

Abstract
The in vivo metabolic clearance in human has been successfully predicted by using in vitro data of metabolic stability in cryopreserved preparations of human hepatocytes. In the predictions by human hepatocytes, the systematic underpredictions of in vivo clearance have been commonly observed among different datasets. The regression-based scaling factor for the in vitro-to-in vivo extrapolation has mitigated discrepancy between in vitro prediction and in vivo observation. In addition to the elimination by metabolic degradation, the important roles of transporter-mediated hepatic uptake and canalicular excretion have been increasingly recognized as a rate-determining step in the hepatic clearance. It has been, therefore, proposed that the in vitro assessment should allow the evaluation of clearances for both transporter(s)-mediated uptake/excretion and metabolic degradation. This review first outlines the limited ability of subcellular fractions such as liver microsomes to predict hepatic clearance in vivo. It highlights the advantages of cryopreserved human hepatocytes as one of the versatile in vitro systems for the prediction of in vivo metabolic clearance in human at the early development stage. The following section discusses the mechanisms underlying the systematic underprediction of in vivo intrinsic clearance by hepatocytes. It leads to the proposal for the assessment of hepatic uptake clearance as one of the kinetically important determinants for accurate predictions of hepatic clearance in human. The judicious combination of advanced technologies and understandings for the drug disposition allows us to rationally optimize new chemical entities to the drug candidate with higher probability of success during the clinical development.
Key words: hepatic clearance, human hepatocytes, human PK prediction, OATP1B1
INTRODUCTION
It has become critically important to discover more bioavailable drug candidates in many therapeutic targets where the drug would be preferably given per oral to the patients. At the early discovery stage in the development of new drug, the in vitro metabolic stability is routinely examined in a high-throughput manner at the pharmaceutical companies. The rationale of this strategy is that the in vitro metabolic stability in the preparations from human should reasonably well predict in vivo clearance in human. In parallel with the in vitro assessment for the metabolic stability in preclinical species as well as human, the pharmacokinetics of new chemical entities is often examined in order to identify the major route(s) of elimination in animals from in vitro/in vivo correlation analysis. These data assure that any in vitro data using hepatic derived systems would be meaningful for the quantitative prediction in human. Predictions based on the metabolic clearance from in vitro samples have been able to account for the potential interspecies differences in the metabolism. However, much evidence such as drug–drug interactions through the inhibition of transporters (1–3) and interindividual variations of pharmacokinetics through the genetic polymorphism in the transporters (4,5) has also suggested that the transporter-mediated uptake and excretion processes, often or partly at least, become a rate-determining step as a determinant of hepatic clearance and pharmacokinetics in human for many drugs (4,6–8). Therefore, the accuracy of prediction for the hepatic clearance in human will be certainly improved by integrating transporter-mediated process(es) into the existing paradigm for the prediction, based on the understanding of rate-determining step in the hepatic clearance in human.
PREDICTION OF HEPATIC CLEARANCE: FROM LIVER MICROSOMES TO HEPATOCYTES
Based on the accumulated data for the quantitative prediction of hepatic clearance from in vitro studies with rat liver microsomes and isolated rat hepatocytes, the prediction strategy was proposed by Houston in 1994 (9) and 1997 (10). Following those reports, many results were published and consistently indicated that the in vivo hepatic (or total body) clearance could be reasonably well predicted from the metabolic (intrinsic) clearance determined in the in vitro system on the basis of existing pharmacokinetic premises (9–26). Most studies in early years used rat liver microsomes and/or rat hepatocytes for the prediction of clearance due to the feasibility of in vitro experiments and limited availability of human samples (9–17,21,24). The prediction strategy established in rat has been extended to that for human as the in vitro samples (liver microsomes, liver slices, and freshly isolated hepatocytes) from human have become prevalent since early 1990s (18–20,22,23,25–27). The prediction of pharmacokinetics in human with reasonable level of confidence is one of the crucial components in the assessment of probability of success (POS) of drug candidate at the preclinical evaluation stage. Therefore, the in vitro studies with widely and routinely available human preparations certainly contributed to the reduction of attrition rate attributable to the inappropriate pharmacokinetics in human during the clinical development at the pharmaceutical companies (28,29).
It has been long recognized that the isolated rat hepatocytes generally provide better in vitro grounds for the quantitative/qualitative predictions of in vivo metabolism than liver slices and subcellular fractions (10). The advantages of using isolated rat hepatocytes over other in vitro preparations have been well documented in the context of quantitative predictions of in vivo metabolism. For example, the predictions by isolated rat hepatocytes for the inhibitory effects of metabolite(s) on the disposition of parent compounds (i.e., product inhibition) were quantitatively successful for diazepam (12) and phenytoin (17) in rat: the overwhelming accumulation of inhibitory metabolite(s) in the liver microsomes underpredicted in vivo metabolism of parent drugs due to the lack of conjugation activities. In vitro preparations with activated liver microsomes also underpredicted in vivo UDP-glucuronosyltransferase activities in rat (30) and human (31,32), likely due to the insufficient activation of conjugative activity in liver microsomes to the extent relevant to in vivo (31) and/or to the potent competitive inhibitions by unsaturated long-chain fatty acids (oleic, linoleic, and arachidonic acids) released during microsomal incubations (33); in contrast, the metabolic clearance primarily catalyzed by glucuronidation in hepatocytes generally well predicted in vivo clearance in human (31,34,35). It has been consistently demonstrated that the accuracy of predictions for hepatic clearance by hepatocytes was superior to that by liver microsomes for a wide range of drugs in rat (10,21) and for the drugs predominantly metabolized by glucuronidation in human (31).
Despite the data suggesting that the preparation of freshly isolated hepatocytes is a superior in vitro tool for the prediction of metabolic clearance in human to that of subcellular fractions, the unpredictable availability of fresh liver from human precluded this in vitro system from routine applications. Cryopreserved preparation of human hepatocytes has instead become prevalent and versatile in vitro system alternative to freshly isolated human hepatocytes. Cryopreserved human hepatocytes have been reported to retain qualitatively and quantitatively comparable metabolic activities with those in hepatocytes from fresh liver (36–42). However, the comparisons between predicted and observed metabolic clearance in human in different datasets (27,35,41,43,44) have indicated that the in vitro metabolic clearance obtained from both human liver microsomes (43) and cryopreserved human hepatocytes (27,35,41,44,45) systematically underpredicted in vivo metabolic clearance by ∼9 and 3∼6-fold, respectively. Consequently, in order to mitigate the quantitative discrepancy between in vitro prediction and in vivo observation in human, the empirical and regression-based scaling factors specifically obtained from the in vitro preparation used for the prediction, instead of biological (anatomical) scaling factors such as microsomal content and hepatocellularity, were employed for the extrapolation of in vitro data to in vivo (27,35,43,45). Empirical (or regression-based) scaling factor represents the discrepancy (observed/predicted) between calculated metabolic (or intrinsic clearance) from in vivo hepatic clearance and those predicted from in vitro metabolic clearance in the cryopreserved human hepatocytes by using hepatocellularity or in human liver microsomes by using liver microsomal content as a biological scaling factor. For example, regression analysis indicated that the calculated metabolic clearance from in vivo was approximately threefold larger (8.5 × 109 cells per kilogram body weight, solid line in Fig. 1) than those predicted from hepatocellularity (3.1 × 109 cells per kilogram body weight, dotted line in Fig. 1) (27). Similar regression analyses for different datasets found that the empirical scaling factors necessary for accurate predictions were 5.2-fold (35) and 5.6-fold larger (45) than hepatocellularity (2.5–3.1 × 109 cells per kilogram body weight) and ninefold larger than the biological scaling factor based on human liver microsomal content (856-mg microsomal protein per kilogram body weight) (43). In order to examine the potential discrepancy and adequate (regression-based) scaling factor for the prediction in human, the values of in vitro metabolic clearance were determined for seven standard drugs (naloxone, buspirone, verapamil, lidocaine, imipramine, metoprolol, and timolol) in the preparations of cryopreserved human hepatocytes from ten different donors (Fig. 1) (27). Consistent with the observations in other datasets (27,35,41,43–45), large interindividual variations were obtained in the in vitro metabolic clearance among different donors by a maximum of tenfold. Interestingly, the prediction for metabolic clearance in rat from rat liver microsomes and rat hepatocytes did not show any significant systematic bias (21), suggesting that the discrepancy can be only evident in human (27,35,41,43–45). Possible mechanisms for the systematic underprediction by biological scaling factors (41,46) are discussed in the following section. For human tissue preparation, there is an impact from extrinsic factors such as tissue handling and storage procedures on the preparation used for the prediction (43). Therefore, the empirical scaling factor needs to be determined in the human preparation used for the extrapolation of in vitro data to in vivo.
Fig. 1.

Interindividual variation of metabolic clearance among cryopreserved human hepatocytes and an empirical scaling factor for the quantitative prediction. Data are from (27)
Negatively biased interindividual variations of metabolic clearance in the in vitro human preparations suggested that a careful pooling of liver preparations as well as an empirical evaluation of scaling factor would improve accuracy of prediction by minimizing potentially biased coverage of metabolizing enzyme activities in the individual preparation. Figure 2 shows the example of the pooling process for the in vitro preparations consisting of cryopreserved human hepatocytes from multiple donors. The calculated values of metabolic clearances from in vivo data (reported from clinical studies) for seven standard drugs (naloxone, buspirone, verapamil, lidocaine, imipramine, metoprolol, and timolol which are known to undergo the metabolic degradation catalyzed by different enzymes responsible for both phases I and II metabolism in human) were compared with those determined in the individual preparation of cryopreserved human hepatocytes from ten different donors (donor # a to j). As shown in the figure, metabolic activity for imipramine (standard compound e) in the preparations (donor # b and d) deviated from the regression lines between in vivo and in vitro metabolic clearance, suggesting that these two preparations had significantly lower activities for imipramine metabolism than those calculated by the empirical scaling factor (i.e., slope of regression line, cells per kilogram). Therefore, the regression line with better correlation between in vivo and in vitro metabolic clearance represents a better coverage by empirical (regression-based) scaling factor for major metabolizing enzymes in human. In this study, the in vitro metabolic clearance determined in the pooled sample consisting of preparations from donors f and h provided predictions of in vivo metabolic clearance based on the best regression line for the in vitro-to-in vivo extrapolation for 12 drugs [i.e., seven standard drugs (naloxone, buspirone, verapamil, lidocaine, imipramine, metoprolol, and timolol) five test drugs (propranolol, diclofenac, quinidine, phenacetin, and caffeine)].
Fig. 2.

Correlation of metabolic clearance between in vitro and in vivo among cryopreserved hepatocytes from different donors
Metabolic stability is routinely examined for new chemical entities at the discovery stage in conjunction with the early identification of metabolic “soft spot” which facilitates structure modification for the lead optimization. Because of the presence of all hepatic drug-metabolizing enzymes and cofactors at physiological levels, the intact hepatocytes are generally more relevant in vitro experimental system than liver microsomes even for the early screening of metabolic stability (40,47,48). In order to clarify the limited predication ability of the results from metabolic stability data with liver microsomes for the in vivo metabolic clearance, the values of metabolic stability in liver microsomes (as percent remaining) are plotted against those in hepatocytes (as metabolic clearance) based on in-house dataset (Fig. 3). As clearly shown in these figures, the compounds having metabolic stability in liver microsomes >30% remaining could have a wide range of metabolic stability (metabolic clearance) in the corresponding hepatocytes. In contrast, the compounds having metabolic stability in liver microsomes <30% remaining were very unstable also in the hepatocytes with the metabolic clearance of >100 mL/min per kilogram. Dataset with rat liver microsomes and hepatocytes was much larger than that with human samples, while no species difference was observed in the cutoff (30%) for the relation between percent remaining in liver microsomes and metabolic clearance in hepatocytes. Therefore, the usefulness of the screening study with human liver microsomes would be limited to the higher-throughput screening of no promising compounds in terms of metabolic stability which needs to undergo structure modification based on the information on the metabolic “soft spot.”
