP1B1 reduced function allele | ↓ 27.6% (Varea/F) | ↓ 44.3%* (CL/F) | ↑ 29.9% | Biliary | Reduced OATP1B1 uptake into the liver in humans after oral dosing | (33, 34) |
| 20. Procainamide | Cimetidine | ↓ 12.4%* (Varea/F) | ↓ 30.5%* (CL/F); ↓ 43.4% (CLr) | ↑ 26.0% | Renal > Metabolism | Inhibition of OCT-mediated uptake in humans after oral dosing; not likely a gut interaction | (35, 36) |
| 21. Procainamide | Ciprofloxacin | ↔ (Varea and Vss) | ↔ (CL); ↓ 14.8% (CLr) | ↔ | Renal > Metabolism | Inhibition of OCT-mediated uptake in humans after iv dosing | (35, 36) |
| 22. Procainamide | Levofloxacin | ↔ (Varea and Vss) | ↓ 17.2% (CL); ↓ 25.9% (CLr) | ↑ 18.5% | Renal > Metabolism | Inhibition of OCT-mediated uptake in humans after iv dosing | (35, 36) |
| 23. Repaglinide | Cyclosporine | ↓ 59.0%* (Varea/F) | ↓ 59.0%* (CL/F) | ↔ | Metabolism | Inhibition of OATP1B1 uptake into the liver in humans after oral dosing | (37) |
| 24. Rosuvastatin | Cyclosporine | ↓ 90.6%* (Varea/F) | ↓ 80.1%* (CL/F) | ↓ 52.7% | Biliary >> Metabolism | Inhibition of OATP1B1 uptake into the liver in humans after oral dosing; t1/2 is normally reported as lower than it is in this study’s baseline; decrease in t1/2 might be artifactual | (38) |
| 25. Rosuvastatin | Gemfibrozil | ↓ 27.5%* (Varea/F) | ↓ 46.8%* (CL/F) | ↑ 36.3% | Biliary >> Metabolism | Inhibition of OATP1B1 uptake into the liver in humans after oral dosing | (39) |
| 26. Rosuvastatin | OATP1B1 reduced function allele | ↓ 50.9%* (Varea/F) | ↓ 38.3%* (CL/F) | ↔ | Biliary >> Metabolism | Reduced OATP1B1 uptake into the liver in humans after oral dosing | (11) |
| 27. Sotalol | Cimetidine | ↔ (Vss) | ↓ 26.7% | ↔ | Renal | Inhibition of OCT-mediated uptake in rats after iv dosing, likely OCT1, OCT2 | (40, 41) |
| 28. Tacrolimus | mdr1a-/- mice | ↔ (Vss) | ↓ 65.4% | ↑ 99.4% | Metabolism >> Biliary > Renal | Lack of P-gp mediated biliary clearance in mice after iv dosing | (42) |
| 29. Telmisartan | Nisoldipine | ↓ 62.2% (Varea/F) | ↓ 51.2% (CL/F) | ↔ | Metabolism | Inhibition of P-gp efflux of metabolites from the liver in humans after oral dosing; the majority of the effect is likely a bioavailability consideration | (43) |
| 30. Tetracycline | Diclofenac | ↔ (Vss) | ↓ 60.7% (CL); ↓ 61% (CLr) | ↑ 88.2% | Renal | Inhibition of OAT-mediated uptake in rats after iv dosing, likely OAT1, OAT2, OAT4 | (15, 44, 45) |
| 31. Tetracycline | Naproxen | ↔ (Vss) | ↓ 75.0% (CL); ↓ 72.0% (CLr) | ↑ 400% | Renal | Inhibition of OAT-mediated uptake in rats after iv dosing, likely OAT1, OAT2, OAT4 | (15, 44, 45) |
| 32. Tezosentan | Cyclosporine | ↓ 65.2% (Vss) | ↓ 74.8% | ↔ | Biliary | Likely inhibition of P-gp efflux in humans after iv dosing | (46) |
| 33. Topotecan | Probenecid | ↔ (Vss) | ↔ (CL); ↓ 29.6% (CLr) | ↔ | Renal > Biliary >> Metabolism | Inhibition of OAT-mediated uptake in mice after iv dosing | (47) |
| 34. Topotecan | Novobiocin | ↑ 254% (Vss) | ↓ 33.7% | ↑ 341% | Renal > Biliary >> Metabolism | Inhibition of Bcrp efflux in rats after iv dosing | (48) |
| 35. Ulifloxacin | Cyclosporine | ↓ 31.4% (Vss) | ↓ 32.1% (CL); ↓ 66.5% (CLb) | ↔ | Biliary ~ Renal | Inhibition of Oat/Oatp-mediated uptake into the liver in rats after iv dosing | (49-51) |
| 36. Ulifloxacin | EHBR rats | ↓ 34.1% (Vss) | ↔ | ↓ 35.0% | Biliary ~ Renal | Lack of Mrp2 decreases biliary excretion of glucuronide; no change in clearance or percent of dose eliminated in the bile or urine after iv dosing | (49-51) |
| 37. Valsartan | EHBR rats | ↓ 35.8% (V1) | ↓ 94.3% (CL); ↓ 97.7% (CLb) | ↑ 1030%* | Biliary >> Metabolism > Renal | Lack of Mrp2 decreases biliary clearance, leading to increased plasma and liver concentrations after iv dosing | (52) |
* indicates calculated parameter, not reported in reference
CLr: renal clearance, CLb: biliary clearance; EHBR: Eisai hyperbilirubinemic rats
RESULTS AND DISCUSSION
Further analysis of the interactions listed in Table I revealed a few interesting trends:
The magnitude of transporter mediated change in volume of distribution may differ depending on which measure of volume is used.
A transporter mediated change in volume of distribution may be independent of or correlated to a change in the drug’s clearance and the associated half life.
In general, interactions at uptake transporters at the liver lead to a significant decrease in volume of distribution, while those at the renal tubules do not lead to a change in volume of distribution, although there are exceptions.
Interactions with efflux transporters at the liver generally lead to a decrease in volume of distribution, while those at the renal tubules lead to an increase in volume of distribution.
The primary location of the interaction (liver or kidneys) is a more important determinant of the change in distribution volume than the secondary change in tissue distribution, as evidenced by interactions that affect the integrity of the blood-brain barrier.
It is possible to predict the direction of the change in pharmacological effect given the mechanisms of action of the drug and the location of the interaction.
Each of these trends will be discussed in relation to the interactions presented in Table I.
The magnitude of transporter mediated change in volume of distribution may differ depending on which measure of volume is used.
As discussed above, there are three measures of distribution volume. The relative contribution of changes in transporter function to these three measures of volume may differ. The plasma concentration-time data from four studies (24, 30, 49, 52) were extracted and reanalyzed using WinNonlin version 2.1 (Pharsight Corporation, Mountain View, CA). For each study, the three measures of volume were calculated under both control and decreased transporter functionality conditions, as shown in Table II, to ensure consistency in calculation methods between the parameter estimates both within and between the four studies. V1 changes less markedly than Vss and Varea. This is expected since full tissue distribution is not likely to have occurred at the initial time points, and transporters not directly associated with very rapidly equilibrating organs may not have had the chance to exert their effects. In contrast, after all organs are in distribution equilibrium, Vss and Varea show the full effect of transporter inhibition, exhibiting bigger changes than seen for V1. Thus, with respect to transporter effects on equilibrium volume measures, little difference is seen between the effects on Vss and Varea.
Table II.