Fig. 3.

Comparison between liver microsomes and hepatocytes for the metabolic stability screening at discovery stage
One of the goals at drug discovery stage is to identify the drug candidates which likely possess appropriate pharmacokinetics with lower hepatic clearance and higher oral bioavailability in human especially when the drug is favorably given to the patients by oral administration. Shibata et al. (27) reported that the in vivo hepatic clearance in human was successfully predicted with cryopreserved human hepatocytes suspended in 100% human serum for the model drugs which mainly undergo the elimination by the metabolism in human liver. The subsequent reports consistently indicated that the hepatocytes suspended in 100% serum have more accurately predicted in vivo metabolic clearance in rat (49) and human (47,50) than those in the absence of serum. The method has been applied to the prediction of pharmacokinetics (clearance, bioavailability, and terminal half-life) in human for drug candidates, and the accuracy of predictions was evaluated by the comparison of predicted values with the observed pharmacokinetics in first-in-human (FIH) studies (Table I and Fig. 4). The oral clearance (CLoral) was calculated from area under the curve (AUC) and dose after oral administrations of drug candidates in the FIH studies as follows:
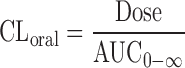 |
1 |
For the prediction of CLoral from in vitro data, the metabolic clearance determined in the cryopreserved human hepatocytes suspended in 100% human serum was extrapolated to that for in vivo (CLmet) with an aid of the regression-based empirical scaling factor for the preparation used as previously described. The hepatic clearance (CLH) and hepatic availability (FH) in human were calculated by the dispersion model (51) incorporating predicted metabolic clearance (CLmet), dispersion number (DN, 0.17) (51), and hepatic blood flow in human (QH, 21 mL/min per kilogram) (52) as follows:
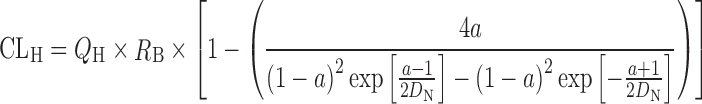 |
2 |
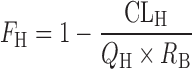 |
3 |
where  ,
, 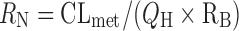 , and RB represents blood-to-plasma concentration ratio. By using the calculated CLH and the mean value of apparent volume of distribution (Vd) from preclinical species corrected by the species difference in the unbound fraction in plasma (18,23,53), the terminal half-life (t1/2) in human was predicted as follows (18):
, and RB represents blood-to-plasma concentration ratio. By using the calculated CLH and the mean value of apparent volume of distribution (Vd) from preclinical species corrected by the species difference in the unbound fraction in plasma (18,23,53), the terminal half-life (t1/2) in human was predicted as follows (18):
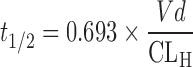 |
4 |
Based on the reports that the rat can serve as a reliable animal model to predict drug absorption in human based on the linear correlation in the percent of absorbed dose between rat and human (54,55), the absorption fraction (fa) in human was predicted from the combined recovery of radioactivity from bile and urine after an oral administration of radiolabeled compound to the bile duct cannulated rat. Assuming that the intestinal metabolism was negligible for the compounds (i.e., FG = 1 in Eq. 5), the predicted fa was then used for the prediction of CLoral as follows (23):
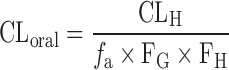 |
5 |
As shown in Fig. 4, the predicted terminal half-lives and CLoral reasonably well agreed with those observed in FIH studies for all ten drug candidates. These data suggested that the in vitro system consisting of cryopreserved human hepatocytes suspended in 100% serum can predict pharmacokinetics in human for the drug candidates which undergo hepatic metabolism as a predominant route of elimination. The prediction has facilitated prioritization/selection of clinical development candidates with POS assessment of human pharmacokinetics at the preclinical stage prior to the entry of clinical studies.
Table I.
Prediction of Terminal Half-life and Oral Clearance in First-in-Human Studies (Data From In-house Database)
| Compound # | Observed in FIH study | Predicted | ||||
|---|---|---|---|---|---|---|
| Dose (mg) | t1/2 (h) | CLoral a (mL/min/kg) | t 1/2 b (h) | f a c | CLoral d (mL/min per kilogram) | |
| 1 | 0.5 | 5.1 | 0.36 | 2–5 | 0.59 | 1.3 |
| 2 | 1 | 11.4 | 5.5 | 4–5 | 0.66 | 6.1 |
| 3 | 0.4 | 9.4 | 1.2 | 6–11 | 0.80 | 0.54 |
| 4 | 5 | 7.9 | 2.4 | 2–3 | 0.84 | 7.9 |
| 5 | 12.5 | 13.1 | 10.9 | 4–10 | 0.82 | 9.3 |
| 6 | 2.5 | 14.1 | 33.4 | 14 | 0.68 | 35.8 |
| 7 | 0.5 | 3.5 | 26.5 | 3–6 | 0.69 | 22.5 |
| 8 | 5 | 40 | 1.0 | 20 | 0.76 | 3.6 |
| 9 | 10 | 19 | 2.1 | 9 | 0.79 | 9.2 |
| 10 | 350 | 8.9 | 12.8 | 9 | 0.50 | 24.6 |
a
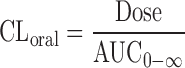
b
 where Vd represents the observed Vd in preclinical species (corrected by unbound fractions) and CLH represents the value predicted from the incubation of cryopreserved human hepatocytes suspended in 100% human serum
where Vd represents the observed Vd in preclinical species (corrected by unbound fractions) and CLH represents the value predicted from the incubation of cryopreserved human hepatocytes suspended in 100% human serum
cCombined fractions of radioactivity (percent of dose) excreted into bile and urine after PO dosing of radiolabeled compound to bile duct cannulated rats
d
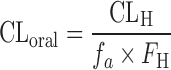 where CLH and F
H represent the values predicted from cryopreserved human hepatocytes suspended in 100% serum.
where CLH and F
H represent the values predicted from cryopreserved human hepatocytes suspended in 100% serum.
Fig. 4.

Comparison of predicted and observed oral clearance in First-in-Human (FIH) studies. Numbers are corresponding to those compounds in Table I
LIMITATION OF IN VITRO METABOLIC CLEARANCE TO PREDICT IN VIVO INTRINSIC CLEARANCE
Systematic underpredictions of in vivo metabolic clearance have been reported for human when the standard biological scaling factors were used to extrapolate the values of metabolic clearance determined in human liver microsomes and isolated (or cryopreserved) human hepatocytes to those in vivo as described in the previous section (22,23,26,27,35,41,43–46). Iwatsubo et al. (26) reported significant discrepancy between in vivo intrinsic clearance and in vitro metabolic clearance for 25 drugs in human (Fig. 5). As shown in the figure, for most outliers having >5-fold difference between in vivo and in vitro, the calculated values of in vivo intrinsic clearance from human were sevenfold to 80-fold higher than that predicted from the metabolic clearance determined in the in vitro studies. Several possible reasons for the discrepancy are discussed in the following sections.
Fig. 5.

Correlation between in vivo intrinsic clearance and in vitro metabolic clearance for 25 drugs in human. Data are taken from (26)
The interindividual variation in the metabolic activities would likely be caused by the intrinsic factors such as genetic polymorphism, smoking, alcohol drinking, or drug administration. However, the intrinsic variability itself would result in a uniform variation, leading to both overpredictions and underpredictions of in vivo clearance by in vitro metabolic clearance. The values of in vitro metabolic clearance of midazolam determined in S-13 fractions prepared from biopsy samples were extrapolated to the hepatic clearance by using standard biological scaling factor (56,57). The predicted clearance was well correlated with in vivo clearance calculated from pharmacokinetic information on the same individuals, irrespective of a large interindividual difference in the CYP3A content between 1.6 and 27.3 pmol/mg of S-13 protein. These results suggested that the extrapolation of in vitro metabolic clearance determined in the in vitro samples prepared under well-controlled condition (e.g., biopsy) well predicts in vivo metabolic clearance and interindividual variation in human by using biological scaling factor (56,57) as observed for rat studies (21). Therefore, the extrinsic factors such as preparation process and/or storage condition of liver samples from human are likely responsible for the potential loss of metabolic activity, resulting in the systematic underprediction by biological scaling factors for human samples (23,27,38,41).
The extrahepatic metabolism for the drug in human has been well documented (58–60), which could theoretically contribute to the discrepancy between in vivo intrinsic clearance and in vitro metabolic clearance (in vivo > in vitro). Metabolizing enzymes in the extrahepatic tissues include CYP isoforms (61), flavin-containing monooxygenases (62,63), carboxylesterase (64–67), and UDP-glucuronosyltransferases (68). Especially for the compounds undergoing CYP3A-mediated metabolism in the liver with negligible urinary excretion as a parent form, the intestinal first-pass metabolism (as a clearance in the intestine, CLG, in Eq. 6) can significantly contribute to the total body clearance (CLtot) as a hidden factor as follows (69–71):
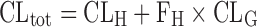 |
6 |
Therefore, the hepatic clearance (and in vivo metabolic clearance in the liver) calculated from total body clearance is likely overestimated by ignoring potential clearance from intestinal metabolism (Eq. 6). However, the contribution of intestinal first-pass metabolism has been reported to be quantitatively significant for CYP3A substrates only when the compound undergoes extensive first-pass metabolism in the liver with the hepatic metabolic clearance for unbound drug of >100 mL/min per kilogram (which corresponds to >4 mL/min per gram liver assuming that the liver and body weights in human are 1,700 g and 70 kg, respectively) (72,73). These data suggested that the overestimation would be only evident for the compounds extensively metabolized by both liver and intestine such as cyclosporine (74), tacrolimus (75), and midazolam (76,77). Generally, the quantitatively important contribution of extrahepatic metabolism to the total body clearance becomes discernible by the specialized surgical procedures applied to assess the AUC values after drug administration and blood sampling at a number of sites relative to the liver and the extrahepatic organs of interest, which is practical only in the experimental animals (30,70,78). Instead, the judicious combination of in vitro metabolic clearance from preclinical animals as well as human and in vivo total body clearance in preclinical animals has been successfully adopted for the prediction of hepatic clearance in human with an aid of empirical and drug-specific scaling factor for the in vitro-to-in vivo extrapolation (43,79,80). Drug-specific scaling factor represents a correction factor for any systematic difference between in vitro and in vivo parameters or systematic underprediction of in vivo clearance caused by indiscernible factor(s) including potential contribution of extrahepatic metabolism determined by the in vitro/in vivo correlation (or IVIVC) approach. Although the mechanism(s) of discrepancy would not be fully understood by the time when the new drug candidate enters the predevelopment stage, the IVIVC method has been helpful for the preliminary prediction of clearance in human.
Systematic underprediction of in vivo metabolic clearance can be also caused by the lack of appropriate correction of nonspecific bindings of compounds to the in vitro incubation matrices such as microsomal lipids and cellular components. It is an important tenet of pharmacokinetics that the only unbound compound, which is assumed to undergo the free exchange and reach rapid equilibrium between outside and inside cells, can be subject to the metabolism and excretion. Therefore, the unbound fractions in blood and/or in the in vitro incubation matrices have been often used to convert the values from enzyme kinetics into that for the unbound compound in order to quantitatively predict pharmacokinetics in experimental animals and human (19,35,43,81,82). The prediction ability of in vitro metabolic data obtained from human liver microsomes was examined for 29 drugs with wide ranges of structure and physicochemical property (19): predictions for the hepatic clearance (CLH) were carried out with “well-stirred” model (83) either disregarding both unbound fractions in the blood (fB) and in the microsomal reaction mixture (fm; Eq. 7), incorporating only fB (Eq. 8) or incorporating both fB and fm (Eq. 9).
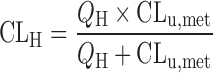 |
7 |
 |
8 |
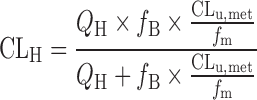 |
9 |
As shown in Fig. 6, the predictions based on the in vitro values reasonably well agreed with human hepatic clearance when both unbound fractions were taken into predictions (shown by open circles in Fig. 6). For these drugs, the systematic underprediction of hepatic clearance was observed if only unbound fraction in the blood was incorporated into the equation (shown by closed triangles in Fig. 6). On the other hand, the discrepancy (as expressed by fold error between predicted and observed hepatic clearance) became indiscernible among prediction models with or without taking unbound fractions into the prediction for the drugs with high hepatic clearance. As the hepatic clearance becomes closer to hepatic blood flow, the clearance is more independent of metabolic (intrinsic) clearance consisting of unbound fractions (in blood/microsomal incubation mixture) and intrinsic clearance for unbound form. Results indicated that the incorporation of both unbound fractions (fB and fm) would provide best predictions of human hepatic clearance based on the in vitro microsomal data regardless of structure, physicochemical property, or hepatic clearance of drugs.
Fig. 6.

Accuracy of prediction for the hepatic clearance in human from in vitro metabolic clearance with or without incorporating unbound fractions. Data in the shaded area are considered to be successful (0.5 < fold error < 2). Data are taken from (19)
The hepatic clearance is described by different equations, depending on the mathematical model used for describing disposition of compounds in the liver, and the relation between hepatic clearance and intrinsic clearance (CLint) depends on the models as shown by Fig. 7. Under the assumption that the drug (or chemical entity) is mixed infinitely well inside the liver, the “well-stirred” model is applicable to the hepatic clearance (83):
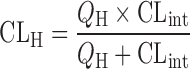 |
10 |
In the opposite extreme case in terms of flow dynamics of solutes in the sinusoidal space of the liver, the drug is mixed only in the infinitely small section along the flow path from input to output of the liver, the “parallel-tube” model is applicable (83):
 |
11 |
In addition to the aforementioned two extreme cases, based on the analysis on the distribution of hepatic residence time of solutes after the bolus injection into the liver, the “dispersion” model (Eq. 2) described in the previous section was introduced as the mathematical model more relevant to the observed flow dynamics in the liver than others (84):
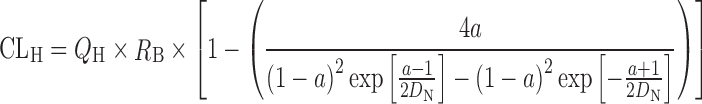 |
12 |
where  and
and 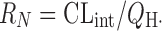 DN is the dispersion number which determines the extent of dispersion of solutes in the liver: the “dispersion” model (Eq. 2) becomes both “well-stirred” (Eq. 10) and “parallel-tube” (Eq. 11) models when the DN approaches infinite (i.e., infinitely mixed condition) and zero (i.e., no mixing condition), respectively. The prediction of in vivo hepatic clearance from the same intrinsic clearance is not significantly different among models, while the calculation of in vivo intrinsic clearance from the observed hepatic clearance is more dependent on the models for the compounds with higher hepatic clearance, closer to the hepatic blood flow rate (Fig. 7a). For example, the values of in vivo intrinsic clearance for the hypothetical compound having hepatic clearance of 20 mL/min per kilogram are calculated to be 420, 93, and 64 mL/min per kilogram by “well-stirred,” “dispersion,” and “parallel-tube” models, respectively, when the hepatic blood flow rate (QH) is assumed to be 21 mL/min per kilogram (52). For the direct comparison of intrinsic (metabolic) clearance between in vivo and in vitro, the “in vivo” intrinsic clearance is often calculated from in vivo clearance by using “well-stirred” model because of the mathematical simplicity. The calculation can yield the “overestimated in vivo” intrinsic clearance, leading to the underprediction by the in vitro intrinsic (metabolic) clearance. On the other hand, the prediction of oral clearance (Fig. 7b), which directly relates hepatic clearance and availability with information on absorption fraction to AUC value after oral administration (Eqs. 1 and 5), highly depends on the model used for the prediction, especially for the compounds with relatively high intrinsic clearance (> 20 mL/min per kilogram). Comparisons of the accuracy in the predictions among different mathematical models have been reported (10,21,79). Results indicated that the three models predicted hepatic clearance with equal levels of accuracy (10,21), while the predictions of in vivo metabolic clearance from in vitro data were comparable between “parallel-tube” and “dispersion” models, and both more accurate with less biased and higher precision than “well-stirred” model (21) or the “dispersion” model provided more reliable prediction for the high-clearance drugs than the other models (20,85–87).
DN is the dispersion number which determines the extent of dispersion of solutes in the liver: the “dispersion” model (Eq. 2) becomes both “well-stirred” (Eq. 10) and “parallel-tube” (Eq. 11) models when the DN approaches infinite (i.e., infinitely mixed condition) and zero (i.e., no mixing condition), respectively. The prediction of in vivo hepatic clearance from the same intrinsic clearance is not significantly different among models, while the calculation of in vivo intrinsic clearance from the observed hepatic clearance is more dependent on the models for the compounds with higher hepatic clearance, closer to the hepatic blood flow rate (Fig. 7a). For example, the values of in vivo intrinsic clearance for the hypothetical compound having hepatic clearance of 20 mL/min per kilogram are calculated to be 420, 93, and 64 mL/min per kilogram by “well-stirred,” “dispersion,” and “parallel-tube” models, respectively, when the hepatic blood flow rate (QH) is assumed to be 21 mL/min per kilogram (52). For the direct comparison of intrinsic (metabolic) clearance between in vivo and in vitro, the “in vivo” intrinsic clearance is often calculated from in vivo clearance by using “well-stirred” model because of the mathematical simplicity. The calculation can yield the “overestimated in vivo” intrinsic clearance, leading to the underprediction by the in vitro intrinsic (metabolic) clearance. On the other hand, the prediction of oral clearance (Fig. 7b), which directly relates hepatic clearance and availability with information on absorption fraction to AUC value after oral administration (Eqs. 1 and 5), highly depends on the model used for the prediction, especially for the compounds with relatively high intrinsic clearance (> 20 mL/min per kilogram). Comparisons of the accuracy in the predictions among different mathematical models have been reported (10,21,79). Results indicated that the three models predicted hepatic clearance with equal levels of accuracy (10,21), while the predictions of in vivo metabolic clearance from in vitro data were comparable between “parallel-tube” and “dispersion” models, and both more accurate with less biased and higher precision than “well-stirred” model (21) or the “dispersion” model provided more reliable prediction for the high-clearance drugs than the other models (20,85–87).
Fig. 7.

Effects of intrinsic clearance (CLint) on hepatic clearance (CLH; a) and oral clearance (CLoral; b) in various mathematical models. Simulations were carried out by “well-stirred” model (Eq. 10), “parallel-tube” model (Eq. 11) and “dispersion” model (Eq. 2). The values of Q
H, f
B, and D
N are 21 (mL/min/kg), 1.0, and 0.17, respectively. The value of CLoral was calculated from CLH and FH (Eq. 3) based on the Eq. 5, assuming 
The expression of intrinsic clearance for the unbound compound involved in the overall elimination in the liver (CLint,all) has been further expanded into the incorporation of distinct processes representing diffusions on the sinusoidal membrane and biliary excretion as well as metabolic degradation (88–90). The general (or expanded) expression of CLint,all (Eq. 13) consists of clearance for the diffusions through membrane in the direction of sinusoid → hepatocytes (PSinf) and that of hepatocytes → sinusoid (PSeff) as well as the clearance for metabolic degradation and/or biliary excretion as a parent form if any (CLint). The equation has become more versatile and crucial as the roles of uptake and efflux transporters as well as diffusion barriers have been increasingly recognized as a rate-determining step in the elimination from the liver (4,6,7).
 |
13 |
Equation 13 indicates that the rate-determining step in the overall elimination in the liver changes according to the relative magnitude of PSeffversus CLint. In the case that the membrane permeability (or PSeff) of the compound is much less than the metabolic clearance (or the clearance for the metabolism plus biliary excretion if any, i.e., PSeff << CLint), CLint,all becomes equal to PSinf as follows:
 |
14 |
This indicates that the calculated in vivo intrinsic clearance from in vivo hepatic clearance does not necessarily represent the clearance for the metabolism and/or biliary excretion. The in vivo intrinsic clearance can be much larger than the predicted intrinsic clearance from in vitro (metabolism) studies if the PSinf is larger than CLint. The value of PSinf is more contributing to the in vivo intrinsic clearance when transporter(s) is (are) more involved in the influx process especially for the less lipophilic compounds in Fig. 5. These compounds likely show larger discrepancies between in vivo (overall) intrinsic clearance and in vitro metabolic clearance (i.e., in vivo intrinsic clearance > in vitro metabolic clearance). In contrast, for the highly membrane permeable compounds (PSinf and PSeff >> CLint), the contribution of transporter(s) to either PSinf or PSeff would be less significant, and the passive diffusion determines the clearance for both directions (PSinf ≈ PSeff). Then, CLint,all becomes equal to CLint as follows:
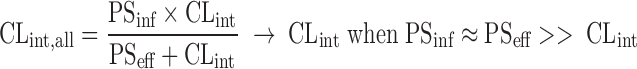 |
15 |
Therefore, under the condition in Eq. 15, the in vivo intrinsic clearance can represent the clearance by metabolism (plus biliary excretion as a parent form if any). One may need to be aware that the relative magnitude of PSeffvs. CLint can also change along with the escalation of dose, due to the decreased CLint by the saturation of metabolism (91,92).
Recent analysis on the relation between plasma protein binding and transporter-mediated hepatic uptake also provided possible explanations for the underprediction of hepatic clearance (93). The trend for the underprediction of in vivo hepatic clearance in human was more evident for the compounds which were highly bound to the plasma proteins (unbound fraction < 5%) and substrates of hepatic uptake transporters (40). These observations suggested that the overestimation of the effect of plasma protein binding can lead to an inaccurate prediction of in vivo intrinsic clearance: the plasma protein binding would not restrict the access of compounds to the hepatocytes as much as anticipated from the assumption of rapid equilibrium between unbound and bound forms of compound in the process of high extraction by a transporter-mediated hepatic uptake (93,94).