Effect of change in transporter function on the distribution volume parameters
| DIGOXIN (Oatp1a4) Reference (24) | (ml/kg) | CONTROL | +RIFAMPIN | % DECREASE |
| V1 | 933 | 454 | 51.2 | |
| Vss | 7140 | 1640 | 77.1 | |
| Varea | 7360 | 1790 | 75.7 | |
| METHOTREXATE (Bcrp) Reference (30) | (L/kg) | CONTROL | +PANTOPRAZOLE | % DECREASE |
| V1 | 417 | 315 | 24.4 | |
| Vss | 933 | 651 | 30.3 | |
| Varea | 1010 | 689 | 31.9 | |
| ULIFLOXACIN (Oat/Oatp) Reference (49) | (ml/kg) | CONTROL | +CYCLOSPORINE | % DECREASE |
| V1 | 831 | 702 | 15.6 | |
| Vss | 4450 | 3070 | 31.0 | |
| Varea | 4880 | 3320 | 31.9 | |
| VALSARTAN (Mrp2) Reference (52) | (ml/kg) | CONTROL | in EHBR rats | % DECREASE |
| V1 | 51.0 | 44.7 | 12.4 | |
| Vss | 258 | 111 | 57.2 | |
| Varea | 420 | 118 | 71.9 |
The plasma concentration-time data from four studies were extracted and reanalyzed using WinNonlin version 2.1 (Pharsight Corporation, Mountain View, CA). For each study, the three measures of volume were recalculated under both control and decreased transporter functionality conditions. These calculated parameters (not those in the reference) are presented.
EHBR: Eisai hyperbilirubinemic rats
A transporter mediated change in volume of distribution can be independent of or correlated to a change in the drug’s clearance and the associated half life.
Half life is considered the most important parameter to the clinician for determining dosing changes due to drug-drug interactions or pharmacogenomic variability, as it is considered the parameter most closely associated with dosing interval and duration of drug effect. In the simplest relation, half life, clearance, and volume of distribution are related by Equation 1:
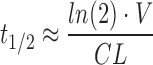 |
1 |
Therefore, the change in half life is proportional to the change in distribution volume, and inversely related to the change in clearance. In this simple single-phase approximation, there will only be one volume term 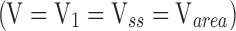 . In reality, most drugs exhibit multiple phases of distribution and/or elimination, and may have many half lives (53). However, changes in this single-phase approximation are still indicative of a general pharmacokinetic trend.
. In reality, most drugs exhibit multiple phases of distribution and/or elimination, and may have many half lives (53). However, changes in this single-phase approximation are still indicative of a general pharmacokinetic trend.
Further, as noted by Sahin and Benet (53), many different single value half lives can be reported for a drug that almost assuredly exhibits multi-compartment kinetics, including the single-phase approximation or the half life for the terminal phase. Therefore, for a number of drugs in Table I, the relationship between clearance, volume, and half life will not follow Equation 1.
In rats, digoxin is primarily metabolized in the liver by Cyp3a. It is a substrate for Oatp1a4 uptake and P-glycoprotein efflux in hepatocytes. When rats were dosed with dexamethasome, Cyp3a, Oatp1a4, and P-gp were induced. Following administration of a single dose of the Oatp-inhibitor rifampin to these dexamethasome induced rats, a decrease in steady state volume of distribution of 70.8% was observed together with a decrease in clearance of 54.2%, while no change in half life was evident in comparison to the induced, but not rifampin inhibited, controls. From previous studies, it is known that the concentration of inhibitor achieved after the single dose had minimal effects on Cyp3a and P-gp. Therefore, inhibition of the uptake transporter led to the pharmacokinetic changes observed (24). Inhibition of Oatp1a4 prevents liver accumulation, decreasing distribution volume. Preventing liver entry also prevents metabolism, leading to the decrease in clearance. Possibly because Oatp1a4 is also expressed at the blood-brain barrier, choroid plexus, ciliary bodies, and retina, in addition to the liver (54), the decrease in volume is greater than the decrease in clearance. In this case, the changes in volume and clearance appear to be correlated.
In humans, however, digoxin is predominantly excreted unchanged in the urine. This process is mediated by P-gp. In patients concomitantly dosed with ritonavir, a P-gp inhibitor, steady state volume increased 76.7%, clearance decreased 41.8%, and half life increased 156% as compared to control (25). Here, inhibition of P-gp prevents efflux of drug from the renal epithelial cells into the urine, decreasing clearance. Inhibition of P-gp also prevents efflux of drug from other tissues protected by P-gp, such as the brain and heart. Therefore, the drug is more widely distributed in the body, and there is less drug in the systemic circulation, making less available to be cleared by the kidneys. Because the clearance rate is also decreased, both factors work towards increasing half life. In this case, the changes in volume and clearance are not correlated.
These examples elucidate mechanisms by which transporter inhibition can lead to significantly different pharmacokinetic patterns for the clinician to consider.
In general, interactions at uptake transporters at the liver lead to a significant decrease in volume of distribution, while those at the renal tubules do not lead to a change in volume of distribution, although there are exceptions.
Of twenty four interactions that involved uptake transporters with decreased function, nine did not cause a significant change in distribution volume. Each of these involved interactions documented at uptake transporters at the renal tubules for drugs that are primarily excreted unchanged in the urine. Three of twelve renal interactions did exhibit decreased volume. Conversely, the twelve interactions attributed to the liver all led to a decreased volume of distribution. A decrease in volume of distribution would be expected in these interactions, as inhibiting an uptake transporter prevents tissue accumulation. These uptake interactions are retabulated as either hepatic or renal in Table III.
Table III.
Summary of uptake transporter-based interactions at the liver and renal tubules
| Drug | Interaction | V | Reference | Drug | Interaction | V | Reference |
|---|---|---|---|---|---|---|---|
| Hepatic Interactions | Renal Interactions | ||||||
| Atorvastatin | Rifampin | ↓ 94.3% (Vss/F) | (10) | Cefazolin | Probenecid | ↔ (V1) | (12-15) |
| Atorvastatin | OATP1B1 reduced function allele | ↓ 57.6%* (Varea/F) | (11) | Ciprofloxacin | Probenecid | ↔ (Vss) | (19) |
| Cerivastatin | Cyclosporine | ↓ 66.7% (V1/F) | (16, 17) | Famotidine | Probenecid | ↓ 39.5% (V1/F) | (26, 27) |
| Digoxin | Rifampin | ↓ 70.8% (Vss) | (24) | Penicillin G | oat3-/- mice | ↓ 33.5% (Vss) | (31) |
| Glyburide | Rifampin | ↓ 67.4% (Vss/F) | (28) | Pilsicainide | Cimetidine | ↔ (V1/F) | (18, 32) |
| Metformin | OCT1 reduced function allele | ↓ 53.9% (Varea/F) | (29) | Procainamide | Cimetidine | ↓ 12.4%* (Varea/F) | (35, 36) |
| Pitavastatin | OATP1B1 reduced function allele | ↓ 27.6% (Varea/F) | (33, 34) | Procainamide | Ciprofloxacin | ↔ (Varea and Vss) | (35, 36) |
| Repaglinide | Cyclosporine | ↓ 59.0%* (Varea/F) | (37) | Procainamide | Levofloxacin | ↔ (Varea and Vss) | (35, 36) |
| Rosuvastatin | Cyclosporine | ↓ 90.6%* (Varea/F) | (38) | Sotalol | Cimetidine | ↔ (Vss) | (40, 41) |
| Rosuvastatin | Gemfibrozil | ↓ 27.5%* (Varea/F) | (39) | Tetracycline | Diclofenac | ↔ (Vss) | (15, 44, 45) |
| Rosuvastatin | OATP1B1 reduced function allele | ↓ 50.9%* (Varea/F) | (11) | Tetracycline | Naproxen | ↔ (Vss) | (15, 44, 45) |
| Ulifloxacin | Cyclosporine | ↓ 31.4% (Vss) | (49-51) | Topotecan | Probenecid | ↔ (Vss) | (48) |
For further detail, see Table I.