PREDICTION OF TARNSPORTER-MEDIATED HEPATIC UPTAKE CLEARANCE FROM IN VITRO DATA
Theoretically, the transporter-mediated uptake often or partly at least becomes a rate-determining step as a determinant of hepatic intrinsic clearance as described in the previous section (4,7,8,95). Much evidence has been accumulated to support the notion that the transporter-mediated hepatic uptake has been playing an important role in the hepatic intrinsic clearance (and hepatic clearance) in human for many drugs (4,7). Such evidence includes the drug–drug interaction by the inhibition of transporters (1–3) and interindividual variation of pharmacokinetics by the genetic polymorphism of transporters (96–100). For example, cyclosporine A significantly increased systemic exposures of cerivastatin (1) and repaglinide (3) by inhibiting OATP1B1-mediated hepatic uptake (2). Genetic polymorphism of OATP1B1 (96–100) caused variation in the pharmacokinetics in human for many drugs such as fexofenadine (101), pitavastatin (102,103), pravastatin (104–110), repaglinide (111,112), rosuvastatin (113), temocapril (109), and valsartan (109). Although these data strongly suggest that the transporters are involved in the elimination process of drugs as much as the pharmacokinetics are affected by the perturbations of transporter activities, the quantitative prediction of overall intrinsic clearance (CLint,all in Eq. 13) by each individual clearance remains to be challenging (90).
For the quantitative extrapolation of human CYP-mediated metabolic activity in the recombinant CYP isoform to that in the liver microsomes, the utilization of relative activity factor (RAF) was proposed by Crespi (114). The RAF represents the ratio of the metabolic activity of the CYP-isoform-specific marker substrate in the human liver microsomes to that in the complementary-DNA-expressed system for the same isoform, and the value quantitatively facilitates bridging metabolic activities between both in vitro systems (115–118). Successful extrapolations of in vitro metabolic clearance obtained in the recombinant CYP isoforms to that in vivo have been well documented for the compounds with less significant contribution of hepatic uptake to overall clearance in human (115,119–121). This approach cannot only provide quantitative information on the relative contribution of CYP isoform(s) involved in the metabolism of the compound of interest but also the preliminary prediction of hepatic clearance attributable to the CYP-mediated metabolism in human at discovery stage (87,116,122,123). Similar methods have been applied to the quantitative assessment for relative contribution of particular transporter to the overall uptake of the compound of interest in hepatocytes by using RAFs with the reference ligands such as taurocholate (124) and estradiol-17β-d-glucuronide (E217βG) (125) to Ntcp and Oatp1a1, respectively. In these approaches, the values of RAF were calculated as the ratios of uptake clearance of test compound to that of reference ligand in the transporter expressing systems (Rexp) and hepatocytes (Rhep). The percentage of contribution was then calculated as follows (124,125):
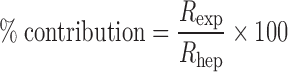 |
16 |
The method has been further extended to the assessment of percentage contributions of OATP1B1- and OATP1B3-mediated hepatic uptakes of pitavastatin (126), E217βG (126), and fexofenadine (127) in human hepatocytes by applying reference ligands, estrone 3-sulfate and cholecystokinin octapeptide for OATP1B1 and OATP1B3, respectively, to the uptake clearance in the transporter-expressing HEK293 cells and cryopreserved human hepatocytes. The concept of RAF with reference ligand to the specific transporter has been successfully utilized to predict percentage contribution of particular transporter to overall hepatic uptake process observed in hepatocytes, and the method warranted potential application to the quantitative predictions for the hepatic clearance in human based on the clearance determined in the specific transporter-expressing cells.
Cryopreserved human hepatocytes have been used for the assessment of hepatic clearance as they have demonstrated to retain the activities of OATP transporters which were reasonably comparable with those in freshly isolated hepatocytes from human (128–130). A typical experimental protocol for the determination of initial uptake rate includes a rapid separation of cellular component from the incubation medium by the centrifugation of cell suspension layered over silicone oil, followed by the determination of compound amount in the cell lysate after the digestion with potassium hydroxide or trichloroacetic acid (131,132). The designated time points for the sampling need to be short enough to calculate the initial slope for the linear uptake into the cell along the incubation time, when the uptake process is assumed to be predominant in the accumulation of compounds in the cell. Also, the radiolabeled compound is often used in order to avoid the potential underestimation of the transported amount in the cell caused by the metabolic degradation. Therefore, most of published studies have examined the hepatic uptake of radiolabeled compounds into the cells through the silicon oil layer by the rapid centrifugation (126,128,133,134), while the method is not practical at the early discovery stage when the radiolabeled compound is not routinely available. Recently, the “media loss” assay in the isolated rat hepatocytes, which determines the loss of parent compound from the incubation medium, has been proposed to quantitatively predict the impact of hepatic uptake on the intrinsic clearance in vivo for the unbound compound in rat (90,135). The values of (unbound) intrinsic clearance for 36 compounds, which were not significantly eliminated into bile or urine as parent forms, were calculated from (1) the initial disappearance rate from whole suspension (conventional method), (2) the AUC of compound concentrations in the incubation medium, and (3) the initial disappearance rate from the incubation medium (Fig. 8). Predicted intrinsic clearance was compared with the observed in vivo values, and the comparison indicated that the conventional prediction (based on the disappearance from whole-cell suspension) yielded the poorest projection (r2 = 0.25 and average ratio = 57) among three approaches, while the prediction from the initial disappearance rate from medium provided the best projection (r2 = 0.72 and average ratio = 3) (135). Large differences (as the fraction of drug in the incubation medium < 0.65) were observed for the majority of compounds between concentrations in the whole-cell suspension (conventional assay) and in the incubation medium (“media loss” assay), suggesting that the incorporation of hepatic uptake clearance is crucial for the accurate prediction of in vivo intrinsic clearance for most of the compounds tested in the study. The values of in vivo intrinsic clearance for the marketed drug transporter substrates (monelukast, bosentan, atorvastatin, and pravastatin) were also predicted well by the in vitro intrinsic clearance based on the initial disappearance rate in the “media loss” assay with freshly isolated human hepatocytes (135) (Table II). Interestingly, the prediction by the “media loss” assay based on the AUC resulted in more significant underprediction (average fold error = 16) than that based on the initial disappearance rate (average fold error = 3). Theoretically, the intrinsic clearance calculated from “media loss” represents the value for overall elimination consisting of diffusions on the sinusoidal membrane and biliary excretion as well as metabolic degradation (Eq. 13) only when the calculation is based on the AUC from the infinite integration of medium concentration. Therefore, the deviation of calculated intrinsic clearance based on the AUC from observed in vivo intrinsic clearance (Fig. 8) suggests potential degradation of cellular activities for transporters/enzymes during incubation for a long time period for the accurate assessment of AUC values (136,137). It was also suggested that the “media loss” assay from the initial disappearance rate may effectively eliminate the terminal phases of polyexponential profiles, the component of which can be affected by the artifacts inherent in the in vitro systems: the in vitro hepatocytes system may have a greater efflux (hepatocytes → incubation medium) clearance by passive diffusion through the larger surface area than the cells only facing to the perisinusoidal space of Disse in the intact liver (138). The concept of “media loss” assay for the accurate assessment of hepatic uptake clearance has been extended to the two-compartment model which enabled accurate predictions of the clearances for hepatic uptake, passive diffusion, and metabolism for atorvastatin, cerivastatin, and indomethacin by simultaneous fittings to the changes of drug concentrations in both incubation medium and cells along the incubation time in rat hepatocytes (139). The hepatic clearances for these drugs were predicted from the obtained parameters, and the predicted values from “media loss” assay well agreed with the observed hepatic clearance, in contrast to that from conventional assay (based on the disappearance from whole-cell suspension). This approach allows the predictions for the contributions of hepatic uptake to both overall hepatic clearance (Eq. 13) and unbound drug concentrations in the hepatocytes. Compartment-model-based analysis has been further extended to the mechanistic assessment of Michelis–Menten parameters (Km and Vmax) responsible for the transporter-mediated hepatic uptake, bidirectional passive diffusion, and nonspecific binding which directly reflect those obtained from in vitro experiments (140). These mechanistic approaches can provide an integrated tool not only for the accurate predictions of pharmacokinetics but also for the predictions of potential drug–drug interactions by transporter(s) and/or CYP(s) and of efficacy by liver-targeted drug candidates.
Fig. 8.

Ratio of intrinsic clearance for unbound drugs calculated from plasma clearance in rat (CLint, ub, in vivo) to that determined in the incubation with isolated rat hepatocytes (CLint, ub, in vitro). Calculations of in vitro intrinsic clearance (CLint, ub, in vitro) were based on (1) the initial disappearance rate in whole suspension [conventional method] (filled circles, with average ratio = 57, dotted line), (2) the AUC in the medium (filled triangles, with average ratio = 16) and (3) the initial disappearance rate in the media (empty circles, with average ratio = 3, solid line) in the isolated rat hepatocytes. Data are taken from (135)
Table II.
Prediction of In Vivo Intrinsic Clearance for Unbound Drugs Using Human Hepatocytes Based on the Initial Disappearance Rate in “Media Loss” Assay (Data From (135))
| Compound | Mean CLint (μL/min/106 cells) | In vivo intrinsic clearance for unbound drug (mL/min per kilogram) | |
|---|---|---|---|
| Predicted | Observed | ||
| Montelukast | 360 ± 250 | 1,100 | 2,700 |
| Prazosin | 12 ± 1.2 | 36 | 50 |
| Pravastatin | 2.1 ± 1.4 | 6.6 | 23 |
| Atorvastatin | 100 ± 19 | 320 | 910 |
| Bosentan | 38 ± 6.5 | 120 | 340 |
Future prediction of hepatic and oral clearance in human judiciously incorporates both information on metabolic clearance and transporter-mediated membrane permeability of drug candidates into the prediction paradigm as described in Fig. 9. Potential contribution of transporter-mediated clearance to overall hepatic clearance would be identified by the evaluation of difference in the concentrations between whole cells and medium in the incubation with cryopreserved human hepatocytes (90,135). Following the identification of positive contribution, the compartment-model based analysis for the time-course of drug concentrations in the hepatocytes (139,140) provides parameters for each transporter-mediated kinetic process in hepatic clearance (Eq. 13). The integrated approach to the in vitro-to-in vivo extrapolation of hepatic clearance by combining in vitro metabolic and transporter-mediated clearance would allow the predictions of hepatic and oral clearance for the drug candidates undergoing kinetically significant transporter-mediated clearance process(es) in human.
Fig. 9.

Lead optimization for the successful development candidate by the predictions of hepatic and oral clearance in human. Future workflow for the incorporation of transporter-mediated hepatic uptake into the optimization strategy is shown by dotted lines
CONCLUSION
With recent advance in technology and continued accumulation of our knowledge for drug transporters (4,7), the identification of transporter(s) responsible for the pharmacokinetics of drug candidates has greatly facilitated both discovery and development of new drug candidates with higher POS from drug metabolism and pharmacokinetics point of view. For example, the transporter(s) involved in the hepatobiliary transport in human can be directly identified (141–143) by using the panels of double transfectant (or quadruple transfectant) such as OATP1B1/MRP2 (142,144), OATP1B1/MDR1 (142), OATP1B1/BCRP (142), OATP1B3/MRP2 (145,146), OATP1B1 OATP1B3 OATP2B1/MRP2 (147) and OCT1/MDR1 (148), which consist of the transporters for sinusoidal uptake/canalicular efflux on the basal/apical membranes in the polarized cell lines. Like the identification of CYP isoform(s) involved in the metabolism of the drug candidate, the information on the identified transporter(s) would be very useful to evaluate the liability of potential drug–drug interaction/genetic polymorphism and to assess the POS for liver-targeted candidates. However, unlike the metabolism study with recombinant CYP isoforms, the quantitative predictions of in vivo clearances from the transcellular transport data in the transporter(s)-transfected cell lines remain to be explored (95,149).
Cryopreserved human hepatocytes have been widely and routinely utilized as the most versatile in vitro system which maintains both metabolizing and transporting activities reasonably comparable with those in the freshly isolated human hepatocytes (27,36,40,42,44,45,47,48). Despite the fact that the values and advantages of hepatocytes for the prediction of hepatic clearance in human have been demonstrated as described herein, the limitation of capability for the quantitative prediction, especially characterized by a systematic underprediction of scaled-up hepatic clearance with a biological scaling factor (hepatocellularity), has emerged from different datasets as a significant concern. In order to mitigate the discrepancy between in vitro predication and in vivo observation, the empirical and regression-based scaling factors have been inevitably employed for the accurate predictions by human hepatocytes (27,34,35,43,45,79,135). The understanding of potential mechanism(s) underlying the systematic underprediction will improve our confidence of prediction by rationalizing the empirical strategy for the in vitro-to-in vivo extrapolation of data from human hepatocytes (40,90).
The hepatic clearance has been recognized as a hybrid parameter consisting of distinct clearance processes such as passive diffusion, hepatic (sinusoidal) uptake, efflux from cell to sinusoid, metabolism, biliary excretion, and (canalicular) transporter-mediated efflux into the bile. Although the equation for (overall) intrinsic clearance in the hepatic elimination (Eq. 13) can be simplified under certain circumstances (Eqs. 14 and 15), it is generally difficult to identify the rate-determining step in the hepatic disposition, especially when the information on relative magnitude of clearance representing each process is limited for the drug candidate of interest at discovery stage. The assay of both concentrations in the incubation medium and whole suspension of hepatocytes along with the incubation time can serve as one of the simple methods to identify the compounds having a significant contribution of hepatic uptake to the in vitro intrinsic clearance (135). The positive data from this assay will provide strong rationale for further studies on the identification of transporter(s) and the quantitative assessment of hepatic uptake process as a potential rate-determining step in hepatic clearance (139,140).
Future prediction of pharmacokinetics in human will increasingly rely on the systematic and integrated approaches to the hepatic and total-body clearance. Successful prediction paradigm will certainly need to incorporate the in vitro piece of quantitative information on physicochemical properties, kinetics of transporter-mediated (sinusoidal and canalicular) transports, and metabolic degradation in the liver as well as qualitative information on the identified transporter(s) and enzyme(s) responsible for the pharmacokinetics in human. Figure 9 shows lead optimization process for the successful development candidate by the predictions of hepatic and oral clearance in human described in this article. Future workflow is also extended by incorporating the evaluations of transporter-mediated hepatic clearance into the optimization strategy as shown by dotted lines. For metabolically promising NCEs, kinetically significant contribution of transporter-mediated clearance process(es) to overall hepatic clearance (Eq. 13) would be subsequently identified by in vitro experiments using cryopreserved human hepatocytes. The information on transporter(s) allows the assessment whether the transporter-mediated clearance would likely be liable for high hepatic clearance/interindividual variation/DDI and the contribution should be mitigated by structure modification; or the transporter-mediated clearance would likely be beneficial (for example, for liver-targeted drug candidates) and the discovery should proceed to further evaluations for the development. A judicious combination of quantitative and qualitative data from in vitro studies can allow the predictions for the potential effects of transporter-mediated and/or CYP-mediated drug–drug interactions and changes of pathophysiological conditions on the unbound drug concentration in the liver and target organ. It also allows a rational optimization of the pharmacokinetic properties of drug candidates, leading to the successful development of ideal drugs with fewer interindividual differences, fewer drug interactions, and more selective delivery to the target organ (7).
References
- 1.Muck W, Mai I, Fritsche L, Ochmann K, Rohde G, Unger S, et al. Increase in cerivastatin systemic exposure after single and multiple dosing in cyclosporine-treated kidney transplant recipients. Clin Pharmacol Ther. 1999;65:251–61. doi: 10.1016/S0009-9236(99)70104-9. [DOI] [PubMed] [Google Scholar]
- 2.Shitara Y, Itoh T, Sato H, Li AP, Sugiyama Y. Inhibition of transporter-mediated hepatic uptake as a mechanism for drug-drug interaction between cerivastatin and cyclosporin A. J Pharmacol Exp Ther. 2003;304:610–6. doi: 10.1124/jpet.102.041921. [DOI] [PubMed] [Google Scholar]
- 3.Kajosaari LI, Niemi M, Neuvonen M, Laitila J, Neuvonen PJ, Backman JT. Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin Pharmacol Ther. 2005;78:388–99. doi: 10.1016/j.clpt.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–46. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Sissung TM, Gardner ER, Gao R, Figg WD. Pharmacogenetics of membrane transporters: a review of current approaches. Methods Mol Biol. 2008;448:41–62. doi: 10.1007/978-1-59745-205-2_4. [DOI] [PubMed] [Google Scholar]
- 6.Mizuno N, Sugiyama Y. Drug transporters: their role and importance in the selection and development of new drugs. Drug Metab Pharmacokinet. 2002;17:93–108. doi: 10.2133/dmpk.17.93. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura S, Maeda K, Sugiyama Y. Recent progresses in the experimental methods and evaluation strategies of transporter functions for the prediction of the pharmacokinetics in humans. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:617–28. doi: 10.1007/s00210-008-0312-9. [DOI] [PubMed] [Google Scholar]
- 8.Maeda K, Suzuki H, Sugiyama Y. Hepatic transport. In: van de Waterbeemd H, Testa B, editors. Drug bioavailability. 2. Weinheim: Wiley; 2008. [Google Scholar]
- 9.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–79. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 10.Houston JB, Carlile DJ. Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab Rev. 1997;29:891–922. doi: 10.3109/03602539709002237. [DOI] [PubMed] [Google Scholar]
- 11.Carlile DJ, Stevens AJ, Ashforth EI, Waghela D, Houston JB. In vivo clearance of ethoxycoumarin and its prediction from in vitro systems. Use of drug depletion and metabolite formation methods in hepatic microsomes and isolated hepatocytes. Drug Metab Dispos. 1998;26:216–21. [PubMed] [Google Scholar]
- 12.Carlile DJ, Zomorodi K, Houston JB. Scaling factors to relate drug metabolic clearance in hepatic microsomes, isolated hepatocytes, and the intact liver: studies with induced livers involving diazepam. Drug Metab Dispos. 1997;25:903–11. [PubMed] [Google Scholar]
- 13.Worboys PD, Brennan B, Bradbury A, Houston JB. Metabolite kinetics of ondansetron in rat. Comparison of hepatic microsomes, isolated hepatocytes and liver slices, with in vivo disposition. Xenobiotica. 1996;26:897–907. doi: 10.3109/00498259609052492. [DOI] [PubMed] [Google Scholar]
- 14.Zomorodi K, Houston JB. Effect of omeprazole on diazepam disposition in the rat: in vitro and in vivo studies. Pharm Res. 1995;12:1642–6. doi: 10.1023/a:1016241000480. [DOI] [PubMed] [Google Scholar]
- 15.Zomorodi K, Carlile DJ, Houston JB. Kinetics of diazepam metabolism in rat hepatic microsomes and hepatocytes and their use in predicting in vivo hepatic clearance. Xenobiotica. 1995;25:907–16. doi: 10.3109/00498259509046662. [DOI] [PubMed] [Google Scholar]
- 16.Hayes KA, Brennan B, Chenery R, Houston JB. In vivo disposition of caffeine predicted from hepatic microsomal and hepatocyte data. Drug Metab Dispos. 1995;23:349–53. [PubMed] [Google Scholar]
- 17.Ashforth EI, Carlile DJ, Chenery R, Houston JB. Prediction of in vivo disposition from in vitro systems: clearance of phenytoin and tolbutamide using rat hepatic microsomal and hepatocyte data. J Pharmacol Exp Ther. 1995;274:761–6. [PubMed] [Google Scholar]
- 18.Obach RS, Baxter JG, Liston TE, Silber BM, Jones BC, MacIntyre F, et al. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J Pharmacol Exp Ther. 1997;283:46–58. [PubMed] [Google Scholar]
- 19.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–9. [PubMed] [Google Scholar]
- 20.Niro R, Byers JP, Fournier RL, Bachmann K. Application of a convective-dispersion model to predict in vivo hepatic clearance from in vitro measurements utilizing cryopreserved human hepatocytes. Curr Drug Metab. 2003;4:357–69. doi: 10.2174/1389200033489334. [DOI] [PubMed] [Google Scholar]
- 21.Ito K, Houston JB. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res. 2004;21:785–92. doi: 10.1023/b:pham.0000026429.12114.7d. [DOI] [PubMed] [Google Scholar]
- 22.Ito K, Iwatsubo T, Kanamitsu S, Nakajima Y, Sugiyama Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu Rev Pharmacol Toxicol. 1998;38:461–99. doi: 10.1146/annurev.pharmtox.38.1.461. [DOI] [PubMed] [Google Scholar]
- 23.Iwatsubo T, Hirota N, Ooie T, Suzuki H, Sugiyama Y. Prediction of in vivo drug disposition from in vitro data based on physiological pharmacokinetics. Biopharm Drug Dispos. 1996;17:273–310. doi: 10.1002/(SICI)1099-081X(199605)17:4<273::AID-BDD961>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 24.Iwatsubo T, Hisaka A, Suzuki H, Sugiyama Y. Prediction of in vivo nonlinear first-pass hepatic metabolism of YM796 from in vitro metabolic data. J Pharmacol Exp Ther. 1998;286:122–7. [PubMed] [Google Scholar]
- 25.Iwatsubo T, Suzuki H, Shimada N, Chiba K, Ishizaki T, Green CE, et al. Prediction of in vivo hepatic metabolic clearance of YM796 from in vitro data by use of human liver microsomes and recombinant P-450 isozymes. J Pharmacol Exp Ther. 1997;282:909–19. [PubMed] [Google Scholar]