* indicates calculated parameter, not reported in reference
From a physiological perspective, the liver is significantly more massive than the kidneys: in the average man, the kidneys weigh about 150 grams each, and the liver weighs about 1.5 kilograms (55). The liver also contains more cellular space available for transporter expression, while considerable kidney mass is interstitial fluid and tubule volume. Similarly, hepatocytes are more available to drug sequestration and storage than kidney epithelial cells. Therefore, preventing drug from entering the hepatocytes will have a greater relative effect on the entire body volume of distribution than will preventing drug from entering the epithelial cells at the renal tubules. Despite substantial decreases in renal clearance and associated increases in systemic concentrations, it appears that volume change due to interactions at the kidney level is not observable.
A few further explanations for this disparity are possible. For one, if the kidney transporters were unique to the renal epithelium, while liver transporters were also expressed at other tissues in the body, inhibition of liver transporters would cause a more significant pharmacokinetic change. However, it seems like the opposite may be true: Table IV shows the tissue distribution of the transporters studied in this analysis. A second possibility is that the transporters that are relatively uniquely expressed at the liver are also more specific for their substrates, while the renal transporters act on a wider range of substrates. In that case, inhibition of a renal transporter would not have much of an effect because another transporter could restore the activity of the dysfunctional transporter. In support of this is the fact that many of the studies focused on the kidney report only that the inhibited transporter is a member of the OAT or OCT family. Further, Sweet et al. (56) report that the OAT transporters, which predominantly mediate clearance at the renal tubules, have significant substrate overlap. An inhibitor would then also inhibit multiple transporters. On the other hand, liver studies often report a specific transporter that is affected. Alternatively, the renal epithelium may be a “looser” barrier than the hepatocyte membranes, implying the transporters may simply have less importance at the renal epithelium. In support of this is the fact that many drugs have different permeability characteristics at the enterocytes of the intestine and at the hepatocytes; certain drugs, such as atorvastatin, may diffuse passively into the intestine, but require an uptake transporter at the liver. Thus, some drugs may also have different permeability characteristics at the renal epithelium and hepatocyte membranes. However, the measured changes in clearance contradict these possibilities: if a single transporter were less important in the kidneys, clearance, in addition to volume, would be unaffected.
Table IV.
Tissue distribution of transporters for which volume modulation has been investigated
| Transporter | Species | Tissue Expression | Reference |
|---|---|---|---|
| A. Efflux | |||
| P-gp | Human | adrenal, sweat glands, blood vessels, liver, kidney, lung > muscle, mammary glands, spleen, gall bladder, heart | (57) |
| P-gp (mdr1a/b) | Rat | brain > kidney > lung > liver | (3) |
| P-gp (mdr1a) | Mouse | adrenal > placenta> kidney, heart > liver, uterus, muscle, spleen, brain, lung | (58) |
| Mrp2 | Rat | liver > kidney, brain | (4) |
| Bcrp | Rat | kidney > liver > gonads > brain > thymus, spleen | (59) |
| Bcrp | Mouse | kidney > liver > gonads > brain > spleen, muscle, lung | (59) |
| B. OATPs | |||
| OATP1B1 | Human | liver | (54) |
| OATP2B1 | Human | liver, placenta, ciliary body | (54) |
| Oatp1a4 | Rat | liver, brain; eye | (45, 54) |
| C. OATs | |||
| OAT1 | Human | kidney > brain | (45) |
| OAT2 | Human | liver > kidney | (60) |
| OAT3 | Human | kidney > brain, | (45) |
| OAT4 | Human | kidney; placenta | (45, 61) |
| Oat1 | Rat | kidney > brain, | (45) |
| Oat2 | Rat | liver > kidney | (45) |
| Oat3 | Rat | liver ~ kidney, brain; eye | (45, 56, 61) |
| Oat4 | Rat | kidney, likely placenta | (62) |
| Oat1 | Mouse | kidney > brain | (45) |
| Oat2 | Mouse | kidney > liver (female > male) | (63) |
| Oat3 | Mouse | kidney > brain | (56) |
| Oat4 | Mouse | kidney, placenta | (62) |
| D. OCTs | |||
| OCT1 | Human | liver > kidney | (45) |
| OCT2 | Human | kidney > brain | (45) |
| OCT3 | Human | liver > kidney > brain | (45) |
| Oct1 | Rat | kidney, liver > brain | (45) |
| Oct2 | Rat | kidney | (45) |
| Oct3 | Rat | kidney, brain | (45) |
Three interactions at the renal tubules presented in Table III do lead to a decrease in volume. For the interaction between procainamide and cimetidine (36), Varea was calculated (as indicated by the asterisk) from the reported data assuming the reported half life was the terminal half life, which may be an incorrect assumption. Moreover, as discussed, the change in Varea is often more extensive than the change in the other volume parameters. However, the decrease in penicillin G Vss in Oat3-/- knockout mice (31) and in famotidine V1 in healthy human volunteers upon co-administration with probenecid (26) seem to be exceptions to the trend.
Interactions with efflux transporters at the liver generally lead to a decrease in volume of distribution, while those at the renal tubules lead to an increase in volume of distribution.
Efflux transporters serve a protective purpose preventing drug distribution at some of the most sensitive tissue sites, such as the brain, lungs, and heart. They are also expressed at the liver canalicular membrane and renal epithelia to facilitate clearance. An increase in distribution volume would be expected after inhibiting an efflux transporter, by increasing penetration to tissues protected by the transporters. Table V highlights the interactions attributed to efflux transporters. Of these thirteen interactions, five lead to an increase in volume, and they are all interactions at the renal tubules. The remaining eight that do not cause a change or lead to a decrease in volume are interactions at the liver.
Table V.
Summary of efflux transporter-based interactions at the liver and renal tubules
| Drug | Interaction | V | Reference | Drug | Interaction | V | Reference |
|---|---|---|---|---|---|---|---|
| Hepatic Interactions | Renal Interactions | ||||||
| Colchicine | SDZ PSC 833 | ↔ (Varea) | (20, 21) | Adriamycin | Verapamil | ↑ 31.2% (Vss) | (8, 9) |
| Daunomycin | Verapamil | ↓ 63.7%* (Varea) | (22, 23) | Cetirizine | Pilsicainide | ↑ 102%* (Vss/F) | (18) |
| Methotrexate | Pantoprazole | ↓ 21.6%* (Varea) | (30) | Digoxin | Ritonavir | ↑ 76.7% (Vss) | (25) |
| Tacrolimus | mdr1a-/- mice | ↔ (Vss) | (42) | Pilsicainide | Cetirizine | ↑ 106%* (Vss/F) | (18) |
| Telmisartan | Nisoldipine | ↓ 62.2% (Varea/F) | (43) | Topotecan | Novobiocin | ↑ 254% (Vss) | (48) |
| Tezosentan | Cyclosporine | ↓ 65.2% (Vss) | (46) | ||||
| Ulifloxacin | EHBR rats (Mrp2-/-) | ↓ 34.1% (Vss) | (49-51) | ||||
| Valsartan | EHBR rats (Mrp2-/-) | ↓ 35.8% (V1) | (52) | ||||
For further detail, see Table I.
* indicates calculated parameters, not reported in reference
EHBR: Eisai hyperbilirubinemic rats
Methotrexate and topotecan are both substrates of the efflux transporter Bcrp, distributed through the blood-brain barrier, liver, and kidneys, among other tissues. When methotrexate was dosed with the Bcrp inhibitor pantoprazole in mice, Varea decreased by 21.6%*, clearance decreased by 45.7%, and the half life increased by 44.4%. Methotrexate is primarily cleared via the bile, where Bcrp has a modulating role (30). On the other hand, when topotecan was dosed with the Bcrp inhibitor novobiocin in rats, Vss increased by 254%, clearance decreased by 33.7%, and half life increased by 341%. Topotecan is primarily eliminated unchanged in the urine, again mediated by Bcrp. In this case, the authors note that increased brain concentrations of topotecan could lead to the increased volume of distribution (48). While increased peripheral tissue distribution is likely in both cases, the effect is not apparent in the liver interaction.
Similarly, the anti-cancer agents daunomycin and adriamycin are both substrates of P-glycoprotein. As daunomycin is eliminated predominantly in the liver by metabolism (23), when dosed with the P-gp inhibitor verapamil in rats, Varea decreased by 63.7%*, clearance decreased by 89.1%, and half life increased by 232% (22). On the other hand, adriamycin is eliminated both through the liver and urine. In rats, Tavoloni and Guarino (9) found that urinary elimination of adriamycin is saturable, while biliary excretion is not. This indicates that P-gp may play a more important role in the kidney than the liver. Upon co-administration with verapamil in humans, Vss increased by 31.2%, clearance decreased by 32.6%, and half life increased by 37.7%. Further, while the values were not reported, the authors do note that the volume of the central compartment decreased, and the volume of the peripheral compartments increased after P-gp inhibition (8). Because Vss is the sum of the volumes of all the compartments, the change in central compartment volume, which most likely includes the kidneys, must be minor compared to the increase in volume of the peripheral compartments.
Therefore, it seems that efflux transporter inhibition leads to a decrease in distribution volume for the central compartment and an increase in distribution volume for the peripheral tissue compartments. The magnitude of the increase in peripheral distribution is greater than the magnitude of the decrease in central compartment volume for a renal interaction, but it is less than the magnitude of the decrease in central compartment volume for a hepatic interaction. So, an increase in total distribution volume is evident for a kidney interaction, but a decrease in total distribution volume is evident for a liver interaction. This conclusion follows the above analysis on the difference in volume changes following uptake transporter interactions in the kidney and liver: the liver is again a greater contributor to distribution volume than the kidneys. Because peripheral distribution does not seem to be a factor in uptake interactions, it is also clear that efflux transporters play a larger role than uptake transporters outside the liver and kidney, despite the fact that uptake transporters are expressed at these other tissues.
A mechanism for a decrease in central compartment volume, however, is not immediately clear. As mentioned in the Introduction, it is possible that when the efflux transporter is inhibited, the eliminating organ is no longer quickly equilibrating with the plasma. Effectually, the plasma concentrations do not reflect the amount of drug at the elimination site because drug is now so highly sequestered in the hepatocytes. As Yates and Arundel (7) derived for a two compartment model, the steady state volume is under-predicted by the value of 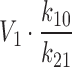 , when elimination is actually from the peripheral compartment, where k10 and k21 are defined for the central compartment elimination model such that k10 is the rate constant of elimination from the central compartment and k21 is the rate constant for flux from the peripheral compartment back in to the central compartment. Thus, the decrease in steady state volume might be a consequence of the pharmacokinetic calculations and may not reflect a “real” volume change. It remains to be elucidated in which cases the central compartment elimination model does not hold, as it is foreseeable that uptake transporter dysfunction will also change the equilibration properties of the eliminating organs.
, when elimination is actually from the peripheral compartment, where k10 and k21 are defined for the central compartment elimination model such that k10 is the rate constant of elimination from the central compartment and k21 is the rate constant for flux from the peripheral compartment back in to the central compartment. Thus, the decrease in steady state volume might be a consequence of the pharmacokinetic calculations and may not reflect a “real” volume change. It remains to be elucidated in which cases the central compartment elimination model does not hold, as it is foreseeable that uptake transporter dysfunction will also change the equilibration properties of the eliminating organs.
The primary location of the interaction (liver or kidneys) is a more important determinant of the change in distribution volume than the secondary change in tissue distribution is, as evidenced by interactions that affect the integrity of the blood-brain barrier.
While transporters function at almost all the major tissues in the body, including the heart, lungs, and muscle, they have been most studied, beside the liver and kidney, at the blood-brain barrier. Here, efflux transporters dominate, where they serve to protect the brain from xenobiotic penetration. Table VI highlights interactions that are associated with an increased distribution of drug to the brain. These five interactions involve P-gp and BCRP, the two transporters most highly implicated in maintaining the integrity of the blood-brain barrier. Despite the increase in brain concentrations in these five studies, there is no common increase in volume of distribution. Instead, it appears the trends discussed above for efflux transporters, that interactions attributed to the renal transporters lead to an increased volume, while interactions at hepatic transporters lead to either a decrease or no change in volume of distribution, generally hold true.
Table VI.
Summary of transporter-based interactions at the blood-brain barrier
| Drug | Interaction | V | Reference |
|---|---|---|---|
| Colchicine | SDZ PSC 833 | ↔ (Varea) | (20, 21) |
| Rhodamine-123 | Cyclosporine | ↔ (V1) | (64, 65) ¶ |
| Tacrolimus | mdr1a-/- mice | ↔ (Vss) | (42) |
| Tezosentan | Cyclosporine | ↓ 65.2% (Vss) | (46) |
| Topotecan | Novobiocin | ↑ 254.5% (Vss) | (47) |
For further detail, see Table I.
¶Not included in Table I. Interaction resulted in increased distribution to the brain due to P-gp inhibition in rats, without significant effect on volume or clearance after iv dosing. Because the interaction did not result in any pharmacokinetic change, it has not been included in Table I.
At the kidney tubules, as discussed, when the Bcrp substrate topotecan was dosed with novobiocin in rats, volume of distribution increased 254%, with increased distribution to the brain (48). At the liver, tacrolimus, a P-gp substrate, was dosed to wild-type and mdr1-/- (P-gp knockout) mice. In these mice, there was no significant change in volume, clearance decreased 65.4%, and half life increased 99.4% as compared to wild-type mice. Knockout mice also exhibited a 33-fold increase in brain concentrations of tacrolimus. Minor increases in liver concentrations were also evident. In mice, tacrolimus is predominantly excreted in the bile (42). Finally, tezosentan, also eliminated into the bile, was also dosed with cyclosporine for inhibition of P-gp in humans. In this study, volume of distribution decreased 65.2%, clearance decreased 74.8%, and half life did not change. The authors note that an increased incidence of adverse events, including headache, hot flushes, and nausea, may have been caused by increased brain distribution of the drug (46).
Thus, while brain distribution may change, even dramatically as in the case of tacrolimus in P-gp knockout mice, these changes do not necessarily manifest in a total body volume of distribution change. It is possible, however, that changes at the other tissues expressing transporters might offer a different conclusion.
It is possible to predict the direction of the change in pharmacological effect given the mechanisms of action of the drug and the location of the interaction.