- 26.Iwatsubo T, Hirota N, Ooie T, Suzuki H, Shimada N, Chiba K, et al. Prediction of in vivo drug metabolism in the human liver from in vitro metabolism data. Pharmacol Ther. 1997;73:147–71. doi: 10.1016/s0163-7258(96)00184-2. [DOI] [PubMed] [Google Scholar]
- 27.Shibata Y, Takahashi H, Chiba M, Ishii Y. Prediction of hepatic clearance and availability by cryopreserved human hepatocytes: an application of serum incubation method. Drug Metab Dispos. 2002;30:892–6. doi: 10.1124/dmd.30.8.892. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy T. Managing the drug discovery/development interface. Drug Discov Today. 1997;2:436–44. [Google Scholar]
- 29.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates. Nat Rev Drug Discov. 2004;3:711–5. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 30.Mistry M, Houston JB. Glucuronidation in vitro and in vivo. Comparison of intestinal and hepatic conjugation of morphine, naloxone, and buprenorphine. Drug Metab Dispos. 1987;15:710–7. [PubMed] [Google Scholar]
- 31.Soars MG, Burchell B, Riley RJ. In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther. 2002;301:382–90. doi: 10.1124/jpet.301.1.382. [DOI] [PubMed] [Google Scholar]
- 32.Boase S, Miners JO. In vitro–in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol. 2002;54:493–503. doi: 10.1046/j.1365-2125.2002.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowland A, Gaganis P, Elliot DJ, Mackenzie PI, Knights KM, Miners JO. Binding of inhibitory fatty acids is responsible for the enhancement of UDP-glucuronosyltransferase 2B7 activity by albumin: implications for in vitro–in vivo extrapolation. J Pharmacol Exp Ther. 2007;321:137–47. doi: 10.1124/jpet.106.118216. [DOI] [PubMed] [Google Scholar]
- 34.Grime K, Riley RJ. The impact of in vitro binding on in vitro–in vivo extrapolations, projections of metabolic clearance and clinical drug–drug interactions. Curr Drug Metab. 2006;7:251–64. doi: 10.2174/138920006776359266. [DOI] [PubMed] [Google Scholar]
- 35.Riley RJ, McGinnity DF, Austin RP. A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab Dispos. 2005;33:1304–11. doi: 10.1124/dmd.105.004259. [DOI] [PubMed] [Google Scholar]
- 36.Li AP. Human hepatocytes: isolation, cryopreservation and applications in drug development. Chem Biol Interact. 2007;168:16–29. doi: 10.1016/j.cbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Li AP, Lu C, Brent JA, Pham C, Fackett A, Ruegg CE, et al. Cryopreserved human hepatocytes: characterization of drug-metabolizing enzyme activities and applications in higher throughput screening assays for hepatotoxicity, metabolic stability, and drug–drug interaction potential. Chem Biol Interact. 1999;121:17–35. doi: 10.1016/s0009-2797(99)00088-5. [DOI] [PubMed] [Google Scholar]
- 38.Li AP. Overview: hepatocytes and cryopreservation—a personal historical perspective. Chem Biol Interact. 1999;121:1–5. doi: 10.1016/s0009-2797(99)00086-1. [DOI] [PubMed] [Google Scholar]
- 39.Li AP, Gorycki PD, Hengstler JG, Kedderis GL, Koebe HG, Rahmani R, et al. Present status of the application of cryopreserved hepatocytes in the evaluation of xenobiotics: consensus of an international expert panel. Chem Biol Interact. 1999;121:117–23. doi: 10.1016/s0009-2797(99)00081-2. [DOI] [PubMed] [Google Scholar]
- 40.Soars MG, McGinnity DF, Grime K, Riley RJ. The pivotal role of hepatocytes in drug discovery. Chem Biol Interact. 2007;168:2–15. doi: 10.1016/j.cbi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 41.Hallifax D, Galetin A, Houston JB. Prediction of metabolic clearance using fresh human hepatocytes: comparison with cryopreserved hepatocytes and hepatic microsomes for five benzodiazepines. Xenobiotica. 2008;38:353–67. doi: 10.1080/00498250701834665. [DOI] [PubMed] [Google Scholar]
- 42.McGinnity DF, Soars MG, Urbanowicz RA, Riley RJ. Evaluation of fresh and cryopreserved hepatocytes as in vitro drug metabolism tools for the prediction of metabolic clearance. Drug Metab Dispos. 2004;32:1247–53. doi: 10.1124/dmd.104.000026. [DOI] [PubMed] [Google Scholar]
- 43.Ito K, Houston JB. Prediction of human drug clearance from in vitro and preclinical data using physiologically based and empirical approaches. Pharm Res. 2005;22:103–12. doi: 10.1007/s11095-004-9015-1. [DOI] [PubMed] [Google Scholar]
- 44.Brown HS, Griffin M, Houston JB. Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab Dispos. 2007;35:293–301. doi: 10.1124/dmd.106.011569. [DOI] [PubMed] [Google Scholar]
- 45.Hallifax D, Rawden HC, Hakooz N, Houston JB. Prediction of metabolic clearance using cryopreserved human hepatocytes: kinetic characteristics for five benzodiazepines. Drug Metab Dispos. 2005;33:1852–8. doi: 10.1124/dmd.105.005389. [DOI] [PubMed] [Google Scholar]
- 46.Rawden HC, Carlile DJ, Tindall A, Hallifax D, Galetin A, Ito K, et al. Microsomal prediction of in vivo clearance and associated interindividual variability of six benzodiazepines in humans. Xenobiotica. 2005;35:603–25. doi: 10.1080/00498250500162870. [DOI] [PubMed] [Google Scholar]
- 47.Blanchard N, Hewitt NJ, Silber P, Jones H, Coassolo P, Lave T. Prediction of hepatic clearance using cryopreserved human hepatocytes: a comparison of serum and serum-free incubations. J Pharm Pharmacol. 2006;58:633–41. doi: 10.1211/jpp.58.5.0008. [DOI] [PubMed] [Google Scholar]
- 48.Jouin D, Blanchard N, Alexandre E, Delobel F, David-Pierson P, Lave T, et al. Cryopreserved human hepatocytes in suspension are a convenient high throughput tool for the prediction of metabolic clearance. Eur J Pharm Biopharm. 2006;63:347–55. doi: 10.1016/j.ejpb.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 49.Blanchard N, Richert L, Notter B, Delobel F, David P, Coassolo P, et al. Impact of serum on clearance predictions obtained from suspensions and primary cultures of rat hepatocytes. Eur J Pharm Sci. 2004;23:189–99. doi: 10.1016/j.ejps.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 50.Blanchard N, Alexandre E, Abadie C, Lave T, Heyd B, Mantion G, et al. Comparison of clearance predictions using primary cultures and suspensions of human hepatocytes. Xenobiotica. 2005;35:1–15. doi: 10.1080/00498250400021820. [DOI] [PubMed] [Google Scholar]
- 51.Roberts MS, Rowland M. Correlation between in-vitro microsomal enzyme activity and whole organ hepatic elimination kinetics: analysis with a dispersion model. J Pharm Pharmacol. 1986;38:177–81. doi: 10.1111/j.2042-7158.1986.tb04540.x. [DOI] [PubMed] [Google Scholar]
- 52.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 53.Sawada Y, Hanano M, Sugiyama Y, Iga T. Prediction of the disposition of nine weakly acidic and six weakly basic drugs in humans from pharmacokinetic parameters in rats. J Pharmacokinet Biopharm. 1985;13:477–92. doi: 10.1007/BF01059331. [DOI] [PubMed] [Google Scholar]
- 54.Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm Res. 1998;15:1792–5. doi: 10.1023/a:1011981317451. [DOI] [PubMed] [Google Scholar]
- 55.Chiou WL, Jeong HY, Chung SM, Wu TC. Evaluation of using dog as an animal model to study the fraction of oral dose absorbed of 43 drugs in humans. Pharm Res. 2000;17:135–40. doi: 10.1023/a:1007552927404. [DOI] [PubMed] [Google Scholar]
- 56.Thummel KE, Shen DD, Podoll TD, Kunze KL, Trager WF, Bacchi CE, et al. Use of midazolam as a human cytochrome P450 3A probe: II. Characterization of inter- and intraindividual hepatic CYP3A variability after liver transplantation. J Pharmacol Exp Ther. 1994;271:557–66. [PubMed] [Google Scholar]
- 57.Thummel KE, Shen DD, Podoll TD, Kunze KL, Trager WF, Hartwell PS, et al. Use of midazolam as a human cytochrome P450 3A probe: I. In vitro–in vivo correlations in liver transplant patients. J Pharmacol Exp Ther. 1994;271:549–56. [PubMed] [Google Scholar]
- 58.Korashy HM, Elbekai RH, El Kadi AO. Effects of renal diseases on the regulation and expression of renal and hepatic drug-metabolizing enzymes: a review. Xenobiotica. 2004;34:1–29. doi: 10.1080/00498250310001638460. [DOI] [PubMed] [Google Scholar]
- 59.Krishna DR, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet. 1994;26:144–60. doi: 10.2165/00003088-199426020-00007. [DOI] [PubMed] [Google Scholar]
- 60.Kapitulnik J, Strobel HW. Extrahepatic drug metabolizing enzymes. J Biochem Mol Toxicol. 1999;13:227–30. doi: 10.1002/(sici)1099-0461(1999)13:5<227::aid-jbt1>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 61.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–73. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 62.Dolphin CT, Cullingford TE, Shephard EA, Smith RL, Phillips IR. Differential developmental and tissue-specific regulation of expression of the genes encoding three members of the flavin-containing monooxygenase family of man, FMO1, FMO3 and FM04. Eur J Biochem. 1996;235:683–9. doi: 10.1111/j.1432-1033.1996.00683.x. [DOI] [PubMed] [Google Scholar]
- 63.Yeung CK, Lang DH, Thummel KE, Rettie AE. Immunoquantitation of FMO1 in human liver, kidney, and intestine. Drug Metab Dispos. 2000;28:1107–11. [PubMed] [Google Scholar]
- 64.Satoh T, Taylor P, Bosron WF, Sanghani SP, Hosokawa M, La Du BN. Current progress on esterases: from molecular structure to function. Drug Metab Dispos. 2002;30:488–93. doi: 10.1124/dmd.30.5.488. [DOI] [PubMed] [Google Scholar]
- 65.Schwer H, Langmann T, Daig R, Becker A, Aslanidis C, Schmitz G. Molecular cloning and characterization of a novel putative carboxylesterase, present in human intestine and liver. Biochem Biophys Res Commun. 1997;233:117–20. doi: 10.1006/bbrc.1997.6413. [DOI] [PubMed] [Google Scholar]
- 66.Imai T. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab Pharmacokinet. 2006;21:173–85. doi: 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- 67.Imai T, Taketani M, Shii M, Hosokawa M, Chiba K. Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug Metab Dispos. 2006;34:1734–41. doi: 10.1124/dmd.106.009381. [DOI] [PubMed] [Google Scholar]
- 68.Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev. 2001;33:273–97. doi: 10.1081/dmr-120000653. [DOI] [PubMed] [Google Scholar]
- 69.Gillette JR, Pang KS. Theoretic aspects of pharmacokinetic drug interactions. Clin Pharmacol Ther. 1977;22:623–39. doi: 10.1002/cpt1977225part2623. [DOI] [PubMed] [Google Scholar]