Glyburide, metformin, and atorvastatin are substrates for uptake transporters in the liver. Following uptake inhibition either via polymorphism or concomitant medication, subjects in the three studies exhibit significantly reduced distribution volumes. However, the direction of the resulting change in pharmacological effect is different.
Glyburide is a hypoglycemic agent indicated for patients with type 2 diabetes. Its main effect is at the pancreatic beta cells, where it stimulates insulin secretion. It is primarily eliminated via metabolism by CYP2C9 and, to a lesser degree, by CYP3A4 in the liver. It is a substrate for uptake mediated by OATP2B1 at the hepatocytes, and subjects show a decrease in steady state volume of 67.4%, a decrease in clearance of 54.6%, without a change in half life, following uptake inhibition via concomitant dosing with rifampin. As would be predicted, inhibition of liver uptake decreases elimination, increasing plasma concentrations. This increases pancreatic beta cells’ access to the drug, increasing the pharmacologic effect. Following a single dose of glyburide and a single dose of rifampin, subjects exhibited significantly decreased blood glucose AUCs over a twelve hour period (28).
Similarly, metformin is the first line therapy for patients with type 2 diabetes. Its pharmacological effect is in the liver hepatocytes, where it prevents gluconeogenesis, effectively decreasing blood glucose levels. It is primarily eliminated via excretion at the renal tubules, a process mediated by OCT2. However, at the liver, it is a substrate for uptake by OCT1. In this unique case, the transporter interaction is not at the primary site of elimination, but because the drug is a substrate for hepatic uptake and it is highly distributed to the liver, the interaction still causes marked pharmacokinetic changes. This further attests to the importance of the liver in the determination of distribution volume in consideration of transporter dysfunction. In patients with polymorphisms in one of their OCT1 alleles, volume of distribution is decreased by 53.9% and clearance is reduced by 37.5%, without a change in half life (29). In this case, a reduced function OCT1 allele decreases hepatocyte access to the drug, decreasing the pharmacologic effect.
Along the same lines, Pasanen et al. (11) and Tachibana-Iimori et al. (66) both studied a single nucleotide polymorphism (SNP) at position 521 in the SLCO1B1 (OATP1B1) gene. Both groups looked at atorvastatin, among other statins, to measure the effect of this polymorphism. Atorvastatin is primarily metabolized by CYP3A4 and by CYP2C9 to a lesser extent, and it is a substrate for OATP1B1 uptake. The drug and its active metabolites are less than 1% excreted in the urine (10). The statins, HMG-CoA reductase inhibitors, decrease cholesterol levels by preventing cholesterol synthesis and increasing clearance of LDL, or “bad”, cholesterol at the hepatocytes. From the pharmacokinetic perspective, Pasanen et al. (11) showed a decrease in Varea of 57.6%* and a decrease in clearance of 59.2%*, without a change in half life, between patients homozygous for either the wild-type or mutant alleles. From a pharmacodynamic perspective, Tachibana-Iimori et al. (66), showed the same patterns held for patients beginning atorvastatin, pravastatin, or simvastatin therapy, all OATP1B1 substrates. Analysis of patients on any of these three drugs found that patients homozygous for the wild-type alleles showed a decrease in total cholesterol of 22.3%, while patients heterozygous for the wild-type and polymorphic allele showed a decrease in total cholesterol of 16.5%, indicating a decreased pharmacological benefit. The difference between wild-type and polymorphic patients is likely to be greater for patients homozygous for the mutant alleles.
In this regard, while the pharmacokinetic consequences of an interaction are important for the clinician to understand, the pharmacodynamic change is also critical to consider before changes to the dosing regimen are made. Within these three examples, although the direction of pharmacokinetic change is the same, a glyburide-rifampin interaction would require a decreased dosing rate to maintain the same pharmacological effect, while patients with polymorphisms in OCT1 or OATP1B1 would require an increased dosing rate of metformin or atorvastatin, respectively, to maintain effect. The potential for toxicity when higher dosing rates are required complicates this issue, and may lead to alternative therapies for patients with such pharmacogenetic variation.
EXPERIMENTAL CONSIDERATIONS
As with any pharmacokinetic study, it is important to understand the experimental conditions and variability that complicate the conclusions that are drawn from transporter interaction studies.
First, there are wide interspecies differences in drugs’ elimination pathways, the expression of transporters, and transporter substrate profiles. For instance, as noted above, digoxin is almost completely metabolized in rats, where it is a substrate for uptake mediated by Oatp1a4 at the liver (24). In humans, however, digoxin is predominantly eliminated in the urine. Similarly, as shown in Table IV, in humans OAT3 is expressed primarily at the kidney, and, to a lesser degree, in the brain, while in rats Oat3 is highly expressed in the liver in addition to the kidneys and brain. For substrates common to both rat and human Oat3/OAT3, this will most likely lead to different tissue distributions and volume calculations between the two species, and indicates that results from an Oat3 focused pharmacokinetic study conducted in rats may not scale to humans. Finally, the interaction between famotidine and probenecid (26) resulting from the inhibition of OAT3 transport at the renal tubules in humans is not reproducible in rats (67). This is likely due to the increased expression of Oct1 in the rat kidney: because famotidine is also a substrate of Oct1 and probenecid does not inhibit Oct1, the transporter serves as an alternate, compensatory route for renal clearance in rats concomitantly dosed with famotidine and probenecid (13). Briefly, these few examples attest to the importance of considering interspecies differences before clinical extrapolations are made from animal data.
Further, while clearance is relatively easily extrapolated from in vitro data to in vivo relevance, the same is not true of volume of distribution. Because volume is focused on the entire body, even ex situ techniques, such as the isolated perfused rat liver (IPRL) or isolated perfused rat kidney, can lead to incorrect approximations of the direction of volume changes. Table VII highlights these discrepancies. Although the published data is sparse, it appears the ex situ results for inhibited uptake transporters in the liver and kidney follow the analysis above (68, 69). However, the IPRL data for inhibited efflux transporters, in particular P-glycoprotein, show an increase in steady-state volume of distribution (70, 71), while the in vivo trend predicts either a decrease or no change in this parameter. It is of interest to note the ex situ data follow what would be generally predicted for uptake and efflux transporter inhibition before the conclusions of the present analysis. A mechanism for this discrepancy remains to be elucidated.
Table VII.
Vss values from ex situ studies in rat
| Interaction | Transporter | Organ | Vss Change | Prediction from Trends | Reference |
|---|---|---|---|---|---|
| Digoxin+Rifampin | Oatp1a4 uptake | Liver | ↓ 17.9% | ↓ | (68) |
| Quinapril+PAH | Oat uptake | Kidney | ↓ 13.7%* | ↔, (↓) | (69) |
| Digoxin+Quinidine | P-gp efflux | Liver | ↑ 95.9% | ↓ | (70) |
| Tacrolimus+GG918 | P-gp efflux | Liver | ↑ 30.1% | ↓ | (70) |
| Talinolol+GG918 | P-gp efflux | Liver | ↑ 74.2% | ↓ | (70) |
| Doxorubicin+GG918 | P-gp efflux | Liver | ↑ 70.2%* | ↓ | (71) |
Italicized interactions are those where the data does not match the prediction from the present analysis.
* indicates calculated parameters, not reported in reference
PAH: p-aminohippurate, GG918: GF120918 (Elacridar)
CONCLUSIONS
Through the above analysis, we show that active drug transporters that modulate tissue distribution act as modifiers of distribution volume. Because transporters can be significantly affected by drug-drug interactions or genetic polymorphisms, changes in drug transporter activity as they affect distribution volume require attention. The above analysis indicates that it is the primary location of the interaction, at the kidneys or the liver, that serves as the major predictor of change in distribution volume. Figure 1 summarizes the trends in effects of transporter dysfunction on distribution volume as discussed above. As knowledge pertaining to the location and function of drug transporters and the substrate status of drugs for these transporters becomes more available, the present analysis provides a framework for understanding future pharmacokinetic interactions rooted in active drug transporters.
Fig. 1.

Summary of trends for predicting effects of transporter inhibition/dysfunction on volume of distribution, depending on whether it is an uptake or efflux transporter and the primary location of the interaction
Acknowledgements
This work was supported in part by NIH grants GM61390 and GM75900, as well as by an unrestricted grant from Amgen, Inc.
References
- 1.Øie S. Drug distribution and binding. J Clin Pharmacol. 1986;26:583–586. doi: 10.1002/j.1552-4604.1986.tb02953.x. [DOI] [PubMed] [Google Scholar]
- 2.Jette L, Beaulieu E, Leclerc JM, Beliveau R. Cyclosporin A treatment induces overexpression of P-glycoprotein in the kidney and other tissues. Am J Physiol. 1996;270:F756–F765. doi: 10.1152/ajprenal.1996.270.5.F756. [DOI] [PubMed] [Google Scholar]
- 3.Brady JM, Cherrington NJ, Hartley DP, Buist SC , Li N , Klaassen CD. Tissue distribution and chemical induction of multiple drug resistance genes in rats. Drug Metab Dispos. 2002;30:838–844. doi: 10.1124/dmd.30.7.838. [DOI] [PubMed] [Google Scholar]
- 4.Cherrington NJ, Hartley DP, Li N, Johnson DR, Klaassen CD. Organ distribution of multidrug resistance proteins 1, 2, and 3 (Mrp1, 2, and 3) mRNA and hepatic induction of Mrp3 by constitutive androstane receptor activators in rats. J Pharmacol Exp Ther. 2002;300:97–104. doi: 10.1124/jpet.300.1.97. [DOI] [PubMed] [Google Scholar]
- 5.Nakashima E, Benet LZ. General treatment of mean residence time, clearance, and volume parameters in linear mammillary models with elimination from any compartment. J Pharmacokinet Biopharm. 1988;16:475–492. doi: 10.1007/BF01062381. [DOI] [PubMed] [Google Scholar]
- 6.Yates JW, Arundel PA. Oral and IV dosing: a method to determine the compartment of drug elimination for two-compartment models. J Pharm Sci. 2008;97:2036–2040. doi: 10.1002/jps.21166. [DOI] [PubMed] [Google Scholar]
- 7.Yates JW, Arundel PA. On the volume of distribution at steady state and its relationship with two-compartmental models. J Pharm Sci. 2008;97:111–122. doi: 10.1002/jps.21089. [DOI] [PubMed] [Google Scholar]
- 8.Kerr DJ, Graham J, Cummings J, Morrison JG, Thompson GG, Brodie MJ, Kaye SB. The effect of verapamil on the pharmacokinetics of adriamycin. Cancer Chemother Pharmacol. 1986;18:239–242. doi: 10.1007/BF00273394. [DOI] [PubMed] [Google Scholar]
- 9.Tavoloni N, Guarino AM. Biliary and urinary excretion of adriamycin in anesthetized rats. Pharmacology. 1980;20:256–267. doi: 10.1159/000137371. [DOI] [PubMed] [Google Scholar]
- 10.Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81:194–204. doi: 10.1038/sj.clpt.6100038. [DOI] [PubMed] [Google Scholar]
- 11.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82:726–733. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- 12.Brown G, Zemcov SJ, Clarke AM. Effect of probenecid on cefazolin serum concentrations. J Antimicrob Chemother. 1993;31:1009–1011. doi: 10.1093/jac/31.6.1009. [DOI] [PubMed] [Google Scholar]
- 13.Shitara Y, Sato H, Sugiyama Y. Evaluation of drug-drug interaction in the hepatobiliary and renal transport of drugs. Annu Rev Pharmacol Toxicol. 2005;45:689–723. doi: 10.1146/annurev.pharmtox.44.101802.121444. [DOI] [PubMed] [Google Scholar]
- 14.Sakurai Y, Motohashi H, Ogasawara K, Terada T, Masuda S, Katsura T, Mori N, Matsuura M, Doi T, Fukatsu A, Inui K. Pharmacokinetic significance of renal OAT3 (SLC22A8) for anionic drug elimination in patients with mesangial proliferative glomerulonephritis. Pharm Res. 2005;22:2016–2222. doi: 10.1007/s11095-005-8383-5. [DOI] [PubMed] [Google Scholar]
- 15.Khamdang S, Takeda M, Babu E, Noshiro R, Onozato ML, Tojo A, Enomoto A, Huang XL, Narikawa S, Anzai N, Piyachaturawat P, Endou H. Interaction of human and rat organic anion transporter 2 with various cephalosporin antibiotics. Eur J Pharmacol. 2003;465:1–7. doi: 10.1016/S0014-2999(03)01381-5. [DOI] [PubMed] [Google Scholar]
- 16.Muck W, Mai I, Fritsche L, Ochmann K, Rohde G, Unger S, Johne A, Bauer S, Budde K, Roots I, Neumayer HH, Kuhlmann J. Increase in cerivastatin systemic exposure after single and multiple dosing in cyclosporine-treated kidney transplant recipients. Clin Pharmacol Ther. 1999;65:251–261. doi: 10.1016/S0009-9236(99)70104-9. [DOI] [PubMed] [Google Scholar]
- 17.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–446. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Tsuruoka S, Ioka T, Wakaumi M, Sakamoto K, Ookami H, Fujimura A. Severe arrhythmia as a result of the interaction of cetirizine and pilsicainide in a patient with renal insufficiency: first case presentation showing competition for excretion via renal multidrug resistance protein 1 and organic cation transporter 2. Clin Pharmacol Ther. 2006;79:389–396. doi: 10.1016/j.clpt.2005.12.302. [DOI] [PubMed] [Google Scholar]
- 19.Jaehde U, Sorgel F, Reiter A, Sigl G, Naber KG, Schunack W. Effect of probenecid on the distribution and elimination of ciprofloxacin in humans. Clin Pharmacol Ther. 1995;58:532–541. doi: 10.1016/0009-9236(95)90173-6. [DOI] [PubMed] [Google Scholar]
- 20.Desrayaud S, Guntz P, Scherrmann JM, Lemaire M. Effect of the P-glycoprotein inhibitor, SDZ PSC 833, on the blood and brain pharmacokinetics of colchicine. Life Sci. 1997;61:153–163. doi: 10.1016/S0024-3205(97)00370-6. [DOI] [PubMed] [Google Scholar]
- 21.Speeg KV, Maldonado AL, Liaci J, Muirhead D. Effect of cyclosporine on colchicine secretion by a liver canalicular transporter studied in vivo. Hepatology. 1992;15:899–903. doi: 10.1002/hep.1840150524. [DOI] [PubMed] [Google Scholar]