- 70.Klippert PJ, Noordhoek J. Influence of administration route and blood sampling site on the area under the curve. Assessment of gut wall, liver, and lung metabolism from a physiological model. Drug Metab Dispos. 1983;11:62–6. [PubMed] [Google Scholar]
- 71.Lin JH, Chiba M, Baillie TA. Is the role of the small intestine in first-pass metabolism overemphasized. Pharmacol Rev. 1999;51:135–58. [PubMed] [Google Scholar]
- 72.Kato M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab Pharmacokinet. 2008;23:87–94. doi: 10.2133/dmpk.23.87. [DOI] [PubMed] [Google Scholar]
- 73.Kato M, Chiba K, Hisaka A, Ishigami M, Kayama M, Mizuno N, et al. The intestinal first-pass metabolism of substrates of CYP3A4 and P-glycoprotein-quantitative analysis based on information from the literature. Drug Metab Pharmacokinet. 2003;18:365–72. doi: 10.2133/dmpk.18.365. [DOI] [PubMed] [Google Scholar]
- 74.Wu CY, Benet LZ, Hebert MF, Gupta SK, Rowland M, Gomez DY, et al. Differentiation of absorption and first-pass gut and hepatic metabolism in humans: studies with cyclosporine. Clin Pharmacol Ther. 1995;58:492–7. doi: 10.1016/0009-9236(95)90168-X. [DOI] [PubMed] [Google Scholar]
- 75.Lampen A, Christians U, Guengerich FP, Watkins PB, Kolars JC, Bader A, et al. Metabolism of the immunosuppressant tacrolimus in the small intestine: cytochrome P450, drug interactions, and interindividual variability. Drug Metab Dispos. 1995;23:1315–24. [PubMed] [Google Scholar]
- 76.Paine MF, Shen DD, Kunze KL, Perkins JD, Marsh CL, McVicar JP, et al. First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 1996;60:14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- 77.Thummel KE, O’Shea D, Paine MF, Shen DD, Kunze KL, Perkins JD, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59:491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- 78.Wang SX, Sutfin TA, Baarnhielm C, Regardh CG. Contribution of the intestine to the first-pass metabolism of felodipine in the rat. J Pharmacol Exp Ther. 1989;250:632–6. [PubMed] [Google Scholar]
- 79.Naritomi Y, Terashita S, Kimura S, Suzuki A, Kagayama A, Sugiyama Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab Dispos. 2001;29:1316–24. [PubMed] [Google Scholar]
- 80.Naritomi Y, Terashita S, Kagayama A, Sugiyama Y. Utility of hepatocytes in predicting drug metabolism: comparison of hepatic intrinsic clearance in rats and humans in vivo and in vitro. Drug Metab Dispos. 2003;31:580–8. doi: 10.1124/dmd.31.5.580. [DOI] [PubMed] [Google Scholar]
- 81.Obach RS. The importance of nonspecific binding in in vitro matrices, its impact on enzyme kinetic studies of drug metabolism reactions, and implications for in vitro–in vivo correlations. Drug Metab Dispos. 1996;24:1047–9. [PubMed] [Google Scholar]
- 82.Obach RS. Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab Dispos. 1997;25:1359–69. [PubMed] [Google Scholar]
- 83.Pang KS, Rowland M. Hepatic clearance of drugs. I. Theoretical considerations of a “well-stirred” model and a “parallel tube” model. Influence of hepatic blood flow, plasma and blood cell binding, and the hepatocellular enzymatic activity on hepatic drug clearance. J Pharmacokinet Biopharm. 1977;5:625–53. doi: 10.1007/BF01059688. [DOI] [PubMed] [Google Scholar]
- 84.Roberts MS, Rowland M. A dispersion model of hepatic elimination. 1. Formulation of the model and bolus considerations. J Pharmacokinet Biopharm. 1986;14:227–60. doi: 10.1007/BF01106706. [DOI] [PubMed] [Google Scholar]
- 85.Ridgway D, Tuszynski JA, Tam YK. Reassessing models of hepatic extraction. J Biol Phys. 2003;29:1–21. doi: 10.1023/A:1022531403741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamamoto T, Suzuki A, Kohno Y, Nagata K, Yamazoe Y. Prediction of drug–drug interactions for AUCoral of high clearance drug from in vitro data: utilization of a microtiter plate assay and a dispersion model. Current Drug Metabolism. 2006;7:135–46. doi: 10.2174/138920006775541570. [DOI] [PubMed] [Google Scholar]
- 87.Yamamoto T, Itoga H, Kohno Y, Nagata K, Yamazoe Y. Prediction of oral clearance from in vitro metabolic data using recombinant CYPs: comparison among well-stirred, parallel-tube, distributed and dispersion models. Xenobiotica. 2005;35:627–46. doi: 10.1080/00498250500159371. [DOI] [PubMed] [Google Scholar]
- 88.Delannoy IAM, Pang KS. Effect of diffusional barriers on drug and metabolite kinetics. Drug Metab Dispos. 1987;15:51–8. [PubMed] [Google Scholar]
- 89.Liu LC, Pang KS. The roles of transporters and enzymes in hepatic drug processing. Drug Metab Dispos. 2005;33:1–9. doi: 10.1124/dmd.104.001149. [DOI] [PubMed] [Google Scholar]
- 90.Webborn PJ, Parker AJ, Denton RL, Riley RJ. In vitro–in vivo extrapolation of hepatic clearance involving active uptake: theoretical and experimental aspects. Xenobiotica. 2007;37:1090–109. doi: 10.3109/00498250701557266. [DOI] [PubMed] [Google Scholar]
- 91.Miyauchi S, Sawada Y, Iga T, Hanano M, Sugiyama Y. Dose-dependent hepatic handling of L-propranolol determined by multiple indicator dilution method—influence of tissue binding of L-propranolol on its hepatic elimination. Biol Pharm Bull. 1993;16:1019–24. doi: 10.1248/bpb.16.1019. [DOI] [PubMed] [Google Scholar]
- 92.Miyauchi S, Sugiyama Y, Sawada Y, Morita K, Iga T, Hanano M. Kinetics of hepatic transport of 4-methylumbelliferone in rats—analysis by multiple indicator dilution method. J Pharmacokinet Biopharm. 1987;15:25–38. doi: 10.1007/BF01062937. [DOI] [PubMed] [Google Scholar]
- 93.Baker M, Parton T. Kinetic determinants of hepatic clearance: plasma protein binding and hepatic uptake. Xenobiotica. 2007;37:1110–34. doi: 10.1080/00498250701658296. [DOI] [PubMed] [Google Scholar]
- 94.Weisiger RA. Dissociation from albumin—a potentially rate-limiting step in the clearance of substances by the liver. Proc Natl Acad Sci USA. 1985;82:1563–7. doi: 10.1073/pnas.82.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maeda K, Sugiyama Y. In vitro–in vivo scale-up of drug transport activities. In: You G, Morris ME, editors. Drug transporters. Hoboken: Wiley; 2007. pp. 557–8. [Google Scholar]
- 96.Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European– and African–Americans. J Biol Chem. 2001;276:35669–75. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- 97.Konig J, Seithel A, Gradhand U, Fromm MF. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch Pharmacol. 2006;372:432–43. doi: 10.1007/s00210-006-0040-y. [DOI] [PubMed] [Google Scholar]
- 98.Mwinyi J, Kopke K, Schaefer M, Roots I, Gerloff T. Comparison of SLCO1B1 sequence variability among German, Turkish, and African populations. Eur J Clin Pharmacol. 2008;64:257–66. doi: 10.1007/s00228-007-0409-y. [DOI] [PubMed] [Google Scholar]
- 99.Pasanen MK, Neuvonen PJ, Niemi M. Global analysis of genetic variation in SLCO1B1. Pharmacogenomics. 2008;9:19–33. doi: 10.2217/14622416.9.1.19. [DOI] [PubMed] [Google Scholar]
- 100.Xu LY, He YJ, Zhang W, Deng S, Li Q, Zhang WX, Liu ZQ, Wang D, Huang YF, Zhou HH, Sun ZQ. Organic anion transporting polypeptide-1B1 haplotypes in Chinese patients. Acta Pharmacol Sin. 2007;28:1693–7. doi: 10.1111/j.1745-7254.2007.00643.x. [DOI] [PubMed] [Google Scholar]
- 101.Niemi M, Kivisto KT, Hofmann U, Schwab M, Eichelbaum M, Fromm MF. Fexofenadine pharmacokinetics are associated with a polymorphism of the SLCO1B1 gene (encoding OATP1B1) Br J Clin Pharmacol. 2005;59:602–4. doi: 10.1111/j.1365-2125.2005.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chung JY, Cho JY, Yu KS, Kim JR, Oh DS, Jung HR, et al. Effect of OATP1B1 (SLCO1B1) variant alleles on the pharmacokinetics of pitavastatin in healthy volunteers. Clin Pharmacol Ther. 2005;78:342–50. doi: 10.1016/j.clpt.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 103.Nishizato Y, Ieiri I, Suzuki H, Kimura M, Kawabata K, Hirota T, et al. Polymorphisms of OATP-C (SLC21A6) and OAT3 (SLC22A8) genes: consequences for pravastatin pharmacokinetics. Clin Pharmacol Ther. 2003;73:554–65. doi: 10.1016/S0009-9236(03)00060-2. [DOI] [PubMed] [Google Scholar]
- 104.Kivisto KT, Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm Res. 2007;24:239–47. doi: 10.1007/s11095-006-9159-2. [DOI] [PubMed] [Google Scholar]
- 105.Niemi M, Neuvonen PJ, Hofmann U, Backman JT, Schwab M, Lutjohann D, et al. Acute effects of pravastatin on cholesterol synthesis are associated with SLCO1B1 (encoding OATP1B1) haplotype *17. Pharmacogenet Genomics. 2005;15:303–9. doi: 10.1097/01213011-200505000-00005. [DOI] [PubMed] [Google Scholar]
- 106.Tachibana-Iimori R, Tabara Y, Kusuhara H, Kohara K, Kawamoto R, Nakura J, et al. Effect of genetic polymorphism of OATP-C (SLCO1B1) on lipid-lowering response to HMG-CoA reductase inhibitors. Drug Metab Pharmacokinet. 2004;19:375–80. doi: 10.2133/dmpk.19.375. [DOI] [PubMed] [Google Scholar]
- 107.Mwinyi J, Johne A, Bauer S, Roots I, Gerloff T. Evidence for inverse effects of OATP-C (SLC21A6) 5 and 1b haplotypes on pravastatin kinetics. Clin Pharmacol Ther. 2004;75:415–21. doi: 10.1016/j.clpt.2003.12.016. [DOI] [PubMed] [Google Scholar]
- 108.Niemi M, Schaeffeler E, Lang T, Fromm MF, Neuvonen M, Kyrklund C, et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1) Pharmacogenetics. 2004;14:429–40. doi: 10.1097/01.fpc.0000114750.08559.32. [DOI] [PubMed] [Google Scholar]
- 109.Maeda K, Ieiri I, Yasuda K, Fujino A, Fujiwara H, Otsubo K, et al. Effects of organic anion transporting polypeptide 1B1 haplotype on pharmacokinetics of pravastatin, valsartan, and temocapril. Clin Pharmacol Ther. 2006;79:427–39. doi: 10.1016/j.clpt.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 110.Zhang W, Chen BL, Ozdemir V, He YJ, Zhou G, Peng DD, et al. SLCO1B1 521T–>C functional genetic polymorphism and lipid-lowering efficacy of multiple-dose pravastatin in Chinese coronary heart disease patients. Br J Clin Pharmacol. 2007;64:346–52. doi: 10.1111/j.1365-2125.2007.02892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niemi M, Backman JT, Kajosaari LI, Leathart JB, Neuvonen M, Daly AK, et al. Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin Pharmacol Ther. 2005;77:468–78. doi: 10.1016/j.clpt.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 112.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics and pharmacodynamics of repaglinide and nateglinide. J Clin Pharmacol. 2008;48:311–21. doi: 10.1177/0091270007311569. [DOI] [PubMed] [Google Scholar]