- 22.Nooter K, Oostrum R, Deurloo J. Effects of verapamil on the pharmacokinetics of daunomycin in the rat. Cancer Chemother Pharmacol. 1987;20:176–178. doi: 10.1007/BF00253975. [DOI] [PubMed] [Google Scholar]
- 23.Yesair DW, Schwartzbach E, Shuck D, Denine EP, Asbell MA. Comparative pharmacokinetics of daunomycin and adriamycin in several animal species. Cancer Res. 1972;32:1177–1183. [PubMed] [Google Scholar]
- 24.Lam JL, Shugarts SB, Okochi H, Benet LZ. Elucidating the effect of final-day dosing of rifampin in induction studies on hepatic drug disposition and metabolism. J Pharmacol Exp Ther. 2006;319:864–870. doi: 10.1124/jpet.106.108282. [DOI] [PubMed] [Google Scholar]
- 25.Ding R, Tayrouz Y, Riedel KD, Burhenne J, Weiss J, Mikus G, Haefeli WE. Substantial pharmacokinetic interaction between digoxin and ritonavir in healthy volunteers. Clin Pharmacol Ther. 2004;76:73–84. doi: 10.1016/j.clpt.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Inotsume N, Nishimura M, Nakano M, Fujiyama S, Sato T. The inhibitory effect of probenecid on renal excretion of famotidine in young, healthy volunteers. J Clin Pharmacol. 1990;30:50–56. doi: 10.1002/j.1552-4604.1990.tb03438.x. [DOI] [PubMed] [Google Scholar]
- 27.Tahara H, Kusuhara H, Chida M, Fuse E, Sugiyama Y. Is the monkey an appropriate animal model to examine drug-drug interactions involving renal clearance? Effect of probenecid on the renal elimination of H2 receptor antagonists. J Pharmacol Exp Ther. 2006;316:1187–1194. doi: 10.1124/jpet.105.094052. [DOI] [PubMed] [Google Scholar]
- 28.Zheng HX, Huang Y, Frassetto LA, Benet LZ. Elucidating rifampin’s inducing and inhibiting effects on glyburide pharmacokinetics and blood glucose in healthy volunteers: unmasking the differential effects of enzyme induction and transporter inhibition for a drug and its primary metabolite. Clin Pharmacol Ther. 2009;85:78–85. doi: 10.1038/clpt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, Giacomini KM. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83:273–280. doi: 10.1038/sj.clpt.6100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Breedveld P, Zelcer N, Pluim D, Sonmezer O, Tibben MM, Beijnen JH, Schinkel AH, van Tellingen O, Borst P, Schellens JH. Mechanism of the pharmacokinetic interaction between methotrexate and benzimidazoles: potential role for breast cancer resistance protein in clinical drug-drug interactions. Cancer Res. 2004;64:5804–5811. doi: 10.1158/0008-5472.CAN-03-4062. [DOI] [PubMed] [Google Scholar]
- 31.VanWert AL, Bailey RM, Sweet DH. Organic anion transporter 3 (Oat3/Slc22a8) knockout mice exhibit altered clearance and distribution of penicillin G. Am J Physiol Renal Physiol. 2007;293:F1332–F1341. doi: 10.1152/ajprenal.00319.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiga T, Hashiguchi M, Urae A, Kasanuki H, Rikihisa T. Effect of cimetidine and probenecid on pilsicainide renal clearance in humans. Clin Pharmacol Ther. 2000;67:222–228. doi: 10.1067/mcp.2000.104018. [DOI] [PubMed] [Google Scholar]
- 33.Chung JY, Cho JY, Yu KS, Kim JR, Oh DS, Jung HR, Lim KS, Moon KH, Shin SG, Jang IJ. Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2005;78:342–350. doi: 10.1016/j.clpt.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol. 2005;68:800–807. doi: 10.1124/mol.105.014019. [DOI] [PubMed] [Google Scholar]
- 35.Bauer LA, Black DJ, Lill JS, Garrison J, Raisys VA, Hooton TM. Levofloxacin and ciprofloxacin decrease procainamide and N-acetylprocainamide renal clearances. Antimicrob Agents Chemother. 2005;49:1649–1651. doi: 10.1128/AAC.49.4.1649-1651.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Somogyi A, McLean A, Heinzow B. Cimetidine-procainamide pharmacokinetic interaction in man: evidence of competition for tubular secretion of basic drugs. Eur J Clin Pharmacol. 1983;25:339–345. doi: 10.1007/BF01037945. [DOI] [PubMed] [Google Scholar]
- 37.Kajosaari LI, Niemi M, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin Pharmacol Ther. 2005;78:388–399. doi: 10.1016/j.clpt.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Simonson SG, Raza A, Martin PD, Mitchell PD, Jarcho JA, Brown CD, Windass AS, Schneck DW. Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin Pharmacol Ther. 2004;76:167–177. doi: 10.1016/j.clpt.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Schneck DW, Birmingham BK, Zalikowski JA, Mitchell PD, Wang Y, Martin PD, Lasseter KC, Brown CD, Windass AS, Raza A. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75:455–463. doi: 10.1016/j.clpt.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 40.Carr RA, Pasutto FM, Foster RT. Influence of cimetidine coadministration on the pharmacokinetics of sotalol enantiomers in an anaesthetized rat model: evidence supporting active renal excretion of sotalol. Biopharm Drug Dispos. 1996;17:55–69. doi: 10.1002/(SICI)1099-081X(199601)17:1<55::AID-BDD938>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 41.Ullrich KJ. Affinity of drugs to the different renal transporters for organic anions and organic cations. In: Amidon GL, Sadee W, editors. Membrane Transporters as Drug Targets. New York: Kluwer Academic/Plenum Publishers; 1999. pp. 159–179. [DOI] [PubMed] [Google Scholar]
- 42.Yokogawa K, Takahashi M, Tamai I, Konishi H, Nomura M, Moritani S, Miyamoto K, Tsuji A. P-glycoprotein-dependent disposition kinetics of tacrolimus: studies in mdr1a knockout mice. Pharm Res. 1999;16:1213–1218. doi: 10.1023/A:1018993312773. [DOI] [PubMed] [Google Scholar]
- 43.Bajcetic M, Benndorf RA, Appel D, Schwedhelm E, Schulze F, Riekhof D, Maas R, Boger BH. Pharmacokinetics of oral doses of telmisartan and nisoldipine, given alone and in combination, in patients with essential hypertension. J Clin Pharmacol. 2007;47:295–304. doi: 10.1177/0091270006297225. [DOI] [PubMed] [Google Scholar]
- 44.Oh YH, Han HK. Pharmacokinetic interaction of tetracycline with non-steroidal anti-inflammatory drugs via organic anion transporters in rats. Pharmacol Res. 2006;53:75–79. doi: 10.1016/j.phrs.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Dresser MJ, Leabman MK, Giacomini KM. Transporters involved in the elimination of drugs in the kidney: organic anion transporters and organic cation transporters. J Pharm Sci. 2001;90:397–421. doi: 10.1002/1520-6017(200104)90:4<397::AID-JPS1000>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 46.van Giersbergen PL, Bodin F, Dingemanse J. Cyclosporin increases the exposure to tezosentan, an intravenous dual endothelin receptor antagonist. Eur J Clin Pharmacol. 2002;58:243–245. doi: 10.1007/s00228-002-0459-0. [DOI] [PubMed] [Google Scholar]