- 113.Choi JH, Lee MG, Cho JY, Lee JE, Kim KH, Park K. Influence of OATP1B1 genotype on the pharmacokinetics of rosuvastatin in Koreans. Clin Pharmacol Ther. 2008;83:251–7. doi: 10.1038/sj.clpt.6100267. [DOI] [PubMed] [Google Scholar]
- 114.Crespi CL. Xenobiotic-metabolizing human cells as tools for pharmacological and toxicological research. Adv Drug Res. 1995;26:179–235. [Google Scholar]
- 115.Nakajima M, Nakamura S, Tokudome S, Shimada N, Yamazaki H, Yokoi T. Azelastine N-demethylation by cytochrome P-450 (CYP)3A4, CYP2D6, and CYP1A2 in human liver microsomes: evaluation of approach to predict the contribution of multiple CYPs. Drug Metab Dispos. 1999;27:1381–91. [PubMed] [Google Scholar]
- 116.Soars MG, Gelboin HV, Krausz KW, Riley RJ. A comparison of relative abundance, activity factor and inhibitory monoclonal antibody approaches in the characterization of human CYP enzymology. Br J Clin Pharmacol. 2003;55:175–81. doi: 10.1046/j.1365-2125.2003.01721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Venkatakrishnan K, von Moltke LL, Court MH, Harmatz JS, Crespi CL, Greenblatt DJ. Comparison between cytochrome P450 (CYP) content and relative activity approaches to scaling from cDNA-expressed CYPs to human liver microsomes: ratios of accessory proteins as sources of discrepancies between the approaches. Drug Metab Dispos. 2000;28:1493–504. [PubMed] [Google Scholar]
- 118.Stormer E, von Moltke LL, Greenblatt DJ. Scaling drug biotransformation data from cDNA-expressed cytochrome P-450 to human liver: a comparison of relative activity factors and human liver abundance in studies of mirtazapine metabolism. J Pharmacol Exp Ther. 2000;295:793–801. [PubMed] [Google Scholar]
- 119.Yamamoto T, Hagima N, Nakamura M, Kohno Y, Nagata K, Yamzoe Y. Prediction of differences in in vivo oral clearance of N,N-dipropyl-2-[4-methoxy-3-(2-phenylethoxy)phenyl]ethylamine monohydrochloride (NE-100) between extensive and poor metabolizers from in vitro metabolic data in human liver microsomes lacking CYP2D6 activity and recombinant CYPs. Xenobiotica. 2004;34:687–703. doi: 10.1080/00498250412331281070. [DOI] [PubMed] [Google Scholar]
- 120.Galetin A, Brown C, Hallifax D, Ito K, Houston JB. Utility of recombinant enzyme kinetics in prediction of human clearance: impact of variability, CYP3A5, and CYP2C19 on CYP3A4 probe substrates. Drug Metab Dispos. 2004;32:1411–20. doi: 10.1124/dmd.104.000844. [DOI] [PubMed] [Google Scholar]
- 121.Proctor NJ, Tucker GT, Rostami-Hodjegan A. Predicting drug clearance from recombinantly expressed CYPs: intersystem extrapolation factors. Xenobiotica. 2004;34:151–78. doi: 10.1080/00498250310001646353. [DOI] [PubMed] [Google Scholar]
- 122.Yamamoto T, Suzuki A, Kohno Y. High-throughput screening to estimate single or multiple enzymes involved in drug metabolism: microtitre plate assay using a combination of recombinant CYP2D6 and human liver microsomes. Xenobiotica. 2003;33:823–39. doi: 10.1080/0049825031000140887. [DOI] [PubMed] [Google Scholar]
- 123.Emoto C, Murase S, Iwasaki K. Approach to the prediction of the contribution of major cytochrome P450 enzymes to drug metabolism in the early drug-discovery stage. Xenobiotica. 2006;36:671–83. doi: 10.1080/00498250600709778. [DOI] [PubMed] [Google Scholar]
- 124.Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y. Contribution of sodium taurocholate co-transporting polypeptide to the uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther. 1998;286:1043–50. [PubMed] [Google Scholar]
- 125.Kouzuki H, Suzuki H, Ito K, Ohashi R, Sugiyama Y. Contribution of organic anion transporting polypeptide to uptake of its possible substrates into rat hepatocytes. J Pharmacol Exp Ther. 1999;288:627–34. [PubMed] [Google Scholar]
- 126.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311:139–46. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 127.Shimizu M, Fuse K, Okudaira K, Nishigaki R, Maeda K, Kusuhara H, et al. Contribution of OATP (organic anion-transporting polypeptide) family transporters to the hepatic uptake of fexofenadine in humans. Drug Metab Dispos. 2005;33:1477–81. doi: 10.1124/dmd.105.004622. [DOI] [PubMed] [Google Scholar]
- 128.Shitara Y, Li AP, Kato Y, Lu C, Ito K, Itoh T, et al. Function of uptake transporters for taurocholate and estradiol 17beta-d-glucuronide in cryopreserved human hepatocytes. Drug Metab Pharmacokinet. 2003;18:33–41. doi: 10.2133/dmpk.18.33. [DOI] [PubMed] [Google Scholar]
- 129.Bi YA, Kazolias D, Duignan DB. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab Dispos. 2006;34:1658–65. doi: 10.1124/dmd.105.009118. [DOI] [PubMed] [Google Scholar]
- 130.Lu C, Li P, Gallegos R, Uttamsingh V, Xia CQ, Miwa GT, et al. Comparison of intrinsic clearance in liver microsomes and hepatocytes from rats and humans: evaluation of free fraction and uptake in hepatocytes. Drug Metab Dispos. 2006;34:1600–5. doi: 10.1124/dmd.106.010793. [DOI] [PubMed] [Google Scholar]
- 131.Iwamoto K, Eaton DL, Klaassen CD. Uptake of morphine and nalorphine by isolated rat hepatocytes. J Pharmacol Exp Ther. 1978;206:181–9. [PubMed] [Google Scholar]
- 132.Schwarz LR, Burr R, Schwenk M, Pfaff E, Greim H. Uptake of taurocholic acid into isolated rat-liver cells. Eur J Biochem. 1975;55:617–23. doi: 10.1111/j.1432-1033.1975.tb02199.x. [DOI] [PubMed] [Google Scholar]
- 133.Yamazaki M, Akiyama S, Nishigaki R, Sugiyama Y. Uptake is the rate-limiting step in the overall hepatic elimination of pravastatin at steady-state in rats. Pharm Res. 1996;13:1559–64. doi: 10.1023/a:1016044032571. [DOI] [PubMed] [Google Scholar]
- 134.Olinga P, Merema M, Hof IH, Slooff MJ, Proost JH, Meijer DK, et al. Characterization of the uptake of rocuronium and digoxin in human hepatocytes: carrier specificity and comparison with in vivo data. J Pharmacol Exp Ther. 1998;285:506–10. [PubMed] [Google Scholar]
- 135.Soars MG, Grime K, Sproston JL, Webborn PJ, Riley RJ. Use of hepatocytes to assess the contribution of hepatic uptake to clearance in vivo. Drug Metab Dispos. 2007;35:859–65. doi: 10.1124/dmd.106.014464. [DOI] [PubMed] [Google Scholar]
- 136.Jones HM, Houston JB. Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab Dispos. 2004;32:973–82. doi: 10.1124/dmd.104.000125. [DOI] [PubMed] [Google Scholar]
- 137.Steinberg P, Fischer T, Kiulies S, Biefang K, Platt KL, Oesch F, et al. Drug metabolizing capacity of cryopreserved human, rat, and mouse liver parenchymal cells in suspension. Drug Metab Dispos. 1999;27:1415–22. [PubMed] [Google Scholar]
- 138.Reinoso RF, Telfer BA, Brennan BS, Rowland M. Uptake of teicoplanin by isolated rat hepatocytes: comparison with in vivo hepatic distribution. Drug Metab Dispos. 2001;29:453–9. [PubMed] [Google Scholar]
- 139.Paine SW, Parker AJ, Gardiner P, Webborn PJ, Riley RJ. Prediction of the pharmacokinetics of atorvastatin, cerivastatin, and indomethacin using kinetic models applied to isolated rat hepatocytes. Drug Metab Dispos. 2008;36:1365–74. doi: 10.1124/dmd.107.019455. [DOI] [PubMed] [Google Scholar]
- 140.Poirier A, Lave T, Portmann R, Brun ME, Senner F, Kansy M, et al. Design, data analysis, and simulation of in vitro drug transport kinetic experiments using a mechanistic in vitro model. Drug Metab Dispos. 2008;36:2434–44. doi: 10.1124/dmd.108.020750. [DOI] [PubMed] [Google Scholar]
- 141.Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y. Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol. 2005;68:800–7. doi: 10.1124/mol.105.014019. [DOI] [PubMed] [Google Scholar]
- 142.Matsushima S, Maeda K, Kondo C, Hirano M, Sasaki M, Suzuki H, et al. Identification of the hepatic efflux transporters of organic anions using double-transfected Madin-Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistance-associated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast cancer resistance protein. J Pharmacol Exp Ther. 2005;314:1059–67. doi: 10.1124/jpet.105.085589. [DOI] [PubMed] [Google Scholar]
- 143.Yamashiro W, Maeda K, Hirouchi M, Adachi Y, Hu Z, Sugiyama Y. Involvement of transporters in the hepatic uptake and biliary excretion of valsartan, a selective antagonist of the angiotensin II AT1-receptor, in humans. Drug Metab Dispos. 2006;34:1247–54. doi: 10.1124/dmd.105.008938. [DOI] [PubMed] [Google Scholar]
- 144.Sasaki M, Suzuki H, Ito K, Abe T, Sugiyama Y. Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/SLC21A6) and multidrug resistance-associated protein 2 (MRP2/ABCC2) J Biol Chem. 2002;277:6497–503. doi: 10.1074/jbc.M109081200. [DOI] [PubMed] [Google Scholar]
- 145.Cui Y, Konig J, Keppler D. Vectorial transport by double-transfected cells expressing the human uptake transporter SLC21A8 and the apical export pump ABCC2. Mol Pharmacol. 2001;60:934–43. doi: 10.1124/mol.60.5.934. [DOI] [PubMed] [Google Scholar]
- 146.Letschert K, Komatsu M, Hummel-Eisenbeiss J, Keppler D. Vectorial transport of the peptide CCK-8 by double-transfected MDCKII cells stably expressing the organic anion transporter OATP1B3 (OATP8) and the export pump ABCC2. J Pharmacol Exp Ther. 2005;313:549–56. doi: 10.1124/jpet.104.081224. [DOI] [PubMed] [Google Scholar]
- 147.Kopplow K, Letschert K, Konig J, Walter B, Keppler D. Human hepatobiliary transport of organic anions analyzed by quadruple-transfected cells. Mol Pharmacol. 2005;68:1031–8. doi: 10.1124/mol.105.014605. [DOI] [PubMed] [Google Scholar]
- 148.Nies AT, Herrmann E, Brom M, Keppler D. Vectorial transport of the plant alkaloid berberine by double-transfected cells expressing the human organic cation transporter 1 (OCT1, SLC22A1) and the efflux pump MDR1 P-glycoprotein (ABCB1) Naunyn Schmiedebergs Arch Pharmacol. 2008;376:449–61. doi: 10.1007/s00210-007-0219-x. [DOI] [PubMed] [Google Scholar]
- 149.Sasaki M, Suzuki H, Aoki J, Ito K, Meier PJ, Sugiyama Y. Prediction of in vivo biliary clearance from the in vitro transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II monolayer expressing both rat organic anion transporting polypeptide 4 and multidrug resistance associated protein 2. Mol Pharmacol. 2004;66:450–9. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]