- 47.Zamboni WC, Houghton PJ, Johnson RK, Hulstein JL, Crom WR, Cheshire PJ, Hanna SK, Richmond LB. Probenecid alters topotecan systemic and renal disposition by inhibiting renal tubular secretion. J Pharmacol Exp Ther. 1998;284:89–94. [PubMed] [Google Scholar]
- 48.Su Y, Hu P, Lee SH, Sinko PJ. Using novobiocin as a specific inhibitor of breast cancer resistant protein to assess the role of transporter in the absorption and disposition of topotecan. J Pharm Pharmaceut Sci. 2007;10:519–536. doi: 10.18433/j3qp4w. [DOI] [PubMed] [Google Scholar]
- 49.Yagi Y, Shibutani S, Hodoshima N, Ishiwata K, Okudaira N, Li Q, Sai Y, Kato Y, Tsuji A. Involvement of multiple transport systems in the disposition of an active metabolite of a prodrug-type new quinolone antibiotic, prulifloxacin. Drug Metab Pharmacokinet. 2003;18:381–389. doi: 10.2133/dmpk.18.381. [DOI] [PubMed] [Google Scholar]
- 50.Aoki M, Iguchi M, Shibasaki S, Kurosawa T, Kato Y, Tsuji A. Transporter-mediated hepatic uptake of ulifloxacin, an active metabolite of a prodrug-type new quinolone antibiotic prulifloxacin, in rats. Drug Metab Pharmacokinet. 2007;22:350–357. doi: 10.2133/dmpk.22.350. [DOI] [PubMed] [Google Scholar]
- 51.Nakashima M, Uematsu T, Kosuge K, Okuyama Y, Morino A, Ozaki M, Takebe Y. Pharmacokinetics and safety of NM441, a new quinolone, in healthy male volunteers. J Clin Pharmacol. 1994;34:930–937. doi: 10.1002/j.1552-4604.1994.tb04007.x. [DOI] [PubMed] [Google Scholar]
- 52.Yamashiro W, Maeda K, Hirouchi M, Adachi Y, Hu Z, Sugiyama Y. Involvement of transporters in the hepatic uptake and biliary excretion of valsartan, a selective antagonist of the angiotensin II AT1-receptor, in humans. Drug Metab Dispos. 2006;34:1247–1254. doi: 10.1124/dmd.105.008938. [DOI] [PubMed] [Google Scholar]
- 53.Sahin S, Benet LZ. The operational multiple dosing half-life: a key to defining drug accumulation in patients and to designing extended release dosage forms. Pharm Res. 2008;25:2869–2877. doi: 10.1007/s11095-008-9787-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/ SLC21 family: phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- 55.Guyton AC, Hall JE. Textbook of Medical Physiology. Philadelphia: Elsevier Saunders; 2006. [Google Scholar]
- 56.Sweet DH, Miller DS, Pritchard JB, Fujiwara Y, Beier DR, Nigam SK. Impaired organic anion transport in kidney and choroid plexus of organic anion transporter 3 (Oat3 (Slc22a8)) knockout mice. J Biol Chem. 2002;277:26934–26943. doi: 10.1074/jbc.M203803200. [DOI] [PubMed] [Google Scholar]
- 57.van der Valk P, van Kalken CK, Ketelaars H, Broxterman HJ, Scheffer G, Kuiper CM, Tsuruo T, Lankelma J, Meijer CJ, Pinedo HM. Distribution of multi-drug resistance-associated P-glycoprotein in normal and neoplastic human tissues. Analysis with 3 monoclonal antibodies recognizing different epitopes of the P-glycoprotein molecule. Ann Oncol. 1990;1:56–64. [PubMed] [Google Scholar]
- 58.Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE. The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol. 1989;9:1346–1350. doi: 10.1128/mcb.9.3.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka Y, Slitt AL, Leazer TM, Maher JM, Klaassen CD. Tissue distribution and hormonal regulation of the breast cancer resistance protein (Bcrp/Abcg2) in rats and mice. Biochem Biophys Res Commun. 2005;326:181–187. doi: 10.1016/j.bbrc.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 60.Pavlova A, Sakurai H, Leclercq B, Beier DR, Yu AS, Nigam SK. Developmentally regulated expression of organic ion transporters NKT (OAT1), OCT1, NLT (OAT2), and Roct. Am J Physiol Renal Physiol. 2000;278:F635–F643. doi: 10.1152/ajprenal.2000.278.4.F635. [DOI] [PubMed] [Google Scholar]
- 61.Sekine T, Cha SH, Endou H. The multispecific organic anion transporter (OAT) family. Eur J Physiol. 2000;440:337–350. doi: 10.1007/s004240000297. [DOI] [PubMed] [Google Scholar]
- 62.Cha SH, Sekine T, Kusuhara H, Yu E, Kim JY, Kim DK, Sugiyama Y, Kanai Y, Endou H. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J Biol Chem. 2000;275:4507–4512. doi: 10.1074/jbc.275.6.4507. [DOI] [PubMed] [Google Scholar]
- 63.Kobayashi Y, Ohshiro N, Shibusawa A, Sasaki T, Tokuyama S, Sekine T, Endou H, Yamamoto T. Isolation, characterization and differential gene expression of multispecific organic anion transporter 2 in mice. Mol Pharmacol. 2002;62:7–14. doi: 10.1124/mol.62.1.7. [DOI] [PubMed] [Google Scholar]
- 64.Wang Q, Yang H, Miller DW, Elmquist WF. Effect of the P-glycoprotein inhibitor, cyclosporin A, on the distribution of rhodamine-123 to the brain: an in vivo microdialysis study in freely moving rats. Biochem Biophys Res Commun. 1995;211:719–726. doi: 10.1006/bbrc.1995.1872. [DOI] [PubMed] [Google Scholar]
- 65.Kunihara M, Nagai J, Murakami T, Takano M. Renal excretion of rhodamine 123, a P-glycoprotein substrate, in rats with glycerol-induced acute renal failure. J Pharm Pharmacol. 1998;50:1161–1165. doi: 10.1111/j.2042-7158.1998.tb03328.x. [DOI] [PubMed] [Google Scholar]
- 66.Tachibana-Iimori R, Tabara Y, Kusuhara H, Kohara K, Kawamoto R, Nakura J, Tokunaga K, Kondo I, Sugiyama Y, Miki T. Effect of genetic polymorphism of OATP-C (SLCO1B1) on lipid-lowering response to HMG-CoA reductase inhibitors. Drug Metab Pharmacokinet. 2004;19:375–380. doi: 10.2133/dmpk.19.375. [DOI] [PubMed] [Google Scholar]
- 67.Lin JH, Los LE, Ulm EH, Duggan DE. Kinetic studies on the competition between famotidine and cimetidine in rats. Evidence of multiple renal secretory systems for organic cations. Drug Metab Dispos. 1988;16:52–56. [PubMed] [Google Scholar]
- 68.Lau YY. Examining the regulation of hepatic drug disposition and metabolism by organic anion transporting peptide, p-glycoprotein, and multidrug resistance-associated protein 2 [dissertation] San Francisco: University of California; 2006. [Google Scholar]
- 69.Kugler AR, Olson SC, Smith DE. Tubular transport mechanisms of quinapril and quinaprilat in the isolated perfused rat kidney: effect of organic anions and cations. J Pharmacokinet Biopharm. 1996;24:349–368. doi: 10.1007/BF02353517. [DOI] [PubMed] [Google Scholar]
- 70.Wu CY. The interactive roles of p-glycoprotein and cytochrome P-450 3A in intestinal and hepatic drug disposition [dissertation] San Francisco: University of California; 2003. [Google Scholar]
- 71.Booth CL, Brouwer KR, Brouwer KL. Effect of multidrug resistance modulators on the hepatobiliary disposition of doxorubicin in the isolated perfused rat liver. Cancer Res. 1998;58:3641–3648. [PubMed] [Google Scholar